Thermosensitive Self-Assembling Block Copolymers as Drug Delivery Systems



 and
and
Abstract
: Self-assembling block copolymers (poloxamers, PEG/PLA and PEG/PLGA diblock and triblock copolymers, PEG/polycaprolactone, polyether modified poly(Acrylic Acid)) with large solubility difference between hydrophilic and hydrophobic moieties have the property of forming temperature dependent micellar aggregates and, after a further temperature increase, of gellifying due to micelle aggregation or packing. This property enables drugs to be mixed in the sol state at room temperature then the solution can be injected into a target tissue, forming a gel depot in-situ at body temperature with the goal of providing drug release control. The presence of micellar structures that give rise to thermoreversible gels, characterized by low toxicity and mucomimetic properties, makes this delivery system capable of solubilizing water-insoluble or poorly soluble drugs and of protecting labile molecules such as proteins and peptide drugs.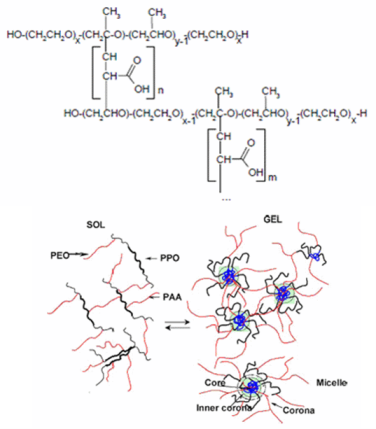
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Amphiphilic block copolymers (AB or ABA-type) with large solubility differences between hydrophilic and hydrophobic moieties, in aqueous medium are able to self-assemble into polymeric micelles characterized by mesoscopic size range. These structures consist of water-insoluble cores and water-soluble shells. Depending on blocks length, the core can assemble into various supramolecular structures characterized by different morphologies [1-3].
In fact, unlike homopolymers made of identical monomeric units, copolymers include two kinds of monomeric units of different solubility. Thus, in solution and at low concentration, these amphiphilic molecules exist as unimers, while at increasing concentrations, aggregation takes place. The final aggregates (called micelles), characterized by spherical shape when the hydrophilic segment is longer than the core block [4], usually include several dozen of this kind of unit. Thus it is possible to define either the Critical Micelle Concentration (CMC) as the concentration of a monomeric amphiphile at which micelles are formed; or the aggregation number as the number of individual molecules forming one micelle. Whereas, for block co-polymers the amphiphile solvation is strongly dependent on temperature, another important parameter to take into account is the Critical Micellization Temperature (CMT), below which the amphiphile exists as unimer, and above which, unimers and aggregates coexist.
Recent studies have focused on thermogelling polymeric micelles made of self-assembling block copolymers such as poloxamers, multiblock copolymers prepared from poly(lactide), polycaprolactone, poly(glycolic acid), and polyether modified poly(acrylic acid) [5-7], These polymers have the ability to form temperature dependent micellar aggregates and, after a further temperature increase, gels due to micelles aggregation or packing. Therefore, with these polymers it is possible to mix drugs in the sol state and at room temperature and the solution can be injected into a target tissue. It forms an in-situ gel depot at body temperature and provides controlled drug release. A formulation which is in the form of injectable liquid at room temperature, but changes into gel at body temperature, with a pH close to neutrality and having a certain biocompatibility and biodegradability surely represents an ideal system.
In general, different kinds of synthetic copolymers are made of poly(ethylene oxide) as hydrophilic block, and of a wider range of hydrophobic blocks. The combination of different PEO-hydrophobic block have given rise to several micelle systems with differing physicochemical properties, such as loading capacity, morphology, biodistribution, size, release kinetics and specificity against different kind of drugs.
Sometimes, these colloidal vectors can act as solubilizing agents by means of their hydrophobic core, which behaves as reservoir where drug molecules can be incorporated by chemical, physical or electrostatic interactions depending on physicochemical properties [8].
Obviously, it is of utmost importance to predict gelation behavior after drug loading into a copolymer formulation. In fact, the micellization and gelation transitions could be influenced by the presence of hydrophobic drug solutes: the hydrophobicities of various molecules have been demonstrated to affect Poloxamer 407 phase behavior [9] and liquid to gel transition [10].
The growing interest in polymeric micelles is strictly related to their similarity with natural carriers as viruses and serum lipoproteins. In fact, their hydrophilic shell makes them not identifiable during blood circulation prolonging the in vivo circulation time [11], while their viral-like size (<100 nm) prevents their uptake by the reticuloendothelial system.
The aim of this review is to illustrate the relevance of different self assembling block copolymers used in the pharmaceutical field as drug targeting systems, by means of their chemical synthesis, description of their applications, together with some of the analitycal techniques used for physicochemical characterization.
2. Poloxamers
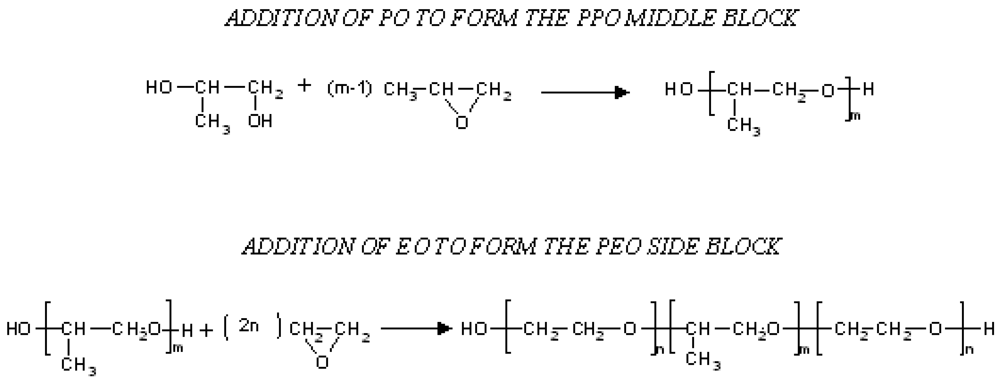
Poloxamers (synonyms Pluronics or Lutrol) are block copolymers of ethylene oxide (EO) and propylene oxide (PO) synthesized by sequential addition of propylene oxide first and then ethylene oxide to a low molecular weight water-soluble propylene glycol. The PEO-PPO-PEO block copolymers are available in a range of molecular weights and PPO/PEO ratios and thus, their physical and surface properties vary over a wide range, and different types (more than 30) are commercially available [12]. Their temperature-dependent self-assembling and thermogelling behavior is well known and, in water, Poloxamers have the ability to change from individual block copolymer molecules (unimers) to self-assembling micelles. For example in water below the CMC, unimers form dispersions while, above this concentration, aggregation phenomena occur giving rise to a process named “micellization”. At the same time, these block copolymers at a given concentration are characterized by specific temperature at which micelles form (CMT). Besides, at elevated temperatures, these systems show phase separation, which is due to the complete dehydration of both PO and EO blocks [13].
Different hypotheses on the mechanism of micelle formation may be proferred. The most reliable is that there is an initial stage of the phase separation with regions that are very rich in Poloxamer and regions that abound more in water. Then a further desolvation process of the PPO part leads to unimer aggregation and to a progressive formation of micelles close to each others, to form clusters. With regard to the sol/gel transition during gelation, partial collapse of PEO's chains in the micellar mantle leads to the formation of a tighter structure [12].
Micelles may have different shapes, namely spherical, cylindrical or lamellar, depending on the length of PO and EO blocks, and the core is always made of hydrophobic blocks, while hydrophilic blocks form external corona. An important property of these aggregates is their ability to incorporate hydrophobic substances being insoluble or poorly soluble in aqueous or hydrophilic environment, thus enhancing solubility. Nevertheless, it is necessary to consider that micellization is sensitive to the presence of water insoluble solutes which can promote a change from spherical to rod or lamellar shape affecting micelle size, aggregation number and different factors involved in drug delivery [14]. Another important characteristic of Poloxamers is their thermogelling behavior: in fact, water dispersions of these polymers are generally liquid at low temperature and form a strong gel at higher temperature, as reported by several authors [15-17]. Different hypothesis have been proposed in order to explain this phenomenon. The sol/gel transition have been correlated to intrinsic changes in micelles properties or to entropic variation in the ordered water molecules close to the PPO segments or to possible formation of a cross-linked and three-dimensional structure able to entrap water in its network [17-20]. Overall, both micellization and gelation depend on different factors, namely temperature, polymer concentration and PEO block length [21]. For example the Poloxamer 407 (branded Lutrol F127) with nominal molecular weight of 12,600 and a PEO/PPO ratio of 2:1 by weight is able to reach the maximum peak in viscosity in a temperature range of between 30° and 50 °C depending on polymer concentration [21].
2.1. Synthesis and Characterization
PEO-PPO-PEO triblock copolymers are synthesized by sequential addition of first propylene oxide (PO) and then ethylene oxide (EO) to a low molecular weight water-soluble propylene glycol. The oxyalkylation steps are carried out in the presence of an alkaline catalyst, generally sodium or potassium hydroxide. The catalyst is then neutralized and removed from the final product (Figure 1) [12].
Different analytical techniques have been utilized to characterize the self assembling behaviour of Poloxamer.
In a previous study, aggregation and phase behavior in water of several Poloxamers were investigated by means of techniques such as static and dynamic light scattering (DLS), small angle neutron scattering (SANS), polarization microscopy, atomic force microscopy (AFM), differential scanning calorimetry (DSC), and 1H- nuclear magnetic resonance (NMR). The sequence of phase behavior observed at increasing concentrations appeared analogous to that shown by normal surfactants, for example, isotropic solution, cubic phase, hexagonal phase and lamellar phase. At the same time, a significant difference compared to normal surfactant was detected, since the phase transition also outlined clearly thermotropic behavior. In fact, a sequence of different mesophases and phase transitions were detected at constant concentration but at increasing temperatures [22,23].
Other techniques were used in order to determine the hydrodynamic radius of micelles, such as pulsed gradient spin echo (PGSE) NMR and fluorescent spectroscopy [24,25].
As mentioned above, micellization phenomenon is related to dehydration of PPO chains, which drives unimer aggregation to form micelles. PPO is soluble in the temperature range of 2–15 °C, while for higher temperatures a precipitation cloud point exists [26]. Thus it is assumed that the hydrophobic PPO is located in the core of the micelle, while the PEO lyophilic block forms the outer corona, as assessed by electron paramagnetic resonance [27,28] (EPR) and the above mentioned techniques. With increasing temperature, micellization becomes more important, and at a definite temperature point, micelles come into contact and no longer move. In addition, the formation of highly ordered structures, such as cubic crystalline phase, has been proposed as the driving force for gel formation [29,30], but this hypothesis has been questioned recently. Thus, packing of micelles and micelle entanglements may be possible mechanisms for Poloxamer solutions gelation at increasing temperature [31].
A complete microstructural characterization of an aqueous solution of Poloxamer 407 has been obtained by combining different techniques such as cryogenic temperature transmission electron microscopy (Cryo-TEM) and small angle neutron scattering (SANS). These different methods allowed measurements of micelles diameters and visualization of their hydrophobic cores and demonstrated the existence of different lyotropic phases for this kind of system [32]. Another technique recently used to characterize Poloxamer 407′s aggregation behavior has been acoustic spectroscopy [33]. Particle size and microrheological extensional moduli (G′ and G″) of the systems were determined from acoustic parameters such as sound attenuation and sound speed. By monitoring particles size and rheological extensional moduli variations at increasing temperatures, it was possible to define and outline Poloxamer 407 transitions.
2.2. Pharmaceutical Application
Poloxamers are a class of surfactants used in different industrial areas such as detergency, foaming, lubrication, dispersion, stabilization, cosmetics, and inks [12]. Preliminary toxicity data indicated that this copolymer is well tolerated [34]. These results have prompted the use of Poloxamer 407 in the design of medical, pharmaceutical and cosmetic systems and therefore they are extensively used in the pharmaceutical field as gel, microemulsions, nanoparticles and solid polymer blends [35]. In particular, the thermogelling behavior and non-toxicity of the Poloxamers lead to different dosage forms being investigated. Early studies evaluated Poloxamer 407 thermosensitive solutions for the treatment of burns [34], topical administration of anticancer agents [36], rectal [37,38], vaginal [39], transdermal [40], and ophthalmic [41,42] administration and sustained delivery of drugs after extravascular parenteral injection [43].
However, the goal of this kind of copolymer is to administer its dispersion when in the sol state and use its ability to transform quickly into strong gels making the formulation of a parenteral delivery system possible. Concerning this route of administration, Poloxamer 407 is usually regarded as non-toxic. In fact after intramuscular injection in rabbits, Poloxamers 238 and 407 displayed musculo irritancy/toxicity comparable to that of traditional intramuscular vehicles, such as saline and peanut oil [44]. Thus, the lack of miotoxicity of these reversible thermal-gelling systems after intramuscular injections to rabbits recently promoted studies on the use of Poloxamer 407 water dispersions as vehicle for sustained release formulations containing proteins by extravascular administration [45,46]. In fact, after parenteral injection, Poloxamer gels can prolong drug release compared to solutions, even though the delivery period rarely exceeds a few days [47,48]. Obviously, drug release control is related to the final stiffness of the thermogel, which depends on Poloxamer 407′s concentration. However it is necessary to consider that increasing copolymer concentration could cause reduction of thermogelation temperature, which would make the system semisolid at room temperature and thus quite difficult to handle.
For these reasons Poloxamer gels have been considered for short-term therapies like pain management [49], infection treatment [48,50], and fertility control [51].
On the other hand, other studies [52] demonstrated that Poloxamer 407 injected intraperitoneally into rats (1.5 g/kg) resulted in sustained hypercholesterolemia and hypertriglyceridemia (96 h). The predominant mechanism for this effect could be attributed to the inhibition of heparin-releasable lipoprotein lipase [53].
Blonder et al. [54] studied how different doses of Poloxamer 407 gels could induce hyperlipidemia in rabbits. They found that the highest dose (137.5 mg/kg) significantly increased serum triglycerides and cholesterol in both male and female rabbits, while lower doses (5.5–27.5 mg/kg) did not alter serum lipids. Thus, the amount of administered polymer should be kept to a minimum, especially when repeated dosing is required.
Another important aspect is the possibility to solubilize hydrophobic drugs thanks to their amphiphilic nature. In a recent paper, Sezgin et al. [55] investigated the solubilization behavior of three poorly water soluble anticancer agents in some Poloxamers (F127, F68, P85) and asserted that these systems are effective new drug carriers, especially for anti-cancer molecules.
On the other hand Poloxamer gels can have potential disadvantages including their weak mechanical strength, rapid erosion (i.e., dissolution from the surface), and non-biodegradability, which prevents the use of high molecular weight polymers that cannot be eliminated by renal excretion [56].
3. PEG-PLA, PEG-PLGA Block Copolymers
A novel concept, which combines thermogelation, biodegradability, and no toxicity, has been proposed for injectable gel systems with improved safety and longer gel duration [57].
With this aim, new injectable biodegradable polymers possessing reverse gelation properties have been synthesized [58,59]. These polymers are triblock copolymers composed of A-blocks and B-blocks arranged as ABA or BAB, where A is polyethylene glycol (PEG) and B can be the poly(dl-lactide, PLA) or the poly(dl-lactide-co-glycolide, PLGA). PLA and PLGA are characterized by good degradability and biocompatibility. PEG presents unique physicochemical and biological properties including biocompatibility, low immunogenicity, water solubility, and can be eliminated from the animal's body when the molar mass is below 30,000 [60]. The final copolymers are soluble in water at or below room temperature but become hydrogels at the injection site, forming depots that degrade over a period of 4–6 weeks [61]. As the polymers are biodegradable, they overcome the problem of carrier removal after the drug depot is exhausted.
Concerning the PLA/PEG/PLA block copolymers, their composition, morphology and crystallinity strongly influence mechanical properties and rate of biodegradation. By adjusting the block copolymer segment sizes through the polymerization parameters, it is possible to modulate the materials characteristics in order to suit a particular application.
Mothé et al. described the preparation and the thermal properties evaluation of biodegradable poly(l-lactide)-b-poly(ethylene glycol)-b-poly(l-lactide) (PLA-PEG-PLA), prepared from two different PEG prepolymers (Mn = 4,000 or 600 g mol−1) and various PLA segment sizes. The PLA degree of polymerization varied from 1.2 up to 145.6. The prepared block copolymers presented wide range of molecular weights (800–25,000 g mol−1) and compositions (16–80 wt.% of PEG) [62].
Lee and coworkers [63] used a low molecular weight PEG (600 daltons) and the total molecular weight of PEG/PLA multiblock copolymer was varied to study the effect of this parameter on the sol-gel transition of the aqueous polymer solution. Then the molecular weight of poly(L-lactic acid) (PLA) was varied to see the effect of hydrophobicity of the PEG/PLA multiblock copolymer on the sol-gel transition.
Changing the polymer composition further, particularly the middle block composition, the block length, and the block ratio, produced the next generation of polyethylene glycol-b-L-lactic acid-coglycolide-b-ethylene glycol) (PEG-PLGA-PEG) triblock copolymers. The aqueous polymer solution is a free-flowing sol at room temperature and becomes a gel at body temperature [61].
The sol-to-gel transition of an aqueous solution of a PEG-PLGA-PEG triblock copolymer is probably due to micellar expansion which is accompanied by an increase in aggregation number driven by hydrophobic forces [64,65]. In fact, aqueous Poloxamer dispersions are known to undergo the sol-to-gel transition by a change in equilibrium from unimer to micelle reducing the number of unassociated polymers but with a constant aggregation number, whereas the PEG-PLGA-PEG triblock copolymer in water seems to undergo the sol-to-gel transition by micellar growth. Thus, Poloxamer 407 presents a large dependence of its CMC on temperature; while taking as an example the PEG-PLGA-PEG triblock copolymer aqueous solutions, two important issues were noted. First, the decrease in the second virial coefficient indicated that the polymer-polymer attraction increases relative to polymer-solvent interaction. Secondly, micelles grew by an increase in aggregation number, as well as an increase in the diameter of a micelle. Therefore, micellar growth and increase in polymer-polymer attraction could drive the sol to gel transition at a certain concentration, as the temperature increases.
The structure-property relationship in the sol-gel showed that the sol-gel transition temperature and critical gel concentration decreased with increasing hydrophobicity of the triblock copolymers [66].
For the PEG-PLGA-PEG triblock copolymers the sol-gel transition temperature can be controlled by changing molecular parameters such as the PLGA length, the PEG length or LA to GA ratio of the middle block. When the hydrophobic block (i.e., PLGA) length of a PEG-PLGA-PEG triblock copolymer was increased from 2,320 to 2,840 at a fixed PEG length of 550 the sol-to-gel transition temperature (ΔT∼ −10 °C) and the critical gel concentration (CGC) decreased, indicating that this transition was driven by hydrophobic forces. An increasing hydrophobic block length brought to an increase of gel-to-sol transition temperature. Thus, the gel region was controlled by varying the PLGA length.
On increasing PEG length of a PEG-PLGA-PEG triblock copolymer, from 550 to 780, at a fixed PLGA length of 2,300, the different regions of the phase diagrams were shifted to higher temperatures (ΔT∼ 18 °C). The gel region remained almost constant. Therefore the gel strength was mainly determined by the hydrophobic block [67]. The DL-lactic is more hydrophobic than glycolic acid in PLGA. Thus the hydrophobicity was increased by increasing DL-lactic acid to glycolic ratio, from 78:22 to 72:28, of PEG-PLGA-PEG (550-2,900-550) triblock then, the sol-to-gel transition temperature (ΔT ∼ −5°C) and CGC (ΔCGC∼ −5 wt%) decreased.
Furthermore, the gelation temperature of PEG-PLGA-PEG copolymers can be influenced by the presence of additives. The addition of a salting-out salt (1 wt%), such as NaCl, decreased the sol-gel transition temperature by 5 °C, whereas a salting-in salt, i.e., NaSCN, increased the sol-to-gel transition temperature by 5 °C.
Interestingly, a series of low-molecular-weight PLGA-PEG (MW 1,000 or 1,450)-PLGA (BAB type) block copolymers with different LA to GA (i.e., 1.5–4.0) and PEG to PLGA (i.e., 0.34–0.5) ratios also showed similar thermo-reversible sol-gel transitions to ABA-type blocks [68,69].
Gelation may occur by a different mechanism from that of ABA block copolymers because of the PLGA end blocks. The polymer forms micelles where PLGA block are in the core and PEG forms the curved shells, but may also give rise to bridging micelles with increasing concentration and temperature.
3.1. Synthesis and Characterization
Several methods are reported in the literature regarding synthesis of PLA-PEG or PLGA/PEG block copolymers.
In order to synthesize (AB)n type multiblock copolymers of PLLA and PEG, dicarboxylated PLLA was prepared by a direct condensation polymerization. A 50 g of L-lactic acid (85% solution in water) and a predetermined amount of succinic acid, used to introduce a dicarboxylated terminal group, were mixed in a round-bottom flask and then dehydrated at 30 °C under reduced pressure for 12 h. Polymerization was carried out at 150 °C for 24 h under reduced pressure. The polymer was recovered by dissolution of the product in chloroform and by precipitation in excess n-hexane and dried under vacuum for three days at 60 °C.
Dicarboxylated PLLA and PEG were poured into a round-bottom flask containing methylene chloride as solvent. PEG's of 2,000, 4,000, 6,000 and 10,000 number-average molecular weight were purified by precipitation in n-hexane. Dicyclohexylcarbodiimide (DCC) and N-dimethylaminopyridine (DMAP) were then added as polyesterification catalyst at the monomer to catalyst ratio of 0.25. Reaction was carried out at 25 °C for 24 h under nitrogen. Multiblock copolymers synthesized were recovered by precipitation of the reaction product, which contained dicyclohexylurea as a by-product, in excess n-hexane after filtration. Low molecular weight unreacted species were removed by fractionation method [70].
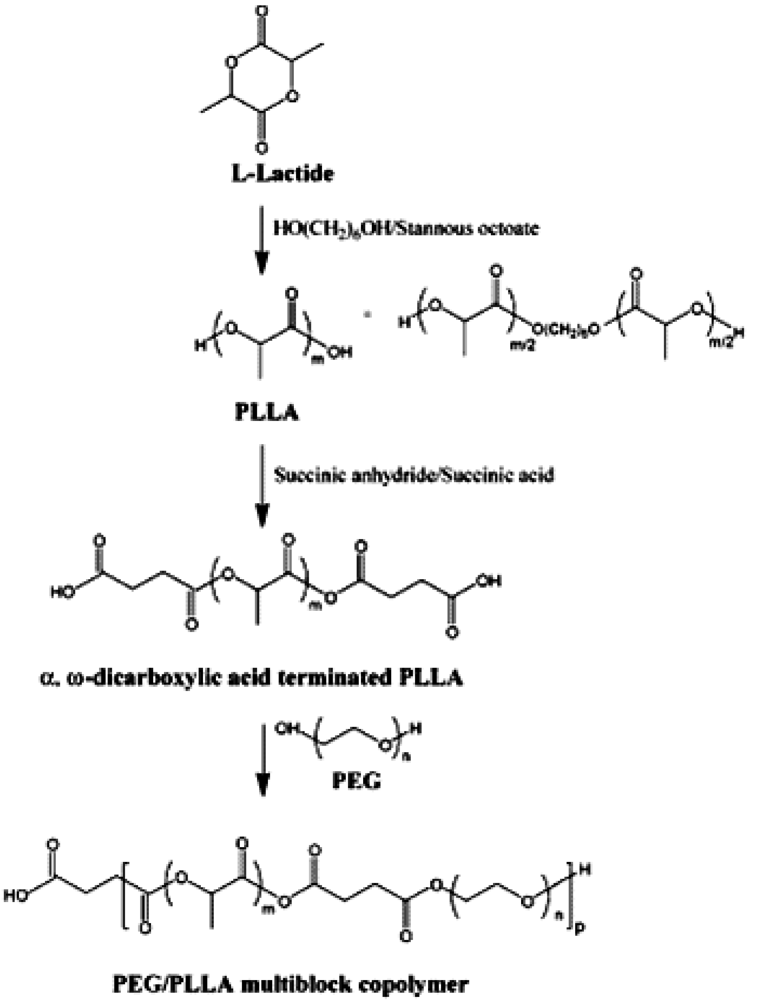
According to Lee's method [63] (Figure 2), the PLA was prepared by typical ring-opening polymerization of L-lactide using stannous octoate as catalyst. 1,6- Hexane diol was used as initiator. The product was isolated by precipitation into diethyl ether/n-hexane. The polymer was redissolved in 30 mL of methylene chloride and precipitated by slowly adding diethyl ether/n-hexane. The residual solvent was removed under vacuum. The succinic anhydride was reacted with PLA to prepare α,ω-dicarboxylic acid terminated PLLA (PLLA-DA) The PEG/PLLA multiblock copolymer was prepared by the coupling reaction between PEG and α,ω--dicarboxylic acid terminated PLA. To control the molecular weight of the PEG/PLLA multiblock copolymer, a little excess amount of the PEG was used.
Mothé et al. introduced in a glove box under dry nitrogen atmosphere, a pre-weighed amount of polyethylene glycol) (Mn = 4,000 or 600 g mol−1) and stannous 2-ethyl-hexanoate in a dry tube. The tube was sealed and immersed into silicon oil bath, at 120 °C for 10 min, and then cooled to room temperature. A pre-weighed amount of l,l-lactide (LA), was introduced into the tube under nitrogen atmosphere, and then sealed and immersed in a bath at 120 °C for 22 h. The copolymer thus obtained was purified by dissolution in chloroform and precipitation in methanol (three times). The prepared block copolymers presented wide range of molecular weights (800&ndash25,000 g mol−1) and compositions (16–80 wt.% of PEG) [62].
In different articles, Li and co-workers reported the synthesis, characterization, and stereocomplex-induced gelation of PLLA/PEG and PDLA/PEG block copolymers. The copolymers were synthesized by ring-opening polymerization of L- or D-lactide in the presence of mono- or dihydroxylated PEG, using zinc metal as catalyst [71,72].
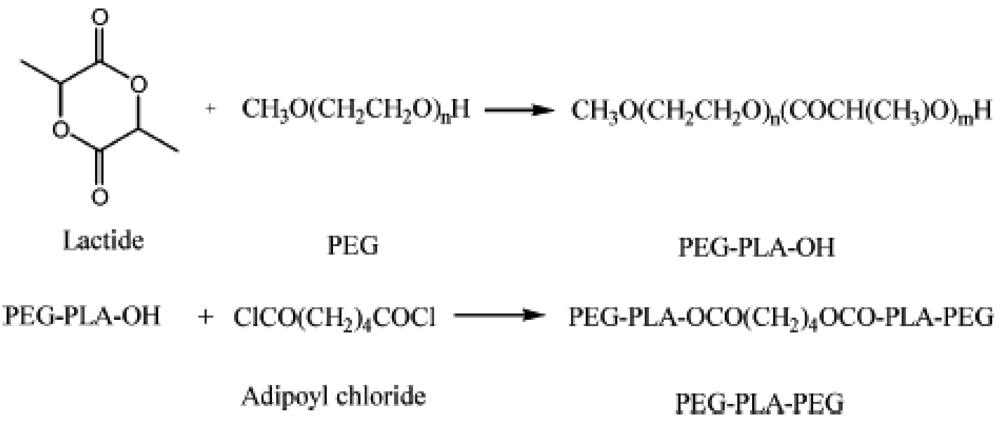
The same author carried out a ring-opening polymerization of D,L-lactide in the presence of monohydroxylated poly(ethyleneglycol) (PEG) with Mn of 2,000 and 5,000, using zinc powder as catalyst. The resulting PEG-b-polylactide (PEG-PLA) diblocks with various ethylene oxide/lactyl (EO/LA) ratios were coupled with adipoyl chloride to yield PEG-PLA-PEG triblock copolymers. N-Dimethylaminopyridine (DMAP) was used as catalyst. This reaction led to formation of an ester bond between PLA and PEG blocks (Figure 3) [71].
Zhang et al. synthesized PLA-PEG-PLA copolymers by ring opening polymerization of L- or d-lactide in the presence of dihydroxyl PEG using zinc lactate as catalyst. In fact zinc lactate was used as catalyst instead of stannous octoate or other catalysts which are rather cytotoxic. Predetermined amounts of PEG and lactide were introduced into a polymerization tube. The initial molar ratio of ethylene oxide to lactate repeat units (EO/LA) was 3/1 or 5/1. Zinc lactate (0.1 wt%) was then added. After degassing, the tube was sealed under vacuum, and polymerization was allowed to proceed at 140 °C. After 24 h, the product was recovered by dissolution in dichloromethane and precipitation in diethyl ether. Finally, the product was washed and dried under vacuum up to constant weight [73].
A poly(ethylene glycol)-poly(D,L-lactide) diblock copolymer (PEG-PLA) having a site specifically protected-sugar group at the PEG chain end was synthesized through a successive ring-opening polymerization of ethylene oxide and D,L-lactide using a methylated protected sugar as an initiator at room temperature under Argon atmosphere. Removal of protective groups from the sugar residue in the block copolymer was quantitatively carried out using 80% trifluoroacetic acid at room temperature, yielding a block copolymer having a glucose or galactose residue at the chain end in a regioselective manner [74].
Various techniques can be utilized to characterize the different diblock copolymers and their water dispersion.
The technique most frequently used to determine molecular weight and molecular weight distribution of synthesized polymers is Gel Permeation Chromatography (GPC) [63,65,67], while Cryo-Transmission Electron Microscopy (Cryo-TEM) [75,76], and dye solubilization method [63,67,75,77,78] are used to verify the formation of core-shell structure polymers [65].
The subsequent step consisted in analysis of self assembling and thermogelation properties of these block copolymers: namely CMC determination and then characterization of micelles and of the sol-gel transition. UV-visible [65] together with fluorescence technique [79] and surface tension [80] were the techniques mostly used in order to determine CMC values, while dynamic light scattering measurements, as a function of temperature [63,67], allowed determination of micelles size.
Regarding the sol-gel transition temperature and behavior the method of election was rheology. In fact both dynamic mechanical and flow curve analyses were used to identify sol-to-gel transitions at increasing temperature [65,71,78].
Another method often used to determine the sol-gel transition, despite the fact that it can be considered subjective and not accurate, was the test tube inverting method (temperature increment selected has been 1 °C per step). Polymer aqueous solutions (i.e., 0.5 g) were prepared in 4 mL vials with inner diameters of 11 mm. The vials were immersed in a water bath at each step for 15 minutes. The sol-gel transition temperature was monitored by inverting the vials, and if there was no flow in 30 s, it was regarded as a gel. The transition temperature was determined with 1 °C accuracy [63,65,78].
Then 1H NMR and 13C NMR analysis were used to study composition and microenvironment change during sol-to-gel transition and structure and composition of the synthesized triblock copolymers [63,65,67,71,78]. As an example, Zhang et al. used 1H NMR and differential scanning calorimetry (DSC) analysis to characterize the synthesized PLLA-PEG-PLLA and PDLA-PEG-PDLA block copolymers. 1H NMR allowed to determine structural characteristics such as EO/LA ratio, number average degree of polymerization (DP), and number average molar masses (Mn) while DSC was used to evaluate the thermal properties, including melting temperature (Tm), melting enthalpy (ΔHm), glass transition temperature (Tg), and cold crystallization temperature (Tc) [73].
Furthermore DSC, IR and X-ray diffraction techniques were also utilized to characterize PEG-PLA-PEG triblock copolymers showing that the synthesized copolymers were semicrystalline with PEG-type crystalline structure, and of decreasing crystallinity at increasing PLA block length [71].
3.2. Pharmaceutical Application
As already mentioned, biodegradable in-situ forming hydrogels represent promising delivery systems.
The different mechanisms of micellization and gelation compared to Pluronics make PEG-PLA or PEG-PLGA copolymers particularly able in controlling drug release. In fact, the Pluronic polymers form micelles that are equilibrated with monomeric polymers. The equilibrium shifts to micelle formation with increasing temperature. Above a CGC, the micelles pack together and occupy the entire volume, resulting in gel formation. According to this mechanism, the formed gel is subject to dissolution upon dilution from its surface, because when diluted, the interaction forces between packed micelles are not strong enough to keep an integrated mass, resulting in dissociation. This allows surface erosion of the gel, which has been utilized as a means for near-zero-order release of incorporated drugs for a short period of time [81]. However, the observation of integrity of the PEG-PLGA-PEG gel in rats even after one month may imply that the gelation mechanism or gel structure is different than that of Poloxamers, or, if it follows the same mechanism, the interactions between packed micelles are in a different order than those found in Poloxamers.
As an example Jeong and co-workers [66] demonstrated that PEG-PLGA-PEG copolymers formed micelles as temperature increased because PEG segments interact with a PLGA core, forming a new intermixed phase between core and shell. As a possible gelation mechanism, it is proposed that, with increased miscibility and increased interphase volume, the micelles start to contact, and the PEG chain in the corona interpenetrates between micelles or the intermixed phase hydrophobically interact with each other, leading to solid micelle packing and preventing the gel from dissolution by dilution. The miscibility between the two blocks still increases with temperature even in the gel phase, leading to unusual turbidity changes with temperature depending on degree of phase mixing.
Due to the mechanism described above, PEG-PLGA-PEG systems give rise to forming gels that maintain their properties (gel shape maintained its three-dimensional form due to the rapid sol-to-gel transition) for more than one month in rats as confirmed by subcutaneous injection of these triblock copolymer aqueous solutions.
Ketoprofen and spironolactone release kinetics were studied from a PEG-PLGA-PEG triblock copolymer hydrogel [82]. The release profile was strongly affected by drug hydrophobicity.
A 10 mM phosphate buffer, containing 0.02 wt.% NaN3 and 0.2 wt.% 3 Tween 20, was used as a drug release medium to improve drug solubility. The release medium pH was maintained between 7 and 7.4 to keep a constant drug solubility. The more hydrophilic ketoprofen was released continuously over two weeks with a first order release profile. The more hydrophobic spironolactone was released over two months with a S-shape release profile: an initial diffusion was followed by a combination of degradation and diffusion at later stage. The more hydrophobic drug partitioned more into the hydrophobic core and the release from the hydrobyphobic PLGA core domain system degradation. Thus, the release of spironolactone could be controlled by initial polymer concentration, loading of drug, and structure of the polymer.
Release of several drugs from PLGA-PEG-PLGA triblock copolymers hydrogel (ReGel®) including protein drugs and paclitaxel was reported [64,68]. ReGel is the trademark of the PLGA-PEG-PLGA copolymers, with a weight average molecular weight (Mw) of approximately 4,200 and an Mw /Mn ratio of 1.3. The unique characteristics of ReGel® hinge on two different main issues. First of all ReGel® is a water soluble, biodegradable polymer at temperatures below gel transition temperature; then ReGel® forms a water-insoluble gel once injected with an increase in viscosity of four orders of magnitude. Thus the gel formed a controlled release drug depot with delivery times ranging from one to six weeks. Another important feature was that this copolymer showed the ability to solubilize (400 to 2,000-fold) and stabilize poorly soluble and sensitive drugs, including proteins. As an example the gel provided excellent control of paclitaxel release for approximately 50 days and showed a flexible approach to protein and small molecule delivery that was simple to process and administer (pGH, G-CSF, insulin, rHbsAg).
Other temperature-responsive PLGA-PEG-PLGA triblock copolymers which have a different dl-lactide/glycolide molar ratio from ReGel® (ranging from 6/1 to 15/1) were also synthesized. Compared with ReGel system, the synthesized copolymers showed higher gelation temperature and longer period of drug release [83].
Another study demonstrated the feasibility to deliver protein (i.e., lysozyme) in biologically active form for longer duration by varying block lengths and concentrations of PLGA-PEG-PLGA triblock copolymers [84].
Yu and co-workers obtained a thermoreversible physical hydrogel by simply mixing an aqueous sol of a block copolymer with a precipitate of a similar copolymer but with different block ratio. The mixture of these two samples with a certain mix ratio underwent a sol-to-gel-to-precipitate transition upon an increase of temperature. This study highlighted how the balance of hydrophobicity and hydrophilicity within this sort of amphiphilic copolymers appeared critical. Then, lysozyme, chosen as model protein, was used to examine whether or not this thermogelling mixture systems could encapsulate and deliver biological substances such as proteins in a biologically active form, for long duration demonstrating that the release rate could also be adjusted by the mix ratios of copolymer mixtures, and an almost zero-order sustained release of lysozymes was achieved up to 50 days. Thus the obtained results outlined that the “mix” method provides a very convenient approach to design injectable thermogelling biomaterials with a broad adjustable window, and the novel copolymer mixture platform can be potentially used in drug delivery and other biomedical applications [85].
Another example was represented by solutions of PLGA-PEG-PLGA containing Calcitonin as model peptide. Zero order release kinetics was achieved for up to 100 hours. No significant burst release effect was observed. Gelation time did not affect the drug release profile of the system and the diffusion was the main mechanism for Calcitonin release from these systems [86]. Besides, calcitonin release kinetics, from a PLGA-PEG-PLGA polymeric solutions (25% w/w), could be controlled by using different excipients such as, for example, sodium lauryl sulfate that showed to reduce drug release rate from the systems [87].
Furthermore, PLGA-PEG-PLGA triblock copolymers were evaluated for sustained release of bee venom peptide. Bee venom peptide was released from copolymer-based hydrogels in the phosphate buffer (pH 7.4) as dissolution medium over 40 days. The hydrogels underwent slower degradation and then faster degradation rate during release process. Accordingly, the mechanism of bee venom peptide was Fickian diffusion during initial stage and then may be a combination of diffusion and degradation, indicating that PLGA-PEG-PLGA copolymer-based hydrogel represented a promising platform for sustained delivery of bee venom peptide [88].
In another study, a temperature-sensitive triblock copolymer PEG-PLGA-PEG was synthesized and blended with an oily phase (Lipiodol(R)) to obtain thermogelling emulsions. The release kinetics of paclitaxel in hydrogel and emulsion formulations were investigated demonstrating the feasibility of the thermogelling emulsions applied for both vascular embolization and sustained release of an antiangiogenic drug [89].
PLGA-PEG-PLGA hydrogel have also been studied for the ocular delivery of dexamethasone acetate. In particular the 20% (w/w) had a low critical solution temperature of 32 °C, which is close to the surface temperature of the eye and demonstrated potential thermosensitive gel-forming properties. This formulation may improve the bioavailability of some eye drugs [90].
Even PLA-PEG block copolymers have been studied for the peptide and protein delivery. A water soluble pentapeptide—TP5, taken as a model drug, was successfully incorporated into PLA-PEG-PLA hydrogels to evaluate the potential of this system as carrier of hydrophilic drugs. Various parameters such as copolymer concentration, drug load and copolymer composition were considered. Higher copolymer concentration led to slower release rate and less burst effect due to more compact structure which disfavored drug diffusion. Similarly, higher molar mass of the copolymers disfavored the release of TP5. In contrast, drug load exhibits little influence on the release profiles due to the high water solubility of TP5. Studies proved the potential of TP5 containing hydrogels, and the morphology of thymus indicated the immunization efficacy of the TP5 release systems based on PLA-PEG-PLA hydrogels [73].
4. PEG-Polycaprolactone Copolymers
Poly(ε-Caprolactone) (PCL) is a highly hydrophobic and crystalline polymer having good biocompatibility and widely used as biomedical material. In fact PCL is one of the most promising synthetic polymers which degrades in aqueous media or when in contact with microorganisms and thus can be used to make compostable polymeric devices [91]. The addition of hydrophilic polyether blocks to PCL chains has been used to enhance hydrophilicity compared to the parent homopolymer. Polyethylene glycol) (PEG) has been used to form various block copolymers with PCL [92,93].
PEG-PCL diblocks having PEG segment blocks of 2000 and PCL segment length of 950–1,500 underwent a sol-to-gel phase transition as the temperature was varied [94].
The main advantage of this system compared with PLGA-based thermogelling copolymers was that the incorporation of PCL led to the formation of polymers in powdery form, instead of a sticky paste making easier the handling of the drug/ polymer formulation for practical applications. In fact PCL triblock copolymers are not only simple to transfer or weigh but also easily dissolved in water.Additionally, these multiblock copolymer solutions were stable as a transparent solution at room temperature, providing practical convenience during drug formulation [95].
The phase transition behavior was mainly determined by PEG/PCL blocks ratio, PCL block length and molecular weight. In fact, a typical phase diagram of these multiblock copolymers in aqueous solution displayed a critical gel concentration (CGC) and a phase-transition temperature, which were mainly determined by these parameters. Phase separation-induced gelation mechanism was advanced to explain the thermoreversible phase transition. The hydrophobic PCL blocks aggregated and formed domains when hydrophilic PEG blocks were hydrated in water. A sol-to-gel transition resulted from the formation of three-dimensional physical network because multi-PCL blocks could diffuse into different domains. A gel-to-sol transition resulted from the melting of these domains and the collapse of physical crosslinkages. These multiblock copolymers have a varied sol-gel transition range near to body temperature with potential applications in injectable drug delivery systems [96].
When a wt of 20% PCL-PEG-PCL triblock copolymer aqueous solution was left overnight at room temperature (20 °C) opaque gel formation was observed. When the same gel was heated up to 50 °C for 30 s and a quickly quenching in an ice bath, it reversibly became a transparent free-flowing sol and when this transparent sol was injected into 37 °C water, it instantaneously became a gel. Thus, different mechanisms occur when opaque gels are formed at 20 °C (low-temperature gel) or for thermogels formed at 37 °C [95].
4.1. Synthesis and Characterization
Perret and Skoulios were the first who synthesized series of PCL/PEG block copolymers [97]. They obtained copolymers by anionic polymerization using naphthalene-sodium complex as catalyst. Later, catalyst-free polymerization was introduced to synthesize the PCL/PEG copolymer by Cerrai et al. [98].
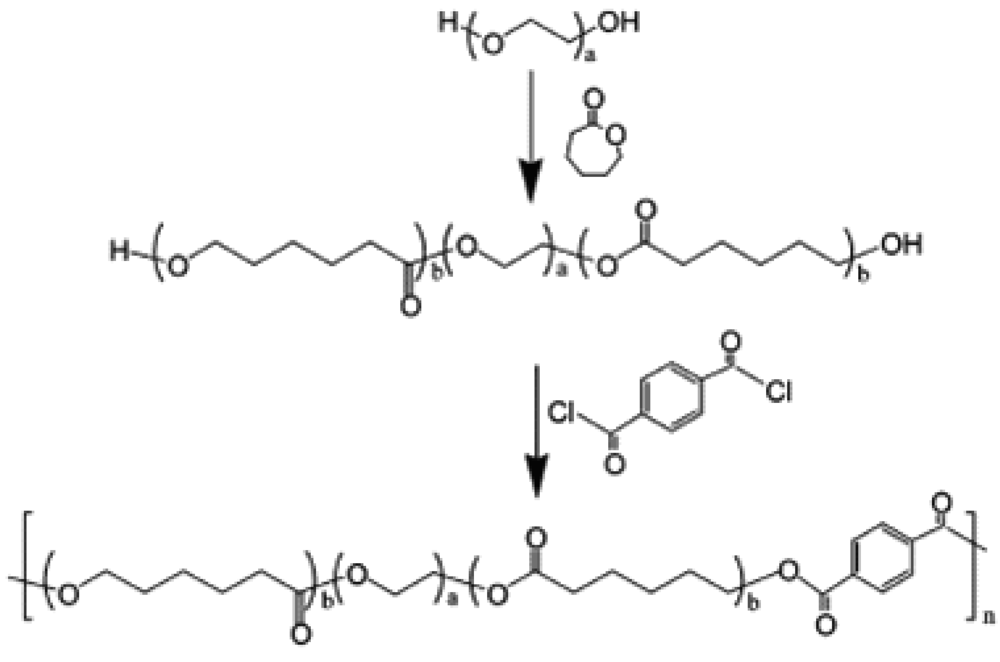
Bae and co-workers prepared PCL-PEG-PCL triblock copolymers by ring-opening polymerization of caprolactone in the presence of PEG (Figure 4) by using stannous octoate as catalyst. For example, to synthesize PCL-PEG-PCL (980-1,000-980) triblock copolymer, PEG (15.0 g, 15.0 mmol, Mn = 1,000) was dissolved in anhydrous toluene (80 mL) and the solvent was distilled off to a final volume of 30 mL to remove the residual water adsorbed by the polymer. Both ε-Caprolactone (23.7 g, 207.6 mmol) and stannous octoate (49 μL, 0.12 mmol) were added to the reaction mixture and precipitated by slowly adding diethyl ether. Terephthaloyl chloride (0.694 g, 3.3 mmol) and triethylamine (1.41 mL, 10.12 mmol) were added to the reaction mixtures and stirred at 60 °C for 24 h. The product was isolated by precipitation into diethyl ether, then the polymer was redissolved in 30 mL of methylene chloride, filtered, and precipitated by slowly adding diethyl ether. The residual solvent was removed under vacuum [95,99-101].
Multiblock copolymers composed of polyethylene glycol)s (PEGs) and biodegradable poly(ε-caprolactone)s (PCLs) were also synthesized through one-step condensation copolymerization with hexamethylene diisocyanate (HDI) as coupling agent. The reacted solution was precipitated in diethyl ether and the resulting copolymer was further purified through dissolution in chloroform and precipitation in diethyl ether. The final product was dried under vacuum at room temperature for over 48 h to yield a translucent solid [96]. The synthesized polymers and their self assembling behavior have been characterized by means of different analytical techniques.
First of all, number average molecular weight and molecular weight distribution of the synthesized PEG-poly(ε-caprolactone) copolymers were determined using gel permeation chromatography (GPC) [96,100-102].
1H NMR measurements were performed to determine molecular structure and composition, such as the PEG/PCL block ratio [95,96,100-102], while 13C NMR has been used to observe spectral changes of PEG/PCL multiblock copolymer (20 wt % in D2O) as a function of temperature [95,101].
Furthermore, dynamic light scattering analyses facilitated studying the size of PEG/PCL multiblock copolymer as a function of temperature [95,101]. Then IR spectra of these multiblock copolymers were performed in order to characterize their molecular structures [96,100].
Then, in order to understand phase behavior of PCL-PEG-PCL triblock copolymer solution and, in particular, the formation of an opaque gel at room temperature (a phenomenon not present in the multiblock copolymer aqueous solution), Bae et al. used X-ray diffraction analysis and Raman spectroscopy as function of time at room temperature. Both techniques highlighted that crystallization of PCL-PEG-PCL triblock copolymer in water is responsible for such phase behavior [95].
Thermal properties, such as melting temperature (Tm) of PEG and PCL homopolymers and multiblock copolymers were measured by differential scanning calorimetry (DSC) [101]. The same technique has been applied to study polymer dispersion and hydrogel [95,96,99].
Samples phase transition were also investigated by using optical microscopy (OM) equipped with a hot plate to monitor samples behavior at increasing temperature [96].
More specifically the sol-gel transition was studied by the test tube inverting method [63,78,95,101] and rheological analyses [95,101].
4.2. Pharmaceutical Application
The PCL-PEG copolymers might have great application in biomedical field. In particular, thanks to their great thermosensitivity and biodegradability, their hydrogels are promising materials to be used as gel-forming controlled drug delivery system.
A new kind of biodegradable and injectable polyethylene glycol)-poly(ε-caprolactone)-poly(ethylene glycol) (PEG-PCL-PEG) hydrogels were prepared in order to develop controlled drug delivery systems. Aqueous solutions of these diblock and three-block copolymers resulted to be a free-flowing sol at either room or below the corresponding critical gel temperatures (CGT), and a gel at body temperature. Thus a mice model was applied to evaluate the gel formation and its subsequent degradation followed by subcutaneous injection of PEG-PCL hydrogels (Figure 5) [103]. In the same work, degradation behavior and drug release behavior of these di- and tri-block copolymers have been investigated. In-vitro release was observed in particular for three different classes of molecules: hydrophilic small-molecular-weight drugs (i.e., Vitamin B12), hydrophobic small-molecular-weight drugs (i.e., honokiol, a multi-functional drug, with optimal potential application for human cancer therapy) and a hydrophilic macromolecular protein drug (i.e., BSA). Thus slow drug release was gained while release profiles were affected to some extent by initial drug loading and hydrogel concentration. Hydrophilic drug were almost completely released from the hydrogel in a week with high release rate (>85% in 24 h) and high initial burst rate (about 30% in 1 h), whereas hydrophobic drug and protein drug could be released slowly over a longer period with lower cumulative release rate (38.8% for honokiol and 27.2% for BSA in 14 days, respectively). Drug release from hydrogels was driven by two forces: diffusion effect and degradation or erosion [103]. Due to good solubility, hydrophilic drugs could diffuse in short time through pores of hydrogel releasing almost all the drug from the hydrogel. But for hydrophobic drugs and proteins in water, low diffusion rates could be seen and strong intermolecular interactions with hydrogel dominated drug release profiles, resulting in low release rate and high residual drug in hydrogels.
Hydrophobic honokiol (HK), as model drug, has also been used in another study [104]. Self-assembled PEG-PCL-PEG micelles encapsulated this drug in order to overcome its poor water solubility and to meet the requirements of intravenous administration without any organic solvent or surfactant. Encapsulation of HK led to sustained release from HK-micelles for up to two weeks while cytotoxicity remained comparable to that of free HK. Thus, HK-micelles were safe, stable, effective, and easy to make and scale up.
PEG-PCL-PEG triblock copolymer was also investigated as in-situ sustained opthalmic drug delivery. Biodegradability within the eye, its effect on cultured human lens epithelia, intraocular pressure, and ocular tissues have been studied. Data indicated that the prepared hydrogel was biocompatible and biodegradable despite the temporary elevated intraocular pressure and slight corneal endothelial damage at specific concentrations. Therefore, this hydrogel showed great biocompatibility, biodegradability, and sustained release property in eyes, making this system a safe candidate for sustained ophthalmic drug delivery [105].
Furthermore, Gong et al. [106] studied a PEG-PCL-PEG hydrogel system for basic fibroblastic growth factor (bFGF) antigen delivery. bFGF encapsulated in this hydrogel resulted in an injectable free-flowing sol at room temperature forming a non-flowing gel at physiological temperature and acting as antigen depot. Furthermore, data about cytotoxicity showed that the hydrogel could be regarded as a safe carrier, and bFGF could be released from the hydrogel system in an extended period. The immunogenicity of bFGF was improved significantly after encapsulating into the hydrogel and the strong humoral immunity created by bFGF-hydrogel was maintained for more than 14 weeks. Thus, these bFGF loaded hydrogels might have great potential as novel vaccine adjuvants for protein antigen.
Compared to PEG-PCL-PEG triblock copolymer, PCL-PEG-PCL triblock copolymer has several advantages: First, the PCL-PEG-PCL triblock copolymer can be synthesized in one step without using any coupling agent; second, PCL-PEG-PCL hydrogels have a wider gel window; third, PCL-PEG-PCL hydrogels can persist for a longer period, about six weeks, compared to PEG-PCL-PEG hydrogels (about two weeks). Both PCL-PEG-PCL and PEG-PCL-PEG are biodegradable, and can sustain drug release in an extended period. One could choose to use PCL-PEG-PCL or PEG-PCL-PEG hydrogels according to the practical needs of different applications [107].
PCL-PEG-PCL block copolymers have been also utilized for protein delivery. In fact, the release behaviors of two model proteins, including bovine serum albumin (BSA) and horseradish peroxidase (HRP), from a gel-forming controlled drug delivery system based on thermosensitive PCL-PEG-PCL polymers were studied in detail. Released HRP was confirmed to preserve its biological activity by specific enzymatic activity assay, in fact gel formation and degradation studies indicated that PCL-PEG-PCL copolymers hydrogels could give a sustained release for at least 45 days by subcutaneous injection. So the great thermosensitivity and biodegradability of these copolymers make them promising gel forming controlled drug delivery system for therapeutic proteins [107].
Biocompatible PEG-PCL micelles has been studied to deliver silicon phthalocyanine Pc 4; a second-generation photosensitizer currently approved by the FDA. This molecule showed promising properties for photodynamic therapy (PDT) in several cancer cells and model tumor systems. Because of its high hydrophobicity, its formulation for delivery and favorable biodistribution is quite difficult. Recently Master et al. demonstrated the efficient encapsulation of Pc 4 in these micelles, their intracellular uptake, and significant cytotoxic effect of the formulation upon photoirradiation. Quantitative estimation of the extent of Pc 4 loading on micelles, and the photocytotoxicity of the micelle-incorporated Pc 4 demonstrated that this system could be used to develop a biocompatible nanomedicine platform for tumor-targeted delivery of Pc 4 for site-selective PDT [108].
4. Polyether Modified Poly(acrylic acid)
Poly(acrylic acid) (PAA) copolymers modified with block-copolymers of poly(ethylene oxide)-poly(propylene oxide)-poly(ethylene oxide) PEO-PPO-PEO (Poloxamers) (Figure 6) show different medicinal applications, including topical and systemic administration, as their components are considered pharmaceutically safe. These copolymers have a unique graft-comb structure (Figure 6) whereby polyether chains (primarily PPO segments with tertiary carbons) are bonded to PAA via C–C bonding [109].
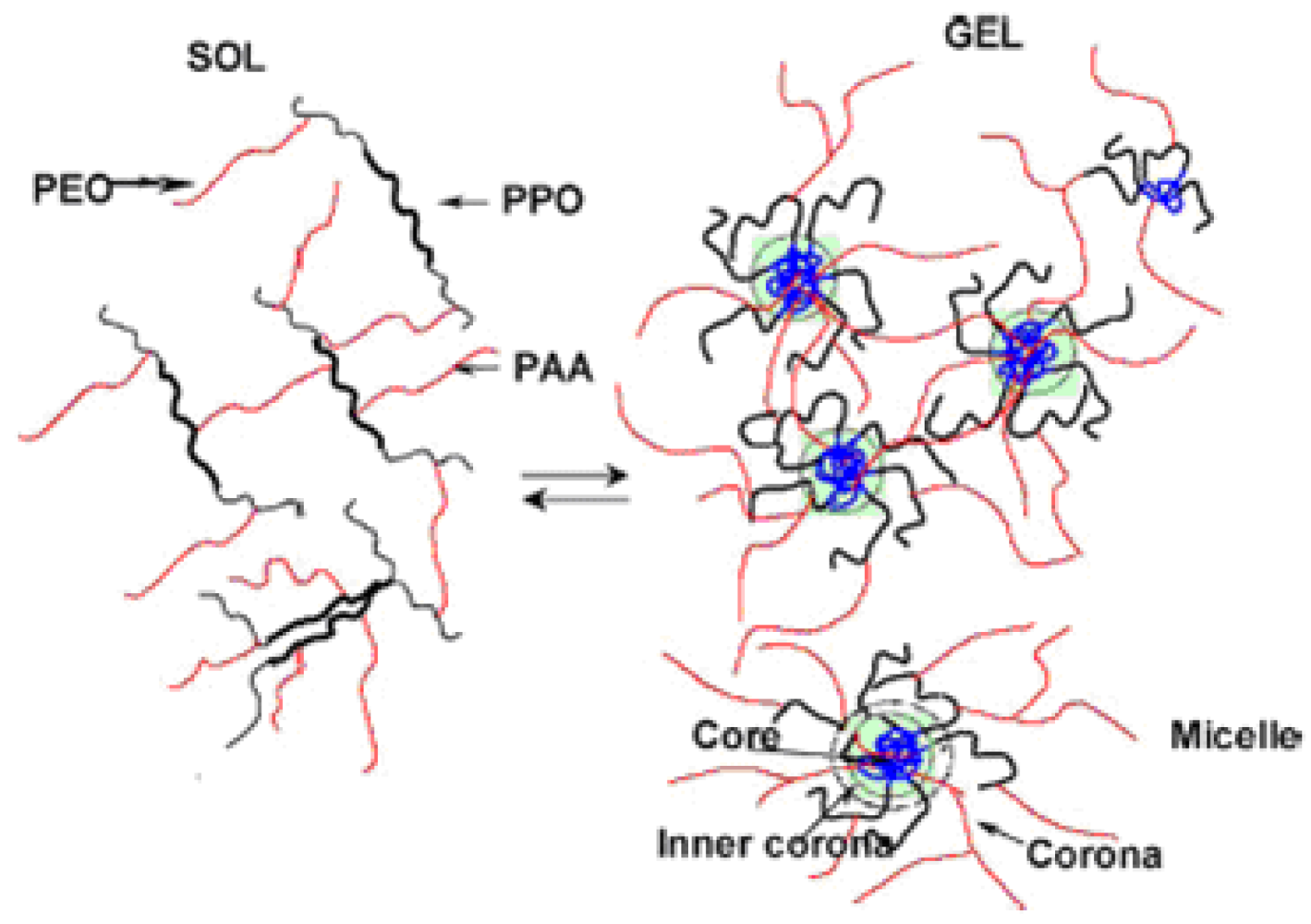
The conformation of PAA in aqueous solutions depends on pH, ionic strength, presence of multivalent ions, while PEO/PPO chains can undergo a transition from a homogeneous solution of random coils to a separated microphase structure depending on temperature and salt concentration [110]. Thus properties of aqueous solutions of graft- or block-copolymers of PAA and polyethers are sensitive to both ionic strength and temperature giving rise to a variety of phase-separated (self-assembled) structures, including micellar aggregates, in the polyether-PAA solutions [110,111]. In fact, attachment of PPO groups onto a polyelectrolyte adds temperature sensitivity to an already pH-sensitive polymer, thus creating a dually responsive material.
Thus, Poloxamer-PAA copolymers [112] are characterized by high molecular weights and high sensitivity to temperature. In semidilute regimes (i.e., 0.01–1 w/v% concentration range [113], Poloxamer-PAA aqueous solutions form reversible gels with significant elastic moduli [114] due to the formation of micelle-like aggregates above a well-defined CMT [114]. In the range of concentration 0.5–3 wt%, PAA-g-Poloxamer and Poloxamer-g-PAA form clear gels when the temperature increased from 4 to 37 ° C at pH 7.4 [115,116]. Gelation of these copolymers is due to an entropically driven self-association of PPO groups to form micelles that provide physical cross-linking points [117]. Besides, the critical gel concentration is considerably lower than that of the parent Poloxamer. However, an increase in polymer concentration results in lower transition temperature and higher gel strength. Thus, polymer concentration, pH and salts influenced gelation properties [118].
In fact, at increasing pH values, the onset of gelation is shifted to lower temperatures, and gel strength increased. These results depends on the fact that gelation is driven by the formation of micelles that act as thermoreversible cross-links (Figure 7). The onset of gelation in poloxamer-g-PAA solutions occur approximately at the critical micellization temperature of the corresponding Poloxamer [116]. A negative change in heat capacity upon gelation suggests a decrease in the exposure of PPO segments to water [114].
4.1. Synthesis and Characterization
Synthesis of the graft copolymer, PAA-g-poloxamer was reported by Hoffman and co-workers [115]. Poloxamer was activated by derivatization with 4-nitrophenyl formate in the presence of triethylamine. After purification, the intermediate was reacted with diaminoethylene to yield an amino terminated Poloxamer which was conjugated to (acrylic acid) via an amide bond using dicyclohexylcarbodiimide (DCC) as a coupling agent. The proposed synthetic route involved a three-step procedure with several intermediate steps of purification, making the process potentially problematic on an industrial scale. In fact, applying Poloxamer derivative animated on both ends would lead to a permanent crosslinking of the poly(acrylic acid) chains with Poloxamer segments by urethane bonding.
Subsequently, the synthesis of another graft copolymer, i.e., poloxamer-g-PAA, was described by Bromberg et al. [112,116]. This single-step synthetic procedure was reproducible and scaleable.
In this procedure the acrylic acid in a 125 mL flask was partially neutralized by addition of 5 M NaOH solution as needed. The typical degree of neutralization of acrylic acid was 6 mol%.
Upon redissolution of the precipitate, Poloxamer was charged into the flask and allowed to completely dissolve in acrylic acid under constant agitation. A 500 mL multinecked, thermostated flanged glass reactor equipped with a mechanical stirrer, syringe sampler, thermometer, programmable heater bath and a gas inlet/outlet [119] was charged with 400 mL of Ganex solution in dodecane and was deoxygenated overnight by nitrogen flow while stirring.
Initiator system formed by a solution of peroxide and/or azo compound in a small amount of acrylic acid was added into Poloxamer solution in acrylic acid under stirring. The resulting solution was deoxygenated by nitrogen flow for 1 h and introduced into the reactor under a nitrogen blanket while stirring. The reactor was equilibrated for 1 h while stirring at 20 °C under nitrogen purge, introduced from the bottom of the reactor. Then at t = 0 the heating began and timing commenced. The reactor was heated to 70 °C at a desired rate (typically 1–2 °C/min) under constant nitrogen flow. At a certain temperature the exothermic reaction began resulting in a rapid temperature rise inside the reactor. Then the heat release subsided, the reactor cooled to 70 °C and it was kept at this temperature for 8–10 h under stirring. The reactor was allowed to equilibrate at 20 °C, the nitrogen flow was discontinued, and the slurry of the resulting polymer was filtered off using Whatman filter paper (retention 10 μm). The polymer was repeatedly washed with excess heptane and then with excess hexane in separation funnels. The resultant white powder was dried under vacuum (10-3 Torr) at 40 °C for 24 h.
To monitor kinetics of polymerization, 0.5 mL samples were withdrawn intermittently from the centre of the reactor. An effective degree of conversion of acrylic acid (AA) into the polymer [F, % = 100 × (concentration of reacted AA)/(initial concentration of AA in the reaction mixture)] was determined by measuring the amounts of acrylic acid monomer left in the reactor during the course of polymerization.
In order to characterize graft-copolymerization of Pluronic and poly(acrylic acid) the effective degree of bonding between Pluronic and poly(acrylic acid) was measured [112].
A sample, withdrawn from the reactor, was filtered using Whatman filter paper (retention 10 μm), dried under vacuum and placed into a Soxhlet extractor charged with 100 mL of dichloromethane, and kept under reflux for 24 h. Then, fresh dichloromethane was added into the extractor and the operation repeated for other 24 h. The samples were collected and evaporated under vacuum. The residual solids were weighed, redissolved in THF, and analyzed for Pluronic content using HPLC and FT-IR. The residual solids were analyzed by HPLC using THF at a 1.0 mL/min flow rate and polyethylene glycol) and poly(acrylic acid) as molecular weight standards. An effective degree of bonding was expressed as B% = 100 × (concentration of Pluronic bonded)/(initial concentration of Pluronic in reaction mixture).
The weight fraction of macroscopic gel particles was measured [116] at 15 °C by filtering 1 w/v% Pluronic-PAA through weighted Acrodisc nylon filters with pore diameters of 0.8 μm. The parameter G,% = 100 × (weight of filtered fraction)/(weight of initial suspension) was measured for each suspension. Weight-average molecular mass of Pluronic-PAA samples typically exceeded 5 × 105 Da, and polydispersity of the fractions subjected to SEC varied from 2.3 to 6.7.
Another important step of characterization is the study of micelles formation. This phenomenon have been analyzed by using different techniques such as light scattering [116], size-exclusion chromatography [116], spin probe techniques [119], and DSC [116]. Temperature sensitive chemical shifts belonging to methyl groups in 13C and 1H NMR provided direct evidence for the aggregation of PPO blocks to be a cause of micelle appearance [120].
Furthermore SANS studies [121] demonstrated that micelle-like aggregates formed in Pluronic-PAA solutions above the CMT are unusually uniformly distributed.
Fluorescence analysis using pyrene I1/I3 emission intensity ratios and DPH fluorescence intensity and depolarization patterns showed that there was a distinct difference in the environment to which probes were exposed depending whether polymer solutions were in the liquid or gelled state. These domains were assumed to be created by the process of grafting the poly(acrylic acids) onto triblock copolymers [117,122]. Together with fluorescence studies, ESR spectroscopy has been utilized by Bromberg and Barr [122] to monitor aggregation phenomena in aqueous solutions of these copolymers. The 12-doxylstearic acid spin probe was used to monitor changes in anisotropic ESR spectra with temperature providing a spectroscopic evidence for the presence of a constrained shell of hydrophobic (PPO) segments near the ionizable (PAA) segments.
Surface tension (Wilhelmy plate method) and rheological measurements (viscosity and dynamic moduli changes) under temperature control were also used to monitor beginning of aggregation and gelation phenomenon in Pluronic-PAA [112,113].
4.2. Pharmaceutical Applications
As both PAA and Poloxamers are considered safe and have been approved by the U.S. Food and Drug Administration as food additives and pharmaceutical ingredients, copolymers based on PAA bonded with Poloxamers could be used advantageously in biomedical applications [123]. Furthermore animal toxicological study showed the non-toxic nature of this copolymer and that these molecules were excreted when administered orally and were not absorbed into the systemic circulation [123].
This poly(oxyethylene-b-oxypropylene-b-oxyethylene)-g-poly(acrylic acid) (trade name Smart Hydrogel) [123] formed clear gels at a critical gel concentration considerably lower than that of the parent Poloxamer.
Moreover, the bioadhesive properties of this system [109] made it interesting for a wide variety of applications. In fact when this system is injected or sprayed as a liquid onto mucosal surfaces it quickly gels. The gelation gives rise to controlled release formulations characterized by lower rate of diffusion and erosion of both polymer and the associated drug, thus enhancing drug retention and bioavailability.
Hydrogels of poloxamer-g-PAA have been proposed for vaginal [124] and topical [125] drug delivery. The pharmacokinetic profile of estradiol after vaginal delivery of poloxamer-g-PAA formulation was equivalent to that of Estrace® vaginal cream, which contains five times more drug. This hydrogel was studied for the treatment of gastrooesophageal reflux by measuring its adherence to the esophageal mucosa [126] revealing that 15% of the administered dose displayed prolonged retention in the esophagus. Furthermore, it was shown that Smart Hydrogel™ could sustain the release of luteinizing hormone-releasing hormone (LHRH) and human insulin [126]. At a polymer concentration of 5% release kinetics of LHRH followed a zero-order rate.
Poloxamer-PAA copolymers can be additionally cross-linked by divinyl cross-linkers, resulting in permanent microgel particles [127].
The obtained swollen microgel particles form viscoelastic, crowded suspensions in water, similar to Carbopol systems and the swelling behavior in water was dependent on pH and temperature. This system showed different advantages. First of all it undergoes transitions in large volumes, in response to changing pH, which is useful in gastrointestinal applications. In fact, since the pH in the stomach is in the 1–2 range, it ensures that the microgels remain collapsed preventing an undesirable drug release in the stomach. The pH in the intestine is 6.2–7.4, leading to highly swollen state of microgels and to release the drug easily from these particles. Besides, micelles incorporated within gel particles are capable of solubilizing hydrophobic molecules including steroid hormones such as megestrol acetate and estradiol, as well as paclitaxel and camptothecin. On the other hand the charged PAA chains of Pluronic-PAA network bonded significant quantities (up to and above stoichiometric [drug]/[COO−] ratios) of basic molecules such as doxorubicin and can be loaded with anticancer agents [127,128]. Moreover, incorporation of different Poloxamer copolymers into microgels strongly influenced structure, swelling, and aggregation behavior of these gel microparticles [127].
As already mentioned these copolymers are characterized by a certain degree of mucoadhesiveness. The mucoadhesive properties were tested by Bromberg and co-workers [109] by measuring fracture strength and work of adhesion between gels and rat intestine correlated with the viscoelastic characteristics of gels such as pseudoequilibrium modulus obtained in creep recovery tests and loss angle measured at fixed oscillatory stress and frequency.
Due to the strong mucoadhesive tendency, formulations based on Pluronic-PAA copolymers, exhibit enhanced retention in esophagus and intestines. In a study on human volunteers, Pluronic (F127)-PAA copolymer solution gelled and formed a protective layer over the esophageal mucosa when administered orally and efficiently adhered to the mucosa of the human esophagus, increasing the micellar gel retention on the mucosa [129].
Furthermore, the fact that these micelles efficiently solubilize hydrophobic drugs, such as paclitaxel and steroids, and protect molecules such as camptothecins from the hydrolytic reactions, makes them good candidates in the oral chemotherapy. Bromberg and coworkers carried out several and studies regarding the oral administration of doxorubicin, paclitaxel, and megestrol acetate using both cross-linked and uncross-linked Poloxamer-PAA copolymer [130,131].
High surface activity of Poloxamer-PAA copolymers in water resulted in interactions with cell membranes and suppression of membrane pumps such as P-glycoprotein. Experiments demonstrated that lightly cross-linked copolymers of PAA and Poloxamer L61, L92, and F127 loaded with antineoplastic drug doxorubicin inhibited P-glycoprotein (P-gp)-mediated doxorubicin efflux from the cells and enhanced passive influx, which increased the overall net cell absorption of doxorubicin. In the same way, micellar formulations of Poloxamer P85 and PAA copolymers appeared to be efficient excipients for oral delivery of paclitaxel in a rodent model. Compared with i.v. delivery, where no considerable change in plasma peak levels (Cmax) was observed when paclitaxel was delivered with cyclosporin A, oral coadministration of paclitaxel and cyclosporin resulted in 8-fold increase in Cmax, 15-fold increase in AUC, and 10-fold enhancement in oral availability of paclitaxel. This great improvement of the pharmacokinetic parameters in oral administration was due to decreased elimination of paclitaxel by the intestinal P-glucoprotein inhibition by means of cyclosporin A and possibly due to the cyclosporin/paclitaxel metabolic competition as substrates for the cytochrome P450 3A4 isozymes, which may reduce the metabolic elimination of the paclitaxel in the gut and the liver [132].
The ionizable carboxyls in the micellar corona facilitate mucoadhesion that enhances residence time of micelles and solubilized drugs in the gastrointestinal tract. Pluronic-PAA micelles were also loaded with weakly basic and water-soluble drugs such as doxorubicin and its analogs, mitomycin C, mitoxantrone, fluorouracil, and cyclophosphamide through electrostatic interactions with the micellar corona [133].
Besides Poloxamer-PAA copolymers have been studied to enhance aqueous solubility and stability of lactone form of camptothecin (CPT). Then, the equilibrium solubility partitioning and hydrolysis of the lactone form of CPT in the presence of Poloxamer-PAA in water and in human serum were studied. CPT solubility in polymer micellar solutions was ca. 3- to 4-fold higher than that in water at pH 5. The amount of CPT solubilized per PPO was considerably greater in Poloxamer-PAA solutions than in the parent Poloxamer solutions, which suggests that the drug is not only solubilized by hydrophobic cores but also by hydrophilic POE-PAA shells of the micelles. Furthermore, the drug loading in micelles of Poloxamer-PAA notably hinders the hydrolytic opening of lactone rings in both alkaline water and human serum [134].
Thus, the pH- and temperature-sensitivity and the ability to solubilize and stabilize hydrophobic steroids, anti-cancer drugs, and proteins together with mucoadhesive properties have made the Poloxamer-PAA gels a feasible vehicle for oral, topical [127] and parenteral drug delivery [135].
5. Conclusion
In the recent past, an impressive number of novel, self-assembling thermosensitive, gel-forming copolymers have been studied. Aqueous solutions of these novel block copolymers characterized by a reversible sol-gel transition behavior allows delivery of drugs in the liquid state, which can form an in-situ gel depot at body temperature providing drug release control. Another important feature is the amphiphilic character of micelles that makes these delivery systems capable of solubilizing water-insoluble or poorly soluble drugs and of protecting labile molecules such as proteins and peptides. The most studied self-assembling copolymers are Poloxamers despite the fact that they have not met initial expectations as pharmaceutical and biomedical implants, mainly due to their non-biodegradability and inability to provide a very long sustained drug delivery. On the other hand, PEO/PLGA and PEG-Polycaprolactone hydrogels represent very attractive systems for pharmaceutical applications. Their biodegradability and their good safety profile make these new polymers very promising as delivery systems able to control drug release over weeks or months by means of parenteral extravascular administration.








References
- Kwon, G.T.O. Polymeric micelles as new drug carriers. Adv. Drug Del. Rev. 1996, 2, 107–116. [Google Scholar]
- Moffitt, M.; Khougaz, K.; Eisenberg, A. Micellization of ionic block copolymers. Acc. Chem. Res. 1996, 2, 95–102. [Google Scholar]
- Tuzar, Z.; Kratochvil, P. Micelles of block- and graft- copolymers in solution. In In Surface Colloid Science; Matijevic, E., Ed.; Plenum Press: New York, NY, USA, 1993; Volume 15. [Google Scholar]
- Zang, L.; Eisenberg, A. Multiple morphologies of “crew-cut” aggregates of polystyrene-b-poly(acrylic acid) block copolymers. Science 1995, 268, 1728–1731. [Google Scholar]
- Lavasanifar, A.; Samuel, J.; Kwon, G. The effect of alkyl core structure on micellar properties of polyethylene oxide)-block-poly(L-aspertamide) derivatives. Colloid. Surface. B 2001, 1, 115–126. [Google Scholar]
- Nuopponen, M.; Ojala, J.; Tenhu, H. Aggregation behaviour of well defined amphiphilic diblock copolymers with poly(N-isopropylacrylammide) and hydrophobic blocks. Polymer 2004, 45, 3643–3650. [Google Scholar]
- Hagan, S.; Coombes, A.; Garnett, M.; Dunn, S.; Davies, M.; Illum, L. Polylactide-poly(ethylene glycol) copolymers as drug delivery system. 1. Characterization od water dispersible micelle-forming system. Langmuir 1996, 12, 2153–2161. [Google Scholar]
- Gaucher, G.; Dufresne, M.; Sant, V.; Kang, N.; Maysinger, D.; Leroux, J. Block copolimer micelles: Preparation, characterization, and application in drug delivery. J. Controll. Rel. 2005, 109, 169–188. [Google Scholar]
- Ivanova, R.; Lindman, B.; Alexandridis, P. Effect of pharmaceutical acceptable glycols on the stability of the liquid crystalline gels formed by poloxamer 407 in water. J. Coll. Interf Sci. 2002, 25, 226–235. [Google Scholar]
- Kwon, K.; Park, M.; Hwang, J.; Char, K. Effect of alcohol addition on gelation in aqueous solution of polyethylene oxide)-poly(propylene oxide)-poly(ethylene oxide) triblock copolymer. Polym. J. 2001, 3, 404–410. [Google Scholar]
- Lukyanov, A.; Gao, Z.; Mazzola, L.; Torchilin, V. Polyethylene glycol-diacyllipid micelles demonstrate increased accumulation in subcutaneous tumors in mice. Pharm. Res. 2002, 19, 1424–1429. [Google Scholar]
- Alexandridis, P.; Hatton, T.A. Polyethylene oxide)-poly(propylene oxide)-poly(ethylene oxide) block copolymer surfactants in aqueous solutions and at interfaces: Thermodynamics, structure, dynamics, and modelling. Colloids Surf. A Physicochem. Eng. Aspects 1995, 96, 1–46. [Google Scholar]
- Goldstein, R. On the theory of lower critical solution pointing hydrogen-bonded mixtures. J. Chem. Phys. 1984, 80, 5340–5341. [Google Scholar]
- Nagarajan, R. Solubilization of hydrocarbons and resulting aggregate shape transition in aqueous solution of Pluronic® (PEO-PPO-PEO) block copolymers. Colloid. Surface. B 1999, 16, 55–72. [Google Scholar]
- Brown, W.; Hvidt, S.; Bahadur, P. Micelle gel formation in Polyethylene oxide)-poly(propylene oxide)-poly(ethylene oxide) triblock copolymer in water solution. Dynamic and static light scattering and oscillatory shear. J. Phys. Chem. 1991, 95, 1850–1858. [Google Scholar]
- Al-Saden, A.A.; Whateley, T.L.; Florence, A.T. Poloxamer association in aqueous solution. J. Colloid Interf. Sci. 1982, 90, 303–309. [Google Scholar]
- Wang, P.; Johnston, T.P. Kinetics of sol-to-gel transition for poloxamer polyol. J. Appl. Polym. Sci. 1991, 43, 283–292. [Google Scholar]
- Rassing, J.; Attwood, D. Ultrasonic velocity and light-scattering studies on the polyoxyethylene-polyoxypropylene copolymer Pluronic F127 in aqueous solution. Int. J. Pharm. 1983, 13, 47–55. [Google Scholar]
- Vadnere, M.; Amido, G.; Lindenbaum, S.; Haslam, J.L. Thermodynamic studies on the gel-sol transition of some pluronic polyols. Int. J. Pharm. 1984, 22, 207–218. [Google Scholar]
- Wanka, G.; Hoffmann, H.; Ulbricht, W. The aggregation behavior of poly(oxyethylene)-poly(oxypropylene)-poly(oxyethylene) block copolymers in aqueous solution. Colloid Polym. Sci. 1990, 268, 101–117. [Google Scholar]
- Pandit, N.; Wang, D. Salt effect on the diffusion and release rate of propanolol from poloxamer 407 gel. Int. J. Pharm. 1998, 167, 183–189. [Google Scholar]
- Alexandridis, P.; Olsson, U.; Lindman, B. A record nine different phases (four cubic, two hexagonal, and one lamellar lyotropic liquid crystalline and two micellar solution) in a ternary isothermal system of an amphiphilic block copolymer and selective solvents (water and oil). Langmuir 1998, 14, 2627–2638. [Google Scholar]
- Wanka, G.; Hoffmann, H.; Ulbricht, W. Phase Diagrams and aggregation behavior of poly(oxyethylene)-poly(oxypropylene)-poly(oxyethylene) triblock copolymers in aqueous solutions. Macromolecules 1994, 27, 4145–4159. [Google Scholar]
- Almgren, M.; Bahadur, P.; Jansson, M.; Li, P.; Brown, W.; Bahadur, A. Static and dynamic properties of a (PEO-PPO-PEO) block copolymer in aqueous solution. J. Colloid Interf. Sci. 1992, 15, 157–165. [Google Scholar]
- Scherlund, M.; Malmsten, M.; Holmqvist, P.; Brodin, A. Thermosetting microemulsion and mixed micellar solutions as drug delivery systems for periodontal anesthesia. Int J. Pharm. 2000, 194, 103–116. [Google Scholar]
- Mortensen, K. Structural studies of aqueous solutions of PEO-PPO-PEO triblock copolymer, their micellar aggregates and mesophases; a small-angle neutron scattering studies. J. Phys. Condens. Matter 1996, 8, 103–124. [Google Scholar]
- Degtyarev, Y.; Schlick, S. Diffusion coefficients of small molecules as guests in various phases of pluronic l64 measured by one-dimensional electron spin resonance imaging. Langmuir 1999, 1, 5040–5047. [Google Scholar]
- Zhou, L.; Schlick, S. Electron spin resonance (ESR) spectra of amphiphilic spin probes in the triblock copolymer EO13PO30EO13 (Pluronic L64): Hydration, dynamics and order in the polymer aggregates. Polymer 2000, 41, 4679–4689. [Google Scholar]
- Mortensen, K.; Pederson, J. Structural study on the micelle formation of poly(ethylene oxide)-poly(propylene oxide)-poly(ethylene oxide) triblock copolymer in aqueous solution. Macromolecules 1993, 26, 805–812. [Google Scholar]
- Schillén, K.; Glatter, O.; Brown, W. Characterization of a PEO-PPOPEO block copolymer system. Progr. Colloid Polym. Sci. 1993, 93, 66–71. [Google Scholar]
- Cabana, A.; Ait-Kadi, A.; Juhasz, J. Study of the gelation process of polyethylene oxide-polypropylene oxide-polyethylene oxide copolymer (poloxamer 407) aqueous solutions. J. Colloid Interface Sci. 1997, 190, 307–312. [Google Scholar]
- Mortensen, K.; Yashayahu, T. Cryo-TEM and SANS microstructural study of pluronic polymer solution. Macromolecules 1995, 2, 8829–8834. [Google Scholar]
- Bonacucina, G.; Misici-Falzi, M.; Cespi, M.; Palmieri, G.F. Characterization of micellar systems by the use of acoustic spectroscopy. J. Pharm. Sci. 2008, 97, 2217–2227. [Google Scholar]
- Schmolka, I. Artificial skin 1. Preparation and properties of Pluronic F-127 gels for treatment of burns. J. Biomed. Mater. Res. 1972, 6, 571–582. [Google Scholar]
- Kabanov, A.V.; Alakhov, V.Y. Pluronic® block copolimer in drug delivery: From micellar nanocontainers to biological response modifiers. Crit. Rev. Ther. Drug Carrier Syst. 2002, 19, 1–73. [Google Scholar]
- Miyazaki, S.; Takeuchi, S.; Yokouchi, C.; Takada, M. Pluronic F-127 gels as a vehicle for topical administration of anticancer agents. Chem. Pharm. Bull. 1984, 32, 4205–4208. [Google Scholar]
- Choi, H.; Jung, J.; Ryu, J.; Yoon, S.; Oh, Y.; Kim, C. Development of in situ-gelling and mucoadhesive acetaminophen liquid suppository. Int. J. Pharm. 1998, 165, 33–44. [Google Scholar]
- Ryu, J.; Chung, S.; Lee, M.; Kim, C.; Shim, C. Increased bioavailability of propranolol in rats by retaining thermally gelling liquid suppositories in the rectum. J. Control. Release 1999, 59, 163–172. [Google Scholar]
- Chang, J.; Oh, Y.; Choi, H.; Kim, Y.; Kim, C. Rheological evaluation of thermosensitive and mucoadhesive vaginals gels in physiological conditions. Int. J. Pharm. 2002, 24, 155–163. [Google Scholar]
- Liaw, J.; Lin, Y. Evaluation of poly(ethylene oxide)-poly(propylene oxide)-poly(ethylene oxide) (PEO-PPO-PEO) gels as a release vehicle for percutaneous fentanyl. J. Control. Release 2000, 68, 273–282. [Google Scholar]
- El-Kamel, A. In vitro and in vivo and evaluation of Pluronic F127- based ocular delivery system for timolol maleate. Int. J. Pharm. 2002, 241, 47–55. [Google Scholar]
- Wei, G.; Xu, H.; Ding, P.; Li, S.; Zheng, J. Thermosetting gels with modulated gelation temperature for ophthalmic use: The rheological and gamma scintigraphic studies. J. Control. Release 2002, 83, 65–74. [Google Scholar]
- Johnston, T.; Miller, S. Inulin disposition following intramuscular administration of an inulin/poloxamer gel matrix. J. Parent. Sci. Technol. 1989, 43, 279–286. [Google Scholar]
- Johnston, T.; Miller, S. Toxicological evaluation of poloxamer vehicles for intramuscular use. J. Parent. Sci. Technol. 1985, 39, 83–88. [Google Scholar]
- Fults, K.; Johnston, T. Sustained release of urease from a Poloxamer gel matrix. J. Parenter. Sci. Technol. 1990, 44, 58–65. [Google Scholar]
- Wang, P.; Johnston, T. Sustained-release interleukin-2 following intramuscular injection in rats. Int. J. Pharm. 1995, 113, 73–81. [Google Scholar]
- Katakam, M.; Ravis, W.; Banga, A. Controlled release of human growth hormone in rats following parenteral administration of poloxamer gels. J. Control. Release 1997, 49, 21–26. [Google Scholar]
- Zhang, L.; Parsons, D.; Navarre, C.; Kompella, U. Development (P407), and in-vitro evaluation of sustained release poloxamer 407 gel formulations of ceftiofur. J. Control. Release 2002, 85, 73–81. [Google Scholar]
- Paavola, A.; Kilpelainen, I.; Yliruusi, J.; Rosenberg, P. Controlled release injectable liposomal gel of ibuprofen for epidural analgesia. Int. J. Pharm. 2000, 199, 85–93. [Google Scholar]
- Veyries, M.; Couarraze, G.; Geiger, S.; Agnely, F.; Massias, L.; Kunzli, B.; Faurisson, F.; Rouveix, B. Controlled release of vancomycin from poloxamer 407 gels. Int. J. Pharm. 1999, 192, 183–193. [Google Scholar]
- Wenzel, J.; Balaji, K.; Koushik, K.; Navarre, C.; Duran, S.; Rahe, C.H.; Kompella, U.B. Pluronic F127 gel formulations of deslorelin and GnRH reduce drug degradation and sustain drug release and effect in cattle. J. Control. Release 2002, 8, 51–59. [Google Scholar]
- Wout, Z.; Pec, E.; Maggiore, J.; Williams, R.; Palicharla, P.; Johnston, T. Poloxamer 407-mediated changes in plasma cholesterol and triglycerides following intraperitoneal injection to rats. J. Parent. Sci. Technol. 1992, 46, 192–200. [Google Scholar]
- Johnston, T.; Palmer, W. Mechanism of poloxamer 407-induced hypertriglyceridemia in the rat. Biochem. Pharmacol. 1993, 46, 1037–1042. [Google Scholar]
- Blonder, J.; Baird, L.; Fulfs, J.; Rosenthal, G. Dose-dependent hyperlipidemia in rabbits following administration of poloxamer 407 gel. Life Sci. 1999, 65, 261–266. [Google Scholar]
- Sezgin, Z.; Yüksel, N.; Baykara, T. Preparation and characterisation of polymeric micelles for solubilization of anticancer drugs. Eur. J. Pharm. Biopharm. 2006, 64, 261–268. [Google Scholar]
- Zhao, X.; Yan, J.; Battle, W.; Allums, S.; Bentley, M. Oligomers of poloxamer 407 as degradable thermal sensitive depot materials for sustained drug release. Proc. Int. Symp. Control. Rel. Bioact. Mater. 2003, 30, 167. [Google Scholar]
- Jeong, B.; Bae, Y.; Lee, D.; Kim, S. Biodegradable block copolymers as injectable drug-delivery systems. Nature 1997, 38, 860–862. [Google Scholar]
- Cha, Y.; Choi, Y.; Bae, Y. Thermosensitive Biodegradable Polymers Based on Poly(ether-ester) Block Copolymers. U.S. Patent 5,702,717, 30 December 1997. [Google Scholar]
- Rathi, R.; Zentner, G. Biodegradable Low Molecular Weight Triblock Poly(lactide-co-glycolide) Polyethylene Glycol Copolymers Having Reverse Thermal Gelation Properties. U.S. Patent 6,004,573, 21 December 1999. [Google Scholar]
- Hu, Y.; Jiang, X.; Ding, Y.; Zhang, L.; Yang, C.; Zhang, J.; Chen, J.; Yang, Y. Preparation and drug release behaviors of nimodipine-loaded poly(caprolactone)-poly(ethylene oxide)-polylactide amphiphilic copolymer nanoparticles. Biomaterials 2003, 24, 2395–2404. [Google Scholar]
- Jeong, B.; Choi, Y.; Bae, Y.; Zentner, G.; Kim, S. New biodegradable polymers for injectable drug delivery systems. J. Control. Release 1999, 62, 109–114. [Google Scholar]
- Mothé, C.; Drumond, W.; Wang, S. Phase behavior of biodegradable amphiphilic poly(l,l-lactide)-b-poly(ethylene glycol)-b-poly(l,l-lactide). Thermochim. Acta 2006, 445, 61–66. [Google Scholar]
- Lee, J.; Bae, Y.H.; Sohn, Y.S.; Jeong, B. Thermogelling aqueous solutions of alternating multiblock copolymers of poly(L-lactic acid) and poly(ethylene glycol). Biomacromolecules 2006, 7, 1729–1734. [Google Scholar]
- Kim, Y.; Choi, S.; Koh, J.; Lee, M.; Ko, K.; Kim, S. Controlled release of insulin from biodegradable triblock copolymer. Pharm. Res. 2001, 18, 548–550. [Google Scholar]
- Jeong, B.; Kibbey, M.R.; Birnbaum, J.C.; Won, Y.; Gutowska, A. Thermogelling biodegradable polymers with hydrophilic backbones: PEG-g-PLGA. Macromolecules 2000, 33, 8317–8322. [Google Scholar]
- Jeong, B.; Bae, Y.H.; Kim, S.W. Thermoreversible gelation of PEG-PLGA-PEG triblock copolymer aqueous solutions. Macromolecules 1999, 32, 7064–7069. [Google Scholar]
- Jeong, B. Biodegradable thermosensitive micelles of PEG-PLGA-PEG triblock copolymers. Colloid. Surface. B 1999, 16, 185–193. [Google Scholar]
- Zentner, G.M.; Rathi, R.; Shih, C.; Mcrea, J.C.; Seo, M.; Oh, H.; Hee, B.G.; Mestecky, J.; Moldoveanu, Z.; Morgan, M.; Weitman, S. Biodegradable block copolymers for delivery of proteins and water-insoluble drugs. J. Control. Release 2001, 7, 203–215. [Google Scholar]
- Lee, D.; Shim, M.; Kim, S.; Lee, H.; Park, I.; Chang, T. Novel thermoreversible gelation of biodegradable PLGA-block-PEO-block-PLGA triblock copolymers in aqueous solution. Macromol. Rapid Commun. 2001, 22, 587–592. [Google Scholar]
- Lee, S.; Chin, I.; Jung, J. Crystallization behavior of poly (L-lactide)-poly (ethylene glycol) multiblock copolymers. Eur. Polym. J. 1999, 35, 2147–2153. [Google Scholar]
- Li, F.; Li, S.; Ghzaoui, A.E.; Nouailhas, H.; Zhuo, R. Synthesis and gelation properties of PEG-PLA-PEG triblock copolymers obtained by coupling monohydroxylated PEG-PLA with adipoyl chloride. Langmuir 2007, 23, 2778–2783. [Google Scholar]
- Li, S. Bioresorbable hydrogels prepared through stereocomplexation between poly(l-lactide) and poly(d-lactide) blocks attached to poly(ethylene glycol). Macromol. BioSci. 2003, 3, 657–661. [Google Scholar]
- Zhang, Y.; Wu, X.; Han, Y.; Mo, F.; Duan, Y.; Li, S. Novel thymopentin release systems prepared from bioresorbable PLA-PEG-PLA hydrogels. Int. J. Pharm. 2010, 386, 15–22. [Google Scholar]
- Yasugi, K.; Nakamura, T.; Nagasaki, Y.; Kato, M.; Kataoka, K. Sugar-installed polymer micelles: Synthesis and micellization of poly (ethylene glycol)-poly (d, l-lactide) block copolymers having sugar groups at the peg chain end. Macromolecules 1999, 32, 8024–8032. [Google Scholar]
- Alexandridis, P.; Holzwarthf, J.; Hatton, T. Micellization of poly(ethy1ene oxide)-poly(propy1ene oxide)-poly(ethy1ene oxide) triblock copolymers in aqueous solutions: Thermodynamics of copolymer association. Macromolecules 1994, 27, 2414–2425. [Google Scholar]
- Discher, B.M.; Won, Y.; Ege, D.; Lee, J.; Bates, F.S.; Discher, D.E.; Hammer, D.A. Polymersomes: Tough vesicles made from diblock copolymers. Science 1999, 284, 1143–1146. [Google Scholar]
- Won, Y.; Davis, H.T.; Bates, F.S. Giant wormlike rubber micelles. Science 1999, 283, 960–963. [Google Scholar]
- Chung, Y.; Simmons, K.L.; Gutowska, A.; Jeong, B. Sol-gel transition temperature of PLGA-g-PEG aqueous solutions. Biomacromolecules 2002, 3, 511–516. [Google Scholar]
- Venkatraman, S.S.; Jie, P.; Min, F.; Yin, B.; Freddy, C.; Leong-Huat, G. Micelle-like nanoparticles of PLA-PEG-PLA triblock copolymer as chemotherapeutic carrier. Polymer 2005, 298, 219–232. [Google Scholar]
- Yang, L.; Zhao, Z.; Wei, J.; Ghzaoui, A.; Li, S. Micelles formed by self-assembling of polylactide/poly(ethylene glycol) block copolymers in aqueoous solutions. J. Colloid Interface Sci. 2007, 31, 470–477. [Google Scholar]
- Blanchard, R.; Bhardwaj, J. Controlled-release delivery system for the A-MSH analog melanotan-I using poloxamer 407. J. Pharm. Sci. 1996, 85, 915–919. [Google Scholar]
- Jeong, B.; Bae, Y.H.; Kim, S.W. Drug release from biodegradable injectable thermosensitive hydrogel of PEG-PLGA-PEG triblock copolymers. J. Control. Release 2000, 63, 155–163. [Google Scholar]
- Qiao, M.; Chena, D.; Ma, X.; Liu, Y. Injectable biodegradable temperature-responsive PLGA-PEG-PLGA copolymers: Synthesis and effect of copolymer composition on the drug release from the copolymer-based hydrogels. Int. J. Pharm. 2005, 294, 103–112. [Google Scholar]
- Chen, S.; Pieper, R.; Webster, D.; Singh, J. Triblock copolymers: synthesis, characterization, and delivery of a model protein. Int. J. Pharm. 2005, 288, 207–211. [Google Scholar]
- Yu, L.; Zhang, Z.; Zhang, H.; Ding, J. Mixing a sol and a precipitate of block copolymers with different block ratios leads to an injectable hydrogel. Biomacromolecules 2009, 10, 1547–1553. [Google Scholar]
- Ghahremankhani, A.A.; Dorkoosh, F.; Dinarvand, R. PLGA-PEG-PLGA tri-block copolymers as an in-situ gel forming system for calcitonin delivery. Polym. Bull. 2007, 59, 637–646. [Google Scholar]
- Ghahremankhani, A.; Dorkoosh, F.; Dinarvand, R. PLGA-PEG-PLGA tri-block copolymers as an in-situ gel forming peptide delivery system: Effect of formulation properties on peptide release. Pharm. Dev. Technol. 2008, 13, 49–55. [Google Scholar]
- Qiao, M.; Chen, D.; Ma, X.; Hu, H. Sustained release of bee venom peptide from biodegradable thermosensitive PLGA-PEG-PLGA triblock copolymer-based hydrogels. Pharmazie 2006, 6, 199–202. [Google Scholar]
- Kan, P.; Lin, X.; Hsieh, M.; Chang, K. Thermogelling emulsions for vascular embolization and sustained release of drugs. J. Biomed. Mater. Res. B Appl. Biomater. 2005, 75, 185–192. [Google Scholar]
- Gao, Y.; Sun, Y.; Ren, F.; Gao, S. PLGA-PEG-PLGA hydrogel for ocular drug delivery of dexamethasone acetate. Drug Dev. Ind. Pharm. 2010, 36, 1131–8. [Google Scholar]
- Jarrett, P.; Benedict, C.; Bell, J.P.; Cameron, J.A.; Huang, S.J. Mechanism of the Biodegradation of Polycaprolactone; Polymers as Biomaterials; Plenum Press: New York, NY, USA, 1984. [Google Scholar]
- Cerrai, P.; Guerra, G.D.; Lelli, L.; Tricoli, M.; Sbarbati Del Guerra, R.; Casone, M.G.; Giusti, P. Block copolymers of L-lactide and polyethylene glycol) for biomedical applications. J. Mater. Sci. Mater. Med. 1994, 5, 33–38. [Google Scholar]
- Li, S.; Garreau, H.; Vert, M.; Manolova, N.; Rashkov, I. Hydrolytic degradation of poly(oxyethylene)-poly-(epsiv-caprolactone) multiblock copolymers. J. Appl. Polym. Sci. 1998, 68, 989–998. [Google Scholar]
- Kim, M.; Seo, K.; Khang, G.; Cho, S.; Hb, L. Preparation of poly(ethylene glycol)-block-poly(caprolactone) copolymers and their applications as thermo-sensitive materials. J. Biomed. Mater. Res. A 2004, 70A, 154–158. [Google Scholar]
- Bae, S.; Joo, M.J.; Jeong, Y. Gelation behavior of polyethylene glycol) and polycaprolactone triblock and multiblock copolymer aqueous solutions. Macromolecules 2006, 39, 4873–4879. [Google Scholar]
- Lee, J.; Hua, F.; Lee, D. Thermoreversible gelation of biodegradable poly(ε-caprolactone) and polyethylene glycol) multiblock copolymers in aqueous solutions. J. Control Rel. 2001, 7, 315–317. [Google Scholar]
- Perret, R.; Skoulios, A. Synthèse et caractérisation de copolymères séquencés polyoxyéthylène/poly-e-caprolactone. Makromol. Chem. 1972, 156, 143–156. [Google Scholar]
- Cerrai, P.; Tricoli, M.; Andruzzi, F.; Paci, M. Polyether-polyester block copolymers by non-catalysed polymerization of -caprolactone with polyethylene glycol). Polymer 1989, 30, 338–343. [Google Scholar]
- Bae, S.J.; Suh, J.M.; Sohn, Y.S.; Bae, Y.H.; Kim, S.W.; Jeong, B. Thermogelling poly(caprolactone-b-ethylene glycol-b-caprolactone) aqueous solutions. Macromolecules 2005, 38, 5260–5265. [Google Scholar]
- Shuai, X.; Ai, H.; Nasongkla, N.; Kim, S.; Gao, J. Micellar carriers based on block copolymers of poly(epsilon-caprolactone) and polyethylene glycol) for doxorubicin delivery. J. Control. Release 2004, 98, 415–426. [Google Scholar]
- Hwang, M.J.; Suh, J.M.; Bae, Y.H.; Kim, S.W.; Jeong, B. Caprolactonic poloxamer analog: PEG-PCL-PEG. Biomacromolecules 2005, 6, 885–890. [Google Scholar]
- Letchford, K.; Zastre, J.; Liggins, R.; Burt, H. Synthesis and micellar characterization of short block length methoxy poly(ethylene glycol)-block-poly(caprolactone) diblock copolymers. Colloid. Surface. B 2004, 35, 81–91. [Google Scholar]
- Gong, C.; Shi, S.; Dong, P.; Kan, B.; Gou, M.; Wang, X.; Li, X.; Luoa, F.; Zhaob, X.; Wei, Y.; Qian, Z. Synthesis and characterization of PEG-PCL-PEG thermosensitive hydrogel. Int. J. Pharm. 2009, 365, 89–99. [Google Scholar]
- Gong, C.; Wei, X.; Wang, X.; Wang, Y. Biodegradable self-assembled PEG-PCL-PEG micelles for hydrophobic honokiol delivery: I. Preparation and characterization. Nanotechnology 2010, 21, 215103. [Google Scholar]
- Yin, H.; Gong, C.; Shi, S.; Liu, X.; Wei, Y.; Qian, Z. Toxicity evaluation of biodegradable and thermosensitive peg-pcl-peg hydrogel as a potential sustained ophthalmic drug delivery system. J. Biomed. Mater. Res. B Appl. Biomater. 2009, 92, 129–137. [Google Scholar]
- Gong, C.; Shi, S.; Peng, X.; Kan, B.; Li Yang, L.; Huang, M.J.; Luo, F.; Zhao, X.; Wei, Y.Q.; Qian, Z.Y. Biodegradable thermosensitive injectable PEG-PCL-PEG hydrogel for bFGF antigen delivery to improve humoral immunity. Growth Factors 2009, 27, 377–383. [Google Scholar]
- Ma, G.; Miao, B.; Song, C. Thermosensitive PCL-PEG-PCL hydrogels: Synthesis, characterization and delivery of proteins. J. Appl. Polym. Sci. 2010, 116, 1985–1993. [Google Scholar]
- Master, A.M.; Rodriguez, M.E.; Kenney, M.E.; Oleinick, N.L.; Gupta, A.S. Delivery of the photosensitizer Pc 4 in PEG-PCL micelles for PDT studies. J. Pharm. Sci. 2010, 99, 2386–2398. [Google Scholar]
- Bromberg, L.; Temchenko, M.; Alakhov, V.; Hatton, T.A. Bioadhesive properties and rheology of polyether-modified poly (acrylic acid) hydrogels. Int. J. Pharm. 2004, 282, 45–60. [Google Scholar]
- Hourdet, D.L.F.; Durand, A.; Lafuma, F.; Audebert, R.; Cotton, J. Small-Angle neutron scattering study of microphase separation in thermoassociative copolymers. Macromolecules 1998, 9297, 5323–5335. [Google Scholar]
- Bromberg, L. Handbook of Surfaces and Interfaces Materials; Nalwa, H.E., Ed.; Academic Press: New York, NY, USA, 2011. [Google Scholar]
- Bromberg, L. Polyether-modified poly(acrylic acid): Synthesis and applications. Ind. Eng. Chem. Res. 1998, 37, 4267–4274. [Google Scholar]
- Bromberg, L. Self-assembly in aqueous solutions of polyether-modified poly(acrylic acid). Solutions 1998, 34, 5806–5812. [Google Scholar]
- Bromberg, L. Properties of aqueous solutions and gels of poly(ethylene oxide)-b-poly(propylene oxide)-b-poly(ethylene oxide)-g-poly(acrylic acid). J. Phys. Chem. B 1998, 102, 10736–10744. [Google Scholar]
- Hoffman, A.; Chen, G.; Wu, X.; Ding, Z.; Kabra, B.; Randeri, K.; Schiller, M.; Ron, E.; Peppas, N.A. Graft copolymers of PEO-PPO-PEO triblock polyethers on bioadhesive polymer backbones: Synthesis and properties. Polym. Prepr. 1997, 38, 524–525. [Google Scholar]
- Bromberg, L. Novel family of thermogelling materials via C–C bonding between poly(acrylic acid) and poly(ethylene oxide)-b-poly(propylene oxide)-b-poly(ethylene oxide). J. Phys. Chem. 1998, 102, 1956–1963. [Google Scholar]
- Ho, A.K.; Bromberg, L.E.; Huibers, P.D.; Connor, A.J.; Perera, J.M.; Stevens, G.; Hatton, T.A. Hydrophobic domains in thermogelling solutions of polyether-modified poly(acrylic acid). Langmuir 2002, 18, 3005–3013. [Google Scholar]
- Bromberg, L. Properties of aqueous solutions and gels of poly(ethylene oxide)-b-poly(propylene oxide)-b-poly(ethylene oxide)-g-poly(acrylic acid). J. Phys. Chem. B 1998, 102, 10736–10744. [Google Scholar]
- Staples, T.L.; Henton, D.E.; Buchholz, F.L. Chemistry of superabsorbent polyacrylates. In Modern Superabsorbent Polymer Technology; Buchholz, F.L., Graham, A.T.E., Eds.; Wiley: New York, NY, USA, 1997. [Google Scholar]
- Bromberg, L.E.; Goldfeld, M.G. Self-assembly in aqueous solutions of hydrophobically modified poly(acrylic acid). Polym. Prepr. 1998, 39, 681–682. [Google Scholar]
- Huibers, P.D.; Bromberg, L.E.; Robinson, B.H.; Hatton, T.A. Reversible gelation in semidilute aqueous solutions of associative polymers: A small-angle neutron scattering study. Macromolecules 1999, 32, 4889–4894. [Google Scholar]
- Bromberg, L.E.; Barr, D.P. Aggregation phenomena in aqueous solutions of hydrophobically modified polyelectrolytes. A probe solubilization study. Macromolecules 1999, 32, 3649–3657. [Google Scholar]
- Bromberg, L.; Ron, E. Temperature-responsive gels and thermogelling polymermatrices for protein and peptide delivery. Adv. Drug. Deliv. Rev. 1998, 3, 197–221. [Google Scholar]
- Ron, E.; Bromberg, L.; Luczak, S.; Kearney, M.; Deaver, D.; Schiller, M. Smart hydrogel: A novel mucosal delivery system. Proc. Int. Symp. Control. Rel. Bioact. Mater. 1997, 2, 407–408. [Google Scholar]
- Ron, E.; Roos, E.; Staples, A.; Bromberg, L.; Schiller, M. Interpenetrating polymer networks for sustained dermal delivery. Proceedings of the International Symposium on the Controlled Release of Bioactive Material Controlled Release Society, Kyoto, Japan, July 1996; 23, pp. 128–129.
- Potts, A.; Jackson, S.; Washington, N.; Gilchrist, P.; Ron, E.; Schiller, M.; Wilson, C.G. In vivo determination of the oesophageal retention of smart hydrogel. Proc. Int. Symp. Control. Rel. Bioact. Mater. 1997, 24, 335–336. [Google Scholar]
- Bromberg, L.; Temchenko, M.; Hatton, T. Dually responsive microgels from polyether-modified poly(acrylic acid): Swelling and drug loading. Langmuir 2002, 18, 4944–4952. [Google Scholar]
- Tian, Y.; Rav, P.; Bromberg, L.; Hatton, T.; Tam, K. Synthesis and aggregation behavior of pluronic F87/poly(acrylic acid) block copolymer in the presence of doxorubicin. Langmuir 2007, 23, 2638–2646. [Google Scholar]
- Bromberg, L.; Ron, E. Protein and peptide release from temperature-responsive gels and thermogelling polymer matrices. Adv. Drug Delivery Rev. 1998, 3, 197–221. [Google Scholar]
- Bromberg, L.; Alakhov, V. Effects of polyether-modified poly(acrylic acid) microgels on doxorubicin transport in human intestinal epithelial Caco-2 cell layers. J. Control. Release 2003, 88, 11–22. [Google Scholar]
- Alakhov, V.; Pietrzynski, G.; Patel, K.; Kabanov, A.; Bromberg, L.; Hatton, T. Pluronic block copolymers and Pluronic poly(acrylic acid) microgels in oral delivery of megestrol acetate. J. Pharm. Pharmacol. 2004, 56, 1233–1241. [Google Scholar]
- Woo, J.; Lee, C.; Shim, C.; Hwang, S. Enhanced oral bioavailability of paclitaxel by coadministration of the P-glucoprotein inhibitor KR30031. Pharm. Res. 2003, 20, 24–30. [Google Scholar]
- Bromberg, L. Polymeric micelles in oral chemotherapy. J. Control. Release 2008, 128, 99–112. [Google Scholar]
- Barreiro-Iglesias, R.; Bromberg, L.; Temchenko, M.; Hatton, T.A.; Concheiro, A.; Alvarez-Lorenzo, C. Solubilization and stabilization of camptothecin in micellar solutions of pluronic-g-poly(acrylic acid) copolymers. J. Control. Release 2004, 97, 537–549. [Google Scholar]
- Kretlow, J.D.; Klouda, L.; Mikos, A.G. Injectable matrices and scaffolds for drug delivery in tissue engineering. Adv. Drug Delivery Rev. 2007, 59, 263–273. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bonacucina, G.; Cespi, M.; Mencarelli, G.; Giorgioni, G.; Palmieri, G.F. Thermosensitive Self-Assembling Block Copolymers as Drug Delivery Systems. Polymers 2011, 3, 779-811. https://doi.org/10.3390/polym3020779
Bonacucina G, Cespi M, Mencarelli G, Giorgioni G, Palmieri GF. Thermosensitive Self-Assembling Block Copolymers as Drug Delivery Systems. Polymers. 2011; 3(2):779-811. https://doi.org/10.3390/polym3020779
Chicago/Turabian StyleBonacucina, Giulia, Marco Cespi, Giovanna Mencarelli, Gianfabio Giorgioni, and Giovanni Filippo Palmieri. 2011. "Thermosensitive Self-Assembling Block Copolymers as Drug Delivery Systems" Polymers 3, no. 2: 779-811. https://doi.org/10.3390/polym3020779
APA StyleBonacucina, G., Cespi, M., Mencarelli, G., Giorgioni, G., & Palmieri, G. F. (2011). Thermosensitive Self-Assembling Block Copolymers as Drug Delivery Systems. Polymers, 3(2), 779-811. https://doi.org/10.3390/polym3020779