3.1. Photopolymerization of MMA Using CuSO4·5H2O/TPMA Catalyst Complex in Various Solvents
Recently, it was published that CuSO
4·5H
2O was able to control the polymerization of methyl methacrylate when a reducing agent, such as ascorbic acid or Fe
0, was used [
20]. In that study, however, a large amount of the copper catalyst, in an equimolar ratio to an alkyl halide initiator, was used. Therefore, in our study, we decided to investigate the possibility of finding the polymerization conditions for photochemically mediated RDRP using only ppm amounts of CuSO
4·5H
2O. UV-vis light with wavelengths over 365 nm was used to photochemically reduce the catalyst without addition of any other reducing agent.
Initially, three different solvents, anisole, DMF and DMSO, were tested to select the best one for all further studies. Based on our previous experience, BPN and TPMA were used as an initiator and ligand, respectively, for these experiments. The CuSO
4·5H
2O catalyst was used at a concentration of 8.5 × 10
−4 mol/L (200 ppm with respect to monomer). It should be noted that even though the amount of CuSO
4·5H
2O/TPMA complex used was completely soluble in pure DMSO, after the addition of MMA it was partially precipitated. On the other hand, in pure anisole, the CuSO
4·5H
2O/TPMA complex did not completely dissolve. As shown in
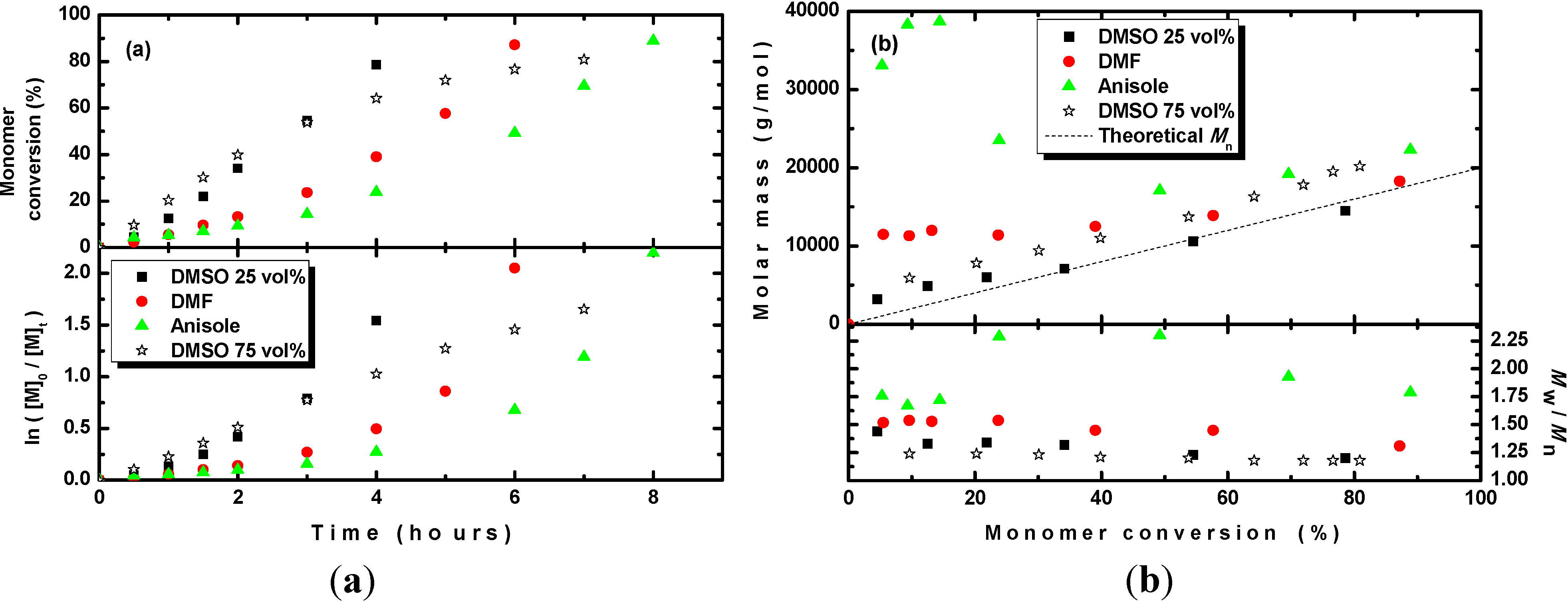
Figure 1a, the photoRDRP of MMA in anisole was the slowest, whereas in DMSO it was much faster than that in either anisole or DMF. In all cases, the polymerization did not follow first-order kinetics, but an increase in polymerization rate with time was observed. This trend could likely be explained by the progressive dissolution of all insoluble parts of the CuSO
4·5H
2O/TPMA complex due to the progressive reduction of the soluble part of the CuSO
4·5H
2O/TPMA complex. When the content of DMSO was increased from 25 vol% to 75 vol% to increase the solubility of the complex, the polymerization followed first-order kinetics (see
Figure 1a). In
Figure 1b, it is shown that although the molar masses agree well with the theoretical ones and a dispersity of approximately 1.2 was obtained in DMSO, in DMF the polymers with slightly higher molar masses and slightly broader dispersities were formed. Contrary to those in anisole, polymers with much higher molar masses and dispersities of approximately 2 were obtained. The reason may be the very low solubility of the CuSO
4·5H
2O/TPMA complex in anisole leading to an insufficient amount of the catalyst in the system and/or the insufficiently fast formation of the CuBr
2/TPMA deactivator.
Figure 1.
(a) Kinetic plots and (b) evolution of the molar mass and Mw/Mn with the conversion of MMA during photoRDRP using CuSO4·5H2O/TPMA catalyst complex in various solvents. BPN was used as an initiator. Experimental conditions: photoRDRP, MMA/BPN/CuII/L = 200/1/0.04/0.16, irradiation at λ > 350 nm; in all experiments: [MMA] = 7.5 M, T = 35 °C, 25 vol% of solvent.
Figure 1.
(a) Kinetic plots and (b) evolution of the molar mass and Mw/Mn with the conversion of MMA during photoRDRP using CuSO4·5H2O/TPMA catalyst complex in various solvents. BPN was used as an initiator. Experimental conditions: photoRDRP, MMA/BPN/CuII/L = 200/1/0.04/0.16, irradiation at λ > 350 nm; in all experiments: [MMA] = 7.5 M, T = 35 °C, 25 vol% of solvent.
To confirm the formation of CuBr
2/TPMA deactivator
in situ after the photochemical reduction of the CuSO
4·5H
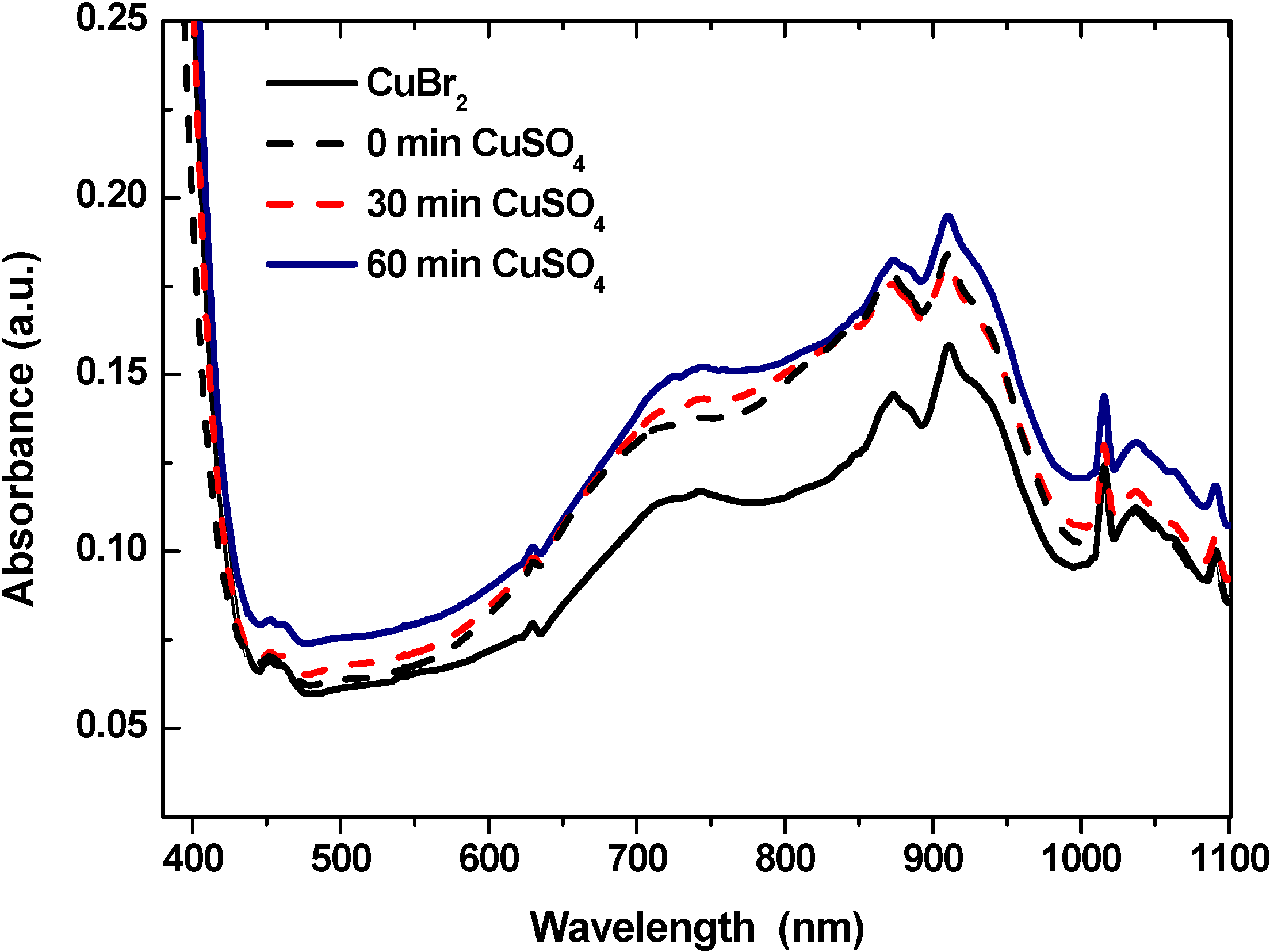
2O/TPMA complex and subsequent abstraction of bromide from the alkyl bromide initiator, changes in the UV-vis spectra after various periods of irradiation in DMSO were monitored. As shown in
Figure 2, the original spectra of the CuSO
4·5H
2O/TPMA complex varied during irradiation. A progressive increase in absorbance in the region of 700–800 nm was observed, whereas a shoulder in this region, typical for the CuSO
4·5H
2O/TPMA complex, transformed after 60 min of irradiation into an absorption peak at λ
max = 742 nm. A similar spectrum featuring an absorption peak at λ
max = 742 nm is typical for CuBr
2/TPMA complex.
3.2. Effect of Ligand Content and Ligand Structure on Photopolymerization of MMA Using CuSO4·5H2O Catalyst
Based on the above-described results, DMSO was chosen as a solvent for all further studies. It was recently reported that excess ligand was essential to the photoRDRP of acrylates when a CuBr
2/Me
6TREN catalytic complex was used in DMSO [
18,
19]. Therefore, we tested how the CuSO
4·5H
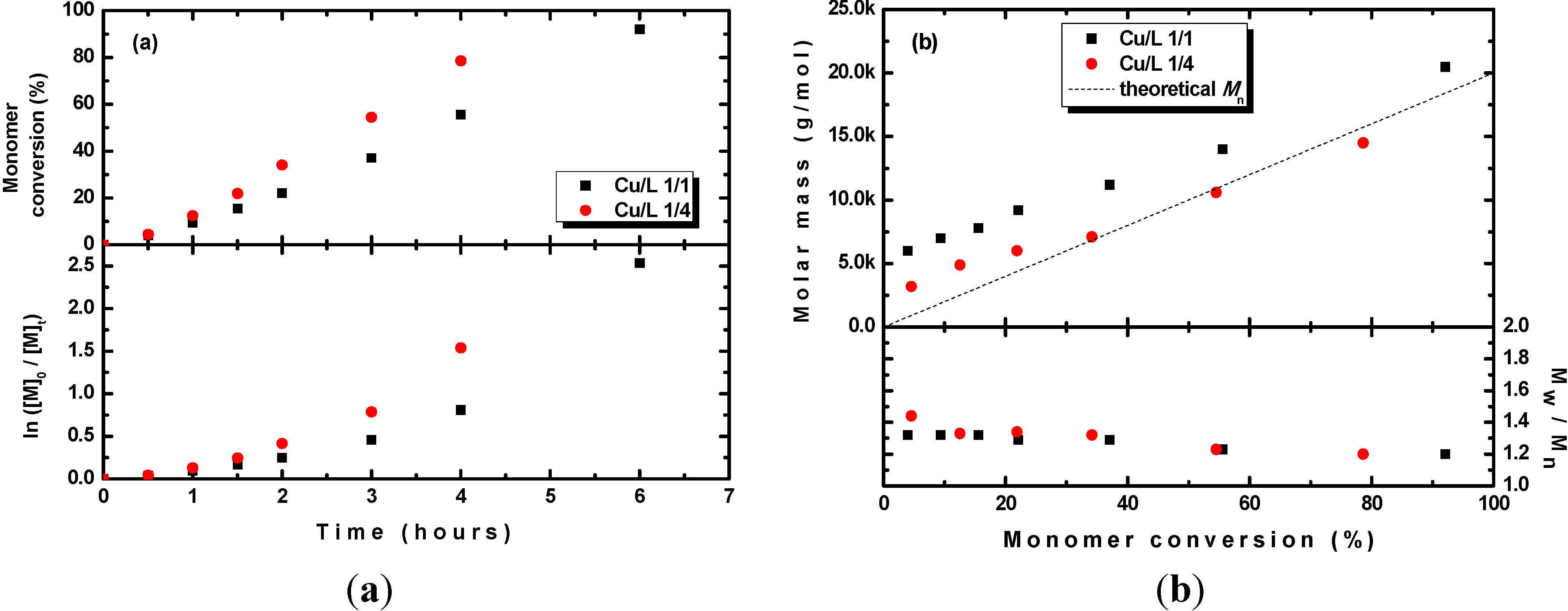
2O/TPMA ratio affected the polymerization of MMA. As shown in
Figure 3, the polymerization was slightly faster when excess ligand was used. In addition, although with excess ligand the molar masses agreed well with the theoretical ones, in the case of an equimolar ratio of ligand to copper catalyst, molar masses slightly higher than the theoretical ones were obtained. On the other hand, in both cases, the dispersities were narrow. Thus, even though the polymerization could also proceed using an equimolar ratio of the ligand, better control over the molar mass and slightly faster polymerization could be achieved using excess ligand. This finding indicates the possible participation of the tertiary amine in the photochemical reduction of the Cu
II catalyst, as recently suggested for CuBr
2 during the photoRDRP of acrylates [
19], even though in the latter case no polymerization of acrylates was observed when an equimolar ratio of CuBr
2 and Me
6TREN was used.
Figure 2.
UV-vis spectra of CuSO4·5H2O/TPMA complex in DMSO before irradiation and after 30 and 60 min of irradiation at λ > 350 nm and UV-vis spectrum of CuBr2/TPMA complex.
Figure 2.
UV-vis spectra of CuSO4·5H2O/TPMA complex in DMSO before irradiation and after 30 and 60 min of irradiation at λ > 350 nm and UV-vis spectrum of CuBr2/TPMA complex.
Figure 3.
(a) Kinetic plots and (b) evolution of the molar mass and Mw/Mn with the conversion of MMA during photoRDRP using CuSO4·5H2O/TPMA ratios of 1/1 and 1/4. BPN was used as an initiator. Experimental conditions: photoRDRP, MMA/BPN/CuII = 200/1/0.04, irradiation at λ > 350 nm; [MMA] = 7.5 M, T = 35 °C, DMSO (25 vol%).
Figure 3.
(a) Kinetic plots and (b) evolution of the molar mass and Mw/Mn with the conversion of MMA during photoRDRP using CuSO4·5H2O/TPMA ratios of 1/1 and 1/4. BPN was used as an initiator. Experimental conditions: photoRDRP, MMA/BPN/CuII = 200/1/0.04, irradiation at λ > 350 nm; [MMA] = 7.5 M, T = 35 °C, DMSO (25 vol%).
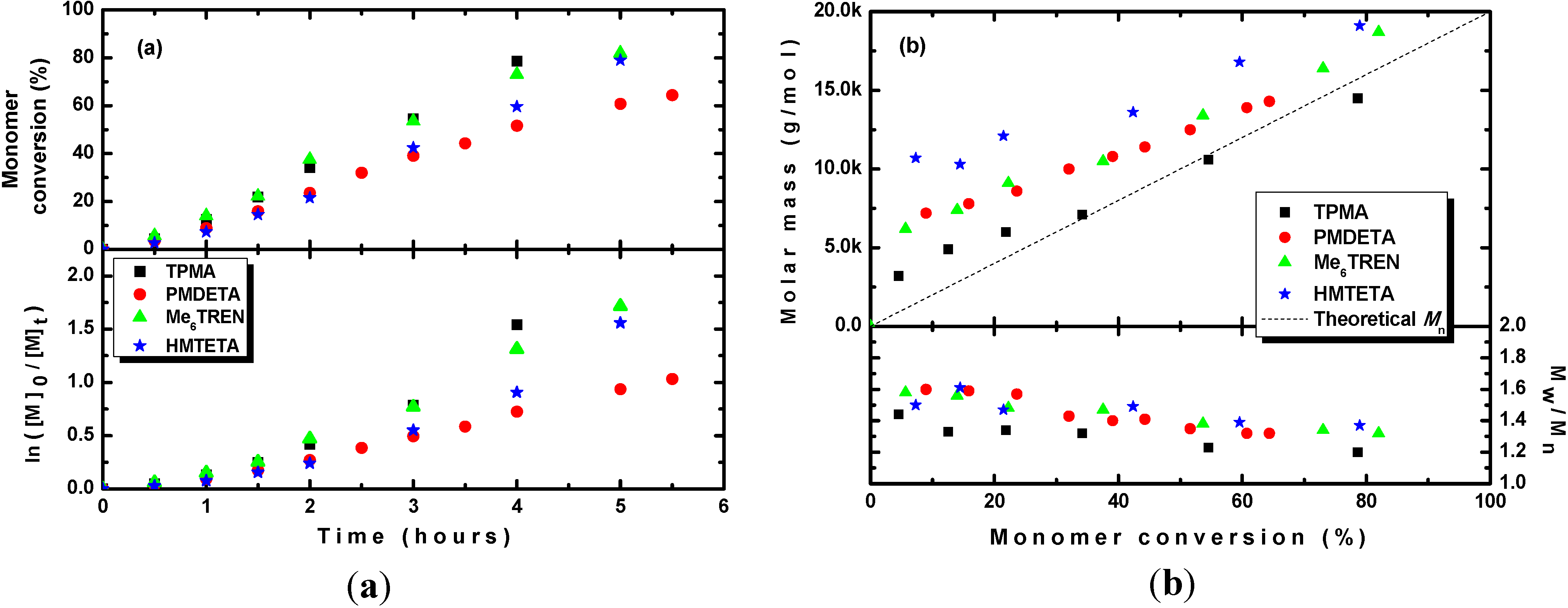
To investigate the effect of the type of ligand used, three other ligands were evaluated. In all cases, the CuSO
4·5H
2O/ligand (L) ratio was maintained at 1/4. As shown in
Table 1, Entries 1–4, after 3 h of irradiation, a monomer conversion similar to that obtained for TPMA was observed for Me
6TREN, which is known to form more active complexes in normal ATRP. In contrast, ligands such as PMDETA and HMTETA, which are known to form less active complexes, yielded lower conversions after 3 h of irradiation. This observation was also confirmed by kinetic studies (see
Figure 4a), in which similar rates of polymerization were obtained for the TPMA and Me
6TREN ligands and slightly lower polymerization rates were obtained for the PMDETA and HMTETA ligands. However, a comparison of the dispersities after 3 h of polymerization (see
Table 1, Entries 1–4) revealed much broader dispersities for all three additionally tested ligands,
i.e., Me
6TREN, PMDETA and HMTETA, than the dispersity observed for TPMA. Kinetic studies also showed that the molar masses were higher than the theoretical ones for the Me
6TREN, PMDETA and HMTETA ligands, and only when TPMA was used did the molar masses agree well with the theoretical ones (see
Figure 4b).
Figure 4.
(a) Kinetic plots and (b) evolution of the molar mass and Mw/Mn with the conversion of MMA during photoRDRP using CuSO4·5H2O catalyst and various ligands (L). BPN was used as an initiator. Experimental conditions: photoRDRP, MMA/BPN/CuII/L = 200/1/0.04/0.16, irradiation at λ > 350 nm; in all experiments: [MMA] = 7.5 M, T = 35 °C, DMSO (25 vol%).
Figure 4.
(a) Kinetic plots and (b) evolution of the molar mass and Mw/Mn with the conversion of MMA during photoRDRP using CuSO4·5H2O catalyst and various ligands (L). BPN was used as an initiator. Experimental conditions: photoRDRP, MMA/BPN/CuII/L = 200/1/0.04/0.16, irradiation at λ > 350 nm; in all experiments: [MMA] = 7.5 M, T = 35 °C, DMSO (25 vol%).
3.3. Effect of CuSO4·5H2O Catalyst Concentration on Photopolymerization of MMA
In this section, the effect of the CuSO
4·5H
2O catalyst concentration in the range of 50–400 ppm was investigated while the concentrations of all other species, including the initiator and ligand, were held constant. A summary of the experiments is provided in
Table 1, Entries 1 and 5–7. The monomer conversion after 3 h was nearly the same regardless of whether 100, 200 or 400 ppm of catalyst with respect to the monomer was used. Nearly the same molar masses and dispersities were also obtained. This result suggests that regardless of the concentration of the catalyst, the solubility of the catalyst could be the limiting factor for both the polymerization rate as well as for the molar mass and dispersity evolution. On the other hand, reducing the catalyst concentration to 50 ppm (see
Table 1, Entry 7) led to a decrease in monomer conversion after 3 h of irradiation. At the same time, the molar mass was slightly higher than the theoretical one and the dispersity was slightly broader, despite still being below 1.3. Because in this case the ligand was in sixteen-fold excess, we further tried to reduce the ligand concentration to only four-fold excess to maintain a copper/ligand ratio of 1/4, as in the studies described in previous sections. As shown in
Table 1, Entry 8, this decrease in the ligand concentration led to a further decrease in the polymerization rate. At the same time, the difference between the experimental and theoretical molar masses was slightly higher. This finding confirmed the effect of the ligand concentration on the polymerization rate and the control of polymerization with respect to molar mass evolution described in the previous section.
Table 1.
Results of photoRDRP of methyl methacrylate. Polymerizations were performed in 25 vol% DMSO at 35 °C with a monomer/initiator ratio of 200/1. Concentration of CuII catalyst in ppm with respect to that of the monomer; 2-bromopropionitrile and 2-chloropropionitrile were used as initiators in Entries 1–13 and 14–15, respectively. Monomer conversion was determined from 1H NMR spectra and molar mass and dispersity by GPC using PMMA standards.
Table 1.
Results of photoRDRP of methyl methacrylate. Polymerizations were performed in 25 vol% DMSO at 35 °C with a monomer/initiator ratio of 200/1. Concentration of CuII catalyst in ppm with respect to that of the monomer; 2-bromopropionitrile and 2-chloropropionitrile were used as initiators in Entries 1–13 and 14–15, respectively. Monomer conversion was determined from 1H NMR spectra and molar mass and dispersity by GPC using PMMA standards.
| Entry | Type of Cu(II) | Cu(II) (ppm) | Ligand | Cu(II)/L ratio | Time [h] | Conv. (%) | Mn, theor (g/mol) | Mn, exp. (g/mol) | Mw/Mn |
|---|
| 1 | CuSO4 | 200 | TPMA | 1/4 | 3 | 55 | 11,130 | 10,600 | 1.23 |
| 2 | CuSO4 | 200 | PMDETA | 1/4 | 3 | 39 | 7,930 | 10,800 | 1.40 |
| 3 | CuSO4 | 200 | HMTETA | 1/4 | 3 | 42 | 8,530 | 13,600 | 1.49 |
| 4 | CuSO4 | 200 | Me6TREN | 1/4 | 3 | 54 | 10,930 | 13,400 | 1.38 |
| 5 | CuSO4 | 400 | TPMA | 1/2 | 3 | 53 | 10,730 | 11,200 | 1.22 |
| 6 | CuSO4 | 100 | TPMA | 1/8 | 3 | 58 | 11,730 | 11,200 | 1.22 |
| 7 | CuSO4 | 50 | TPMA | 1/16 | 3 | 44 | 8,930 | 10,800 | 1.28 |
| 8 | CuSO4 | 50 | TPMA | 1/4 | 3 | 37 | 7,530 | 11,200 | 1.30 |
| 9 | Cu(OTf)2 | 200 | TPMA | 1/4 | 3 | 56 | 11,330 | 12,000 | 1.20 |
| 10 | Cu(OAc)2 | 200 | TPMA | 1/4 | 3 | 62 | 12,530 | 15,800 | 1.16 |
| 11 | Cu(acac)2 | 200 | TPMA | 1/4 | 3 | 56 | 11,330 | 11,800 | 1.31 |
| 12 | CuBr2 | 200 | TPMA | 1/4 | 3 | 58 | 11,730 | 11,600 | 1.13 |
| 13 | CuCl2 | 200 | TPMA | 1/4 | 3 | 61 | 12,330 | 15,200 | 1.15 |
| 14 | CuCl2 | 100 | TPMA | 1/1 | 3 | 5 | 1,130 | 7,800 | 1.47 |
| 15 | CuCl2 | 500 | TPMA | 1/1 | 6 | 18 | 3,730 | 9,600 | 1.33 |
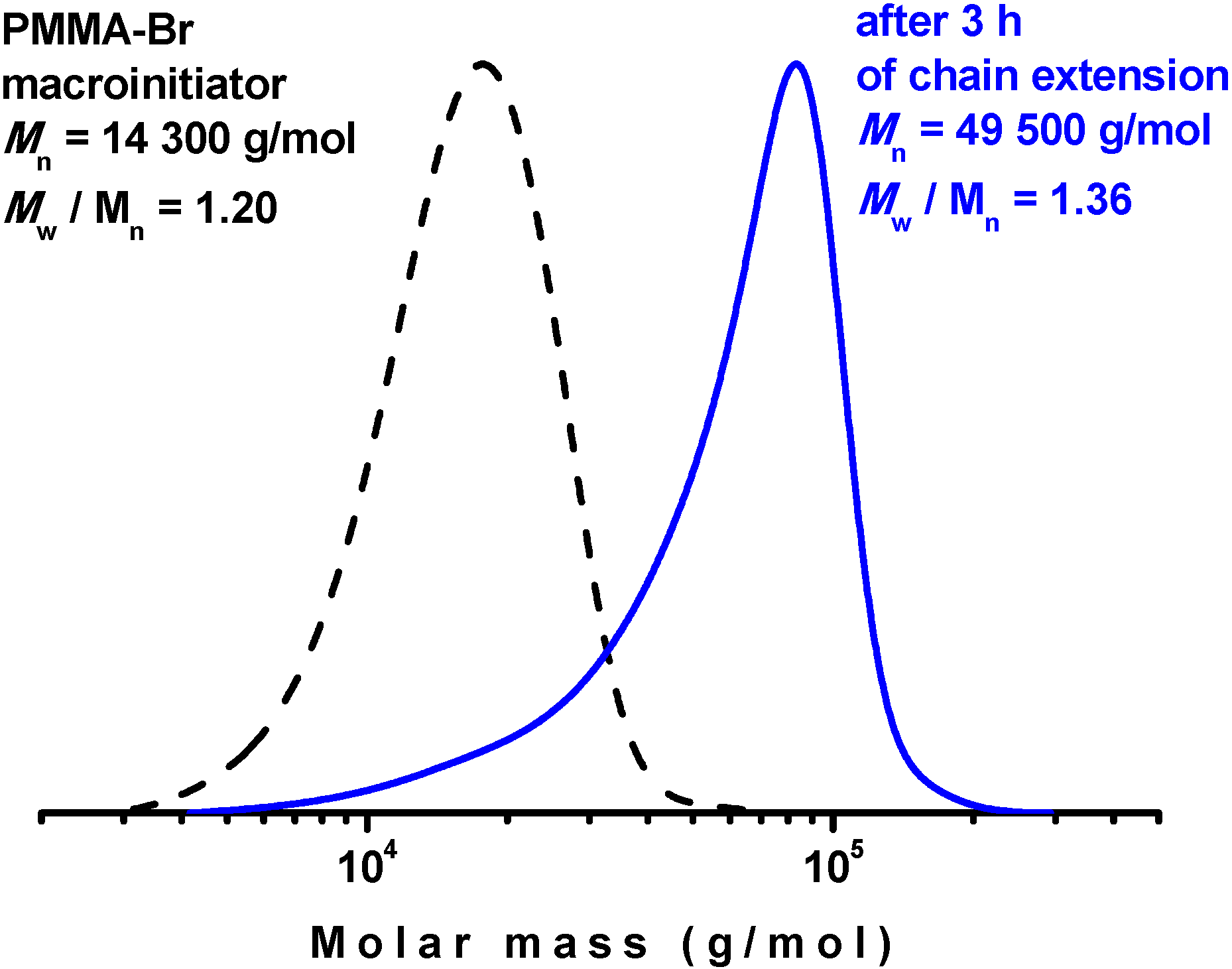
3.4. Chain Extension PhotoRDRP Using CuSO4·5H2O/TPMA Catalyst Complex
The living character of the photoRDRP catalyzed by CuSO
4·5H
2O/TPMA was demonstrated by the chain extension of a PMMA-Br macroinitiator. The PMMA-Br macroinitiator (
Mn = 14,300 g/mol and
Mw/
Mn = 1.20) was prepared by photoRDRP under experimental conditions similar to those described above, with an MMA/BPN/CuSO
4·5H
2O/TPMA ratio of 200/1/0.04/0.16. The polymerization reaction was terminated after 3.5 h at 62% monomer conversion. Chain extension was then performed under identical experimental conditions with an MMA/PMMA-Br/CuSO
4·5H
2O/TPMA ratio of 600/1/0.12/0.48. A higher degree of polymerization was targeted in the chain extension polymerization to provide substantial separation between the macroinitiator and the extended polymer in the GPC traces. After 3 h (73% conversion), the chain-extended PMMA with
Mn = 49,500 g/mol and
Mw/
Mn = 1.36 was obtained. As indicated by the GPC traces of the PMMA before and after the chain extension polymerization (
Figure 5), the molar mass clearly increased after chain extension. A small tail could be observed, which suggests that some non-extended PMMA chains were present in the polymerization mixture. Considering that at the beginning of the polymerization no deactivator was present in the system until it was formed by the reaction of reduced copper with the initiator, some termination reactions can be expected. The range of the termination reactions should be, however, limited by the rather small amount of catalyst used.
Figure 5.
GPC traces from chain extension of PMMA-Br with MMA; (dash black curve) PMMA-Br macroinitiator and (solid blue curve) chain-extended PMMA after 73% conversion of MMA. The preparation of the PMMA-Br macroinitiator and the chain extension were performed by photoRDRP using a CuSO4·5H2O/TPMA catalyst complex. Experimental conditions: preparation of PMMA-Br, MMA/BPN/CuII/TPMA = 200/1/0.04/0.16, DMSO (25 vol%); chain extension: MMA/PMMA–Br/CuII/TPMA = 600/1/0.12/0.48, DMSO (50 vol%); in both polymerizations: [MMA] = 7.5 M, T = 35 °C, irradiation at λ > 350 nm.
Figure 5.
GPC traces from chain extension of PMMA-Br with MMA; (dash black curve) PMMA-Br macroinitiator and (solid blue curve) chain-extended PMMA after 73% conversion of MMA. The preparation of the PMMA-Br macroinitiator and the chain extension were performed by photoRDRP using a CuSO4·5H2O/TPMA catalyst complex. Experimental conditions: preparation of PMMA-Br, MMA/BPN/CuII/TPMA = 200/1/0.04/0.16, DMSO (25 vol%); chain extension: MMA/PMMA–Br/CuII/TPMA = 600/1/0.12/0.48, DMSO (50 vol%); in both polymerizations: [MMA] = 7.5 M, T = 35 °C, irradiation at λ > 350 nm.
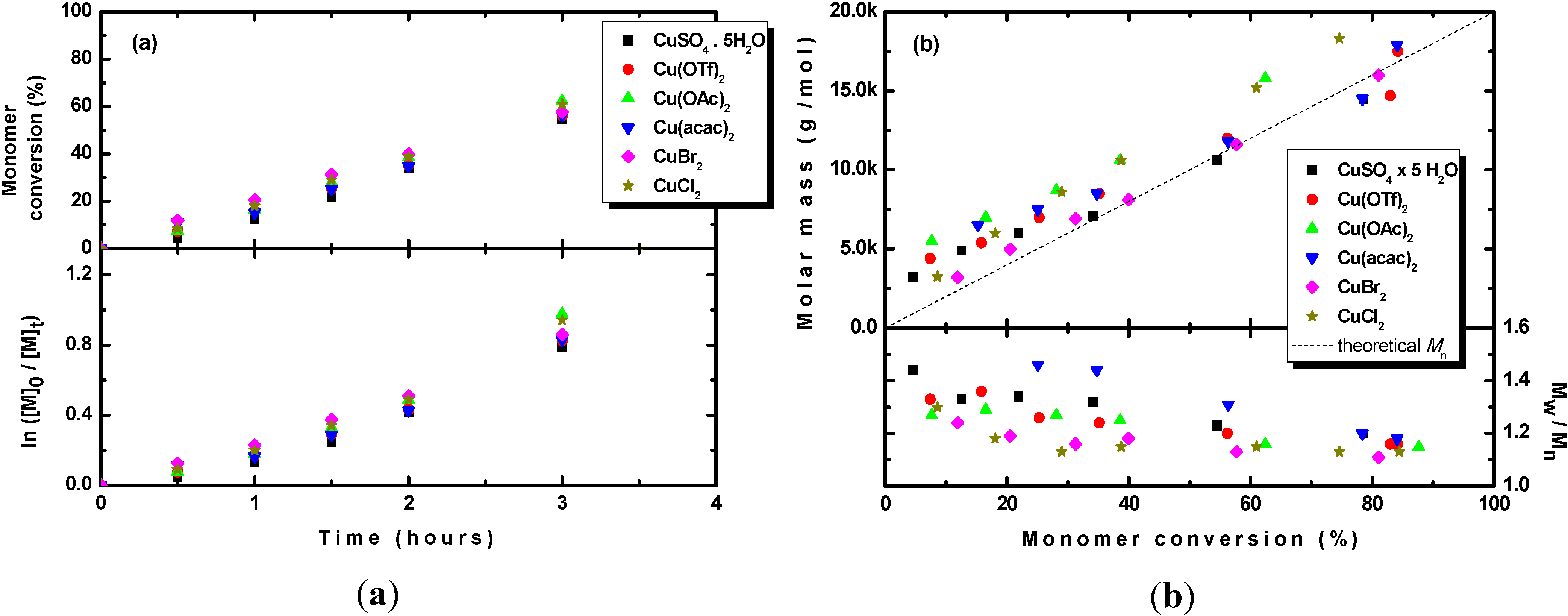
3.5. Effect of Copper Catalyst Structure on Photopolymerization of MMA
Because it was observed that the CuSO
4·5H
2O catalyst was able to control the polymerization of methyl methacrylate under photoRDRP conditions, the applicability of other copper compounds in photoRDRP was further investigated. Three different organic copper compounds, copper acetate (Cu(OAc)
2), copper triflate (Cu(OTf)
2) and copper acetylacetonate (Cu(acac)
2), were investigated and compared with commonly used ATRP catalysts, such as CuBr
2 and CuCl
2. The advantage of the organic copper compounds is their better solubility in organic solvents and thus their ability to support polymerization in homogeneous systems. A concentration of 200 ppm of the catalyst and TPMA in four-fold excess with respect to the catalyst concentration was used in all experiments. A summary of the experiments is provided in
Table 1, Entries 9–13. The results show that for all tested copper catalysts, the monomer conversions after 3 h of irradiation were very similar, falling within the range of 55%–62%. Kinetic studies confirmed that the polymerization rate was almost the same for all tested copper catalysts (see
Figure 6a). In one of the previous sections, it was demonstrated that the CuSO
4·5H
2O catalyst was transformed
in situ into CuBr
2. The same polymerization rate for various copper catalysts confirmed this observation. As suggested by the schematic mechanism shown in
Scheme 1, regardless of type of catalyst used, the Cu
IIX
2/L complex, where X can be 1/2(SO
4), OTf, OAc, acac, Cl or Br, must be photochemically reduced to form the Cu
IX/L complex, which is able to react with alkyl bromide to form an alkyl radical and Cu
IIXBr/L. The Cu
IIXBr/L can subsequently act as a deactivator to form alkyl bromide and Cu
IX/L or can be photochemically reduced to a Cu
IBr/L complex and subsequently abstract another bromide from alkyl bromide to form an alkyl radical and Cu
IIBr
2/L. Ultimately, an equilibrium between the Cu
IBr/L and Cu
IIBr
2/L complexes is reached. Because the CuBr
2 catalyst can be expected to form after the reduction of all of the copper catalysts studied, regardless of their structure, the polymerization will still be controlled by the CuBr/CuBr
2/L catalytic system formed
in situ.
Figure 6.
(a) Kinetic plots and (b) evolution of the molar mass and Mw/Mn with the conversion of MMA during photoRDRP using various copper catalysts. BPN and TPMA were used as the initiator and ligand, respectively. Experimental conditions: photoRDRP, MMA/BPN/CuII/TPMA = 200/1/0.04/0.16, irradiation at λ > 350 nm; in all experiments: [MMA] = 7.5 M, T = 35 °C, DMSO (25 vol%).
Figure 6.
(a) Kinetic plots and (b) evolution of the molar mass and Mw/Mn with the conversion of MMA during photoRDRP using various copper catalysts. BPN and TPMA were used as the initiator and ligand, respectively. Experimental conditions: photoRDRP, MMA/BPN/CuII/TPMA = 200/1/0.04/0.16, irradiation at λ > 350 nm; in all experiments: [MMA] = 7.5 M, T = 35 °C, DMSO (25 vol%).
Scheme 1.
Schematic mechanism of photochemical reduction of various copper(II) compounds and subsequent formation of CuBr/CuBr2 equilibrium ensuring good control in RDRP.
Scheme 1.
Schematic mechanism of photochemical reduction of various copper(II) compounds and subsequent formation of CuBr/CuBr2 equilibrium ensuring good control in RDRP.
Slight differences in the evolution of the molar mass and dispersity were observed, where slightly higher molar masses and slightly broader dispersities were observed, mostly at lower conversions, for the CuSO4·5H2O and organic copper catalysts compared with those obtained for copper halides. The reason for this observation has already been discussed, i.e., the absence of the deactivator at the beginning of the polymerization. However, except for the cases in which CuCl2 or Cu(OAc)2 were used, for the other catalysts, the molar masses agreed well with the theoretical ones, and dispersities below 1.2 were obtained at higher monomer conversions.
It should also be noted that when 100 ppm of CuCl
2/TPMA catalyst complex was used in combination with a chlorinated initiator, such as chloropropionitrile, only 5% monomer conversion was obtained after 3 h of irradiation and the obtained molar mass was much higher than the theoretical one (see
Table 1, Entry 14). Even after increasing the catalyst concentration to 500 ppm, only 18% monomer conversion was obtained after 6 h of irradiation, whereas a much higher molar mass than the theoretical one was obtained (see
Table 1, Entry 15). These results suggest that initiation (activation) of chloride-based initiators is not sufficiently fast to be applicable in the photoRDRP of methyl methacrylate.
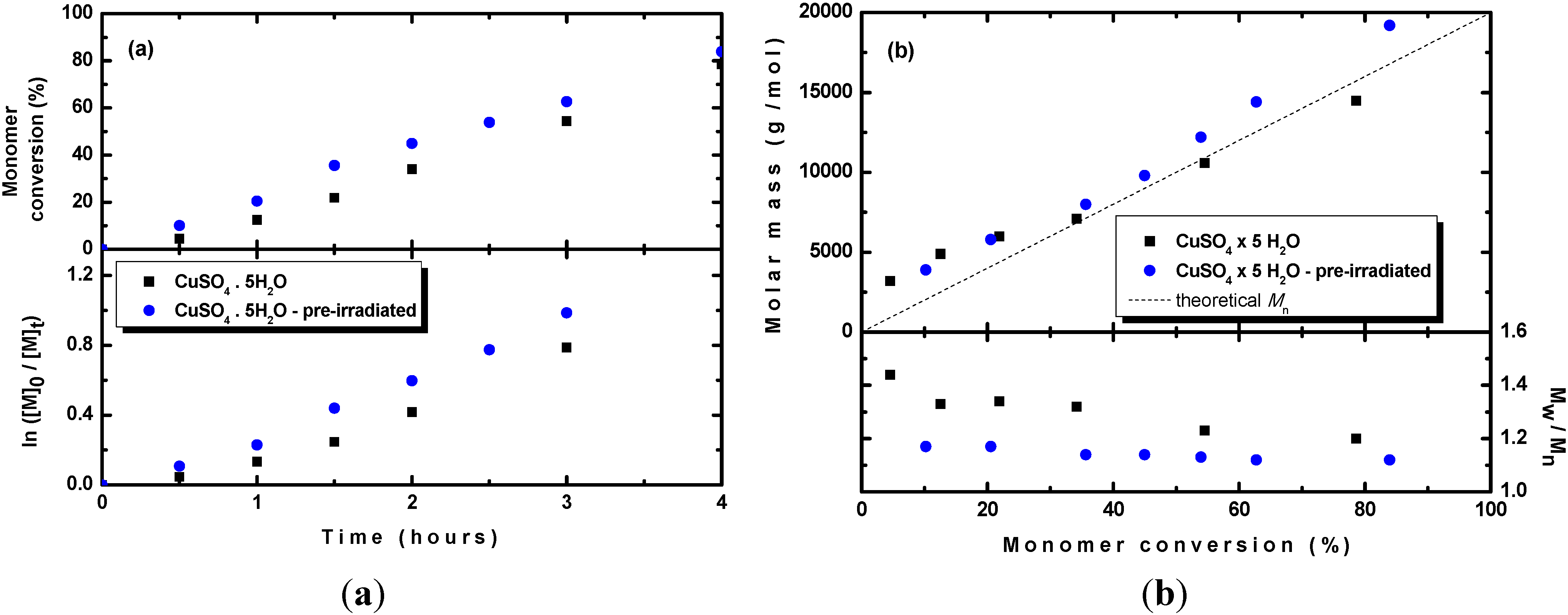
Based on the submitted mechanism, the CuBr/CuBr
2 equilibrium could be obtained by photochemical reduction of any copper catalyst prior to the addition of monomer and thus improved control over molar mass and dispersity could be achieved. Therefore, the CuSO
4·5H
2O/TPMA complex was first pre-irradiated in DMSO in the presence of BPN as an alkyl bromide initiator for 1 h to obtain the CuBr/CuBr
2 equilibrium. Subsequently a MMA was added to initiate the polymerization. As shown in
Figure 7, the kinetic of polymerization was similar whether monomer was added before irradiation or after 1 h of irradiation. In the first case, slight curvature was observed due to progressive dissolving of the catalyst, as already described above, whereas in the case of pre-irradiation, the dependence from the beginning followed the first order kinetic due to achieving a more homogeneous system during pre-irradiation. A larger difference, however, was observed in development of dispersity. In the case of pre-irradiation, the polymer with narrower dispersity was obtained because the pre-irradiation led to formation of CuBr
2 deactivator ensuring better control over the molar mass and dispersity.
Figure 7.
(a) Kinetic plots and (b) evolution of the molar mass and Mw/Mn with the conversion of MMA during photoRDRP using CuSO4·5H2O/TPMA complex. MMA was added either before irradiation or after 1 h of pre-irradiation of the solvent containing catalyst, ligand and initiator. Experimental conditions: photoRDRP, MMA/BPN/CuII/TPMA = 200/1/0.04/0.16, irradiation at λ > 350 nm; in all experiments: [MMA] = 7.5 M, T = 35 °C, DMSO (25 vol%).
Figure 7.
(a) Kinetic plots and (b) evolution of the molar mass and Mw/Mn with the conversion of MMA during photoRDRP using CuSO4·5H2O/TPMA complex. MMA was added either before irradiation or after 1 h of pre-irradiation of the solvent containing catalyst, ligand and initiator. Experimental conditions: photoRDRP, MMA/BPN/CuII/TPMA = 200/1/0.04/0.16, irradiation at λ > 350 nm; in all experiments: [MMA] = 7.5 M, T = 35 °C, DMSO (25 vol%).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
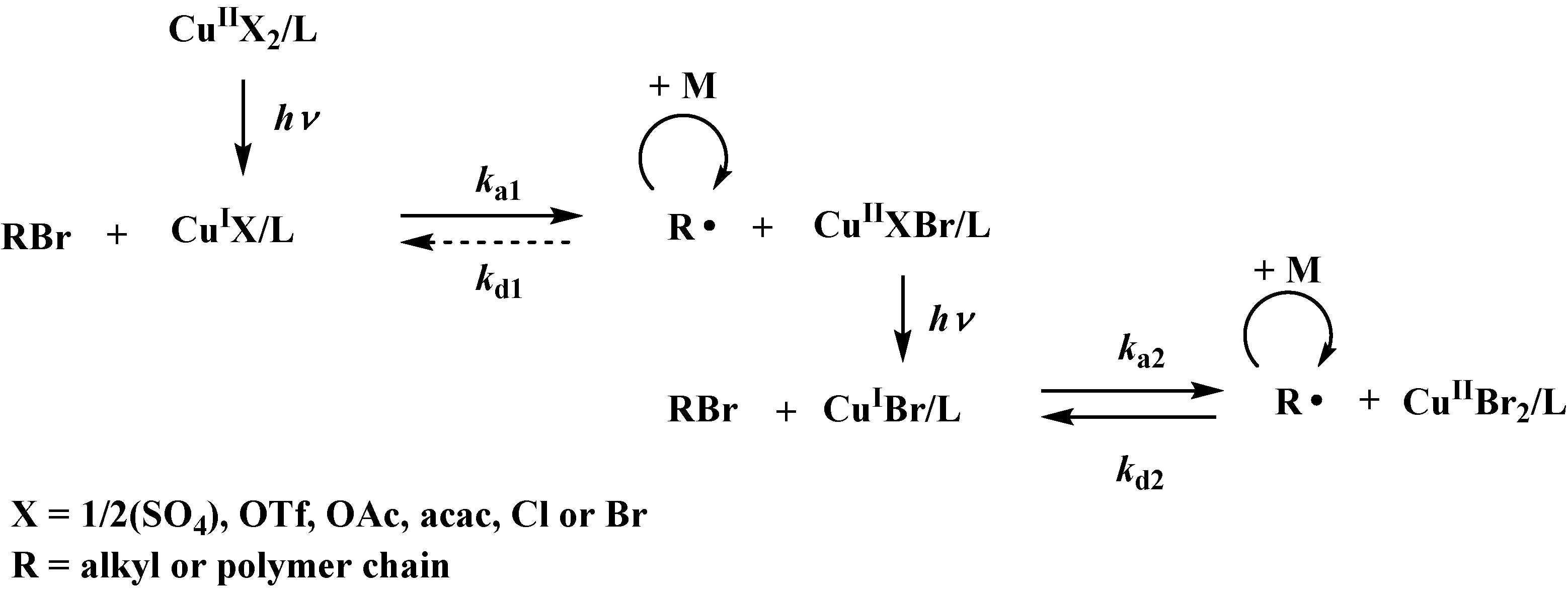{kind=link}



