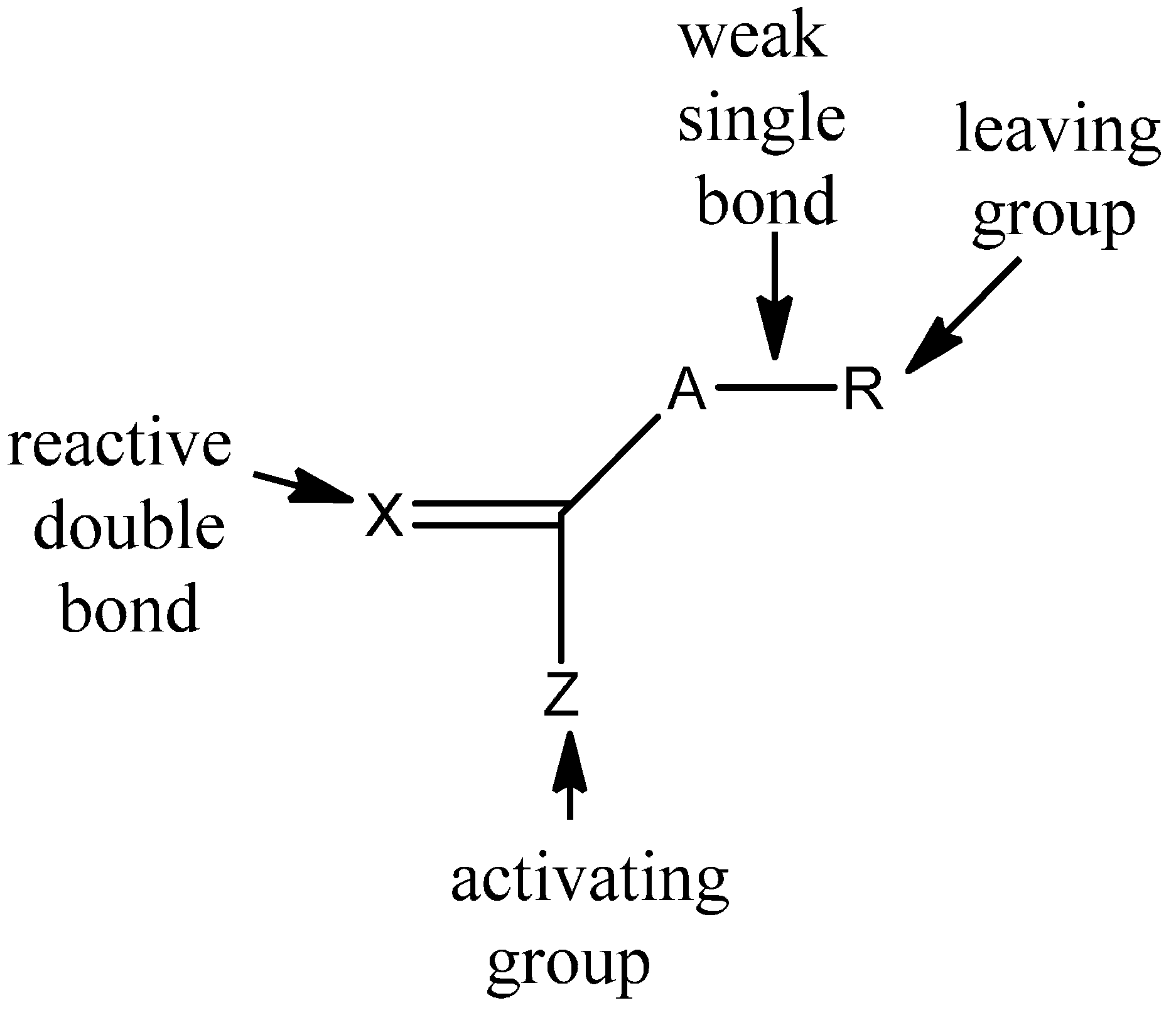
3.1. Theories for Evaluating the Kinetics of Emulsion Polymerization
According to the literature [
8,
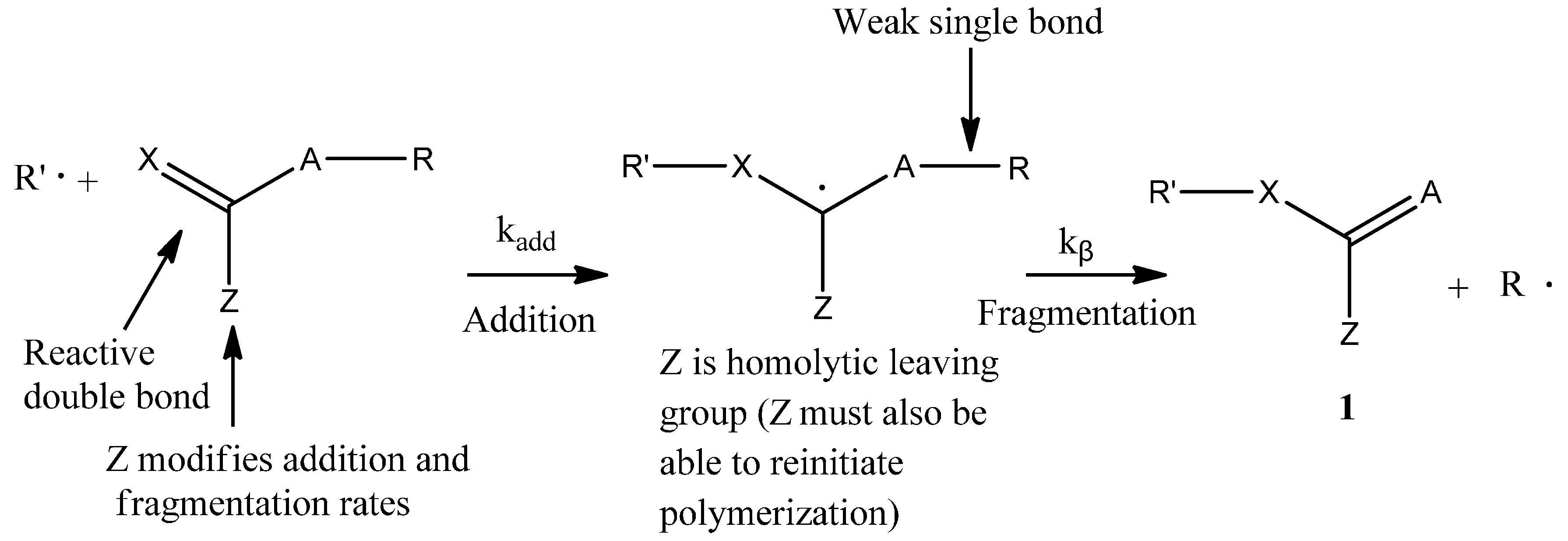
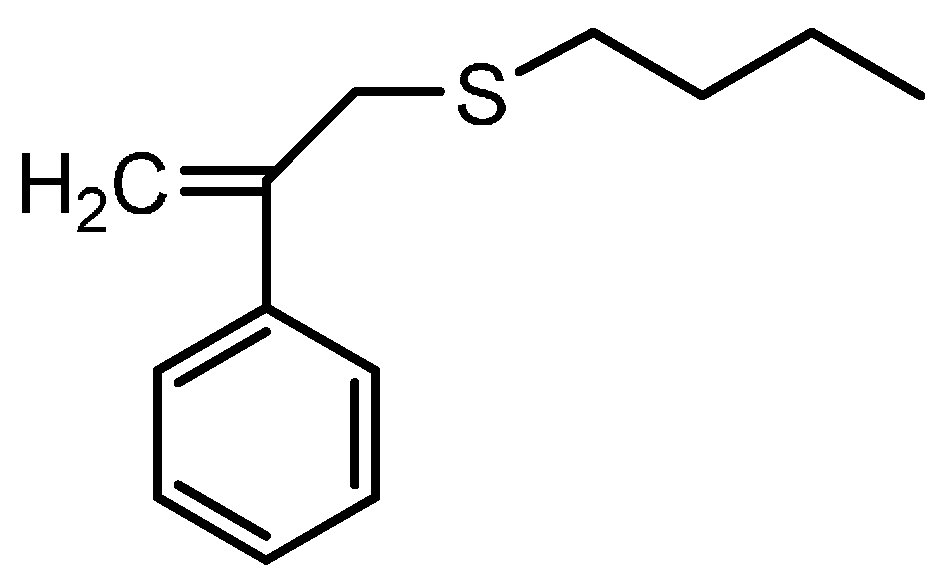
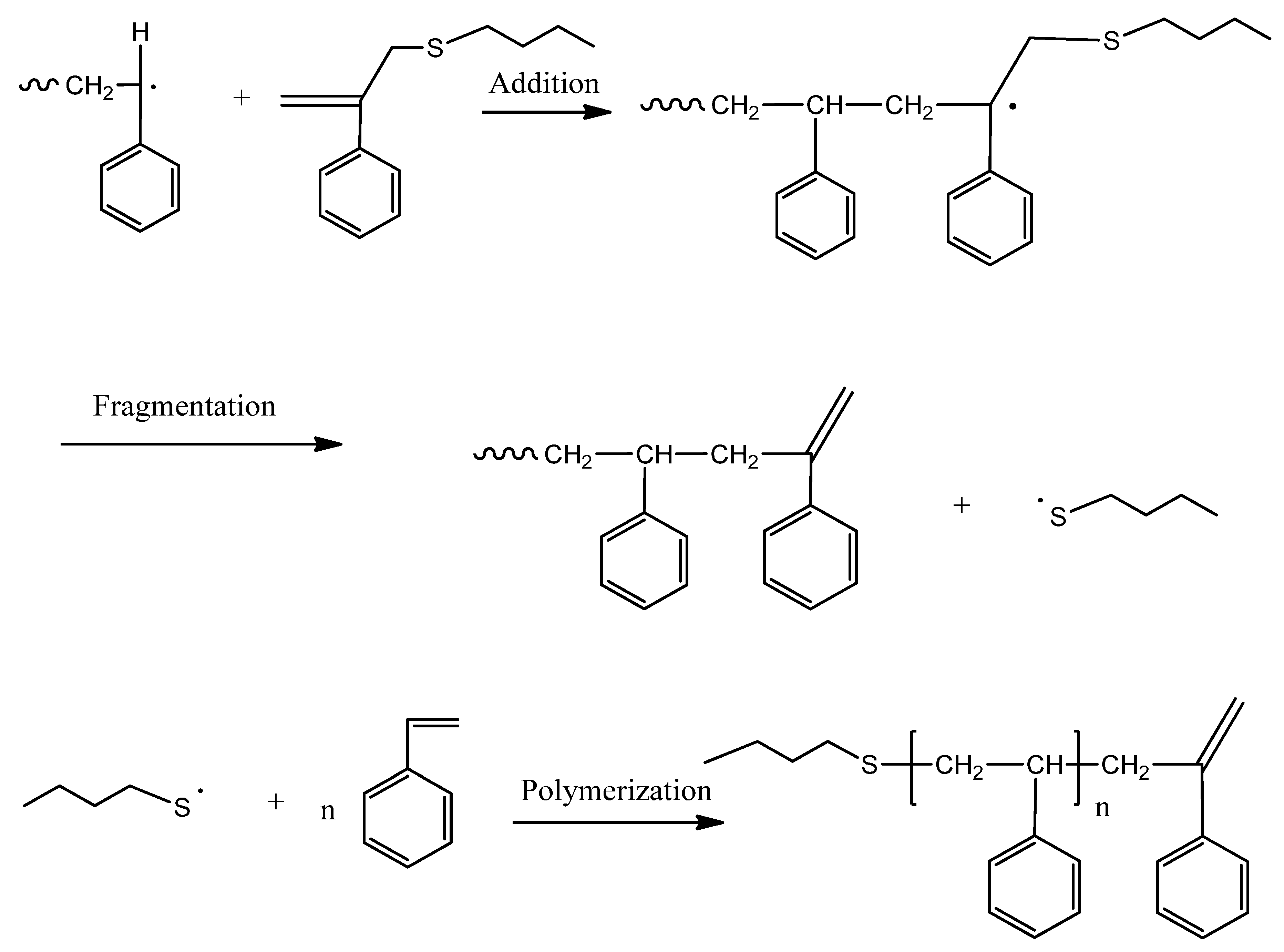
12], BPAS as a CTA used in bulk and solution polymerization does not impede the polymerization kinetics. This means that addition and fragmentation are rapid and irreversible, and with respect to propagation, the polymerization kinetics differ only slightly from those seen in polymerization with conventional chain transfer, such as mercaptans. According to the irreversible AFCT mechanism mentioned in the introduction section, the mechanisms for polymerization of styrene in the presence of BPAS are shown in
Scheme 4.
Scheme 4.
Mechanisms of the polymerization of styrene in the presence of BPAS as the chain transfer agent (CTA).
Scheme 4.
Mechanisms of the polymerization of styrene in the presence of BPAS as the chain transfer agent (CTA).
It should be noted that the polymers with end group structure 1 (
Scheme 2) are macromonomers and have utility as chain transfer agents. This means that with the irreversible AFCT agent, transfer is not completely irreversible because the end group formed is a transfer agent. According to the literature [
23], the styrene macromonomers as chain transfer agents are temperature-dependent for the transfer reaction. For the polymerizations of styrene in the presence of the styrene macromonomer at 80 °C, there was no significant variation in the molecular weight of the polystyrene formed with macromonomer concentration. Thus, if the reaction temperature is lower than 80 °C, then the reversible reaction can be neglected in the kinetics calculation. However, the polymerization reactions at the time of high monomer conversion will generate more macromonomers. As the concentration of macromonomer increases, the probability of the chain transfer reaction by macromonomers will increase. This reaction may form graft polymers or copolymers, leading to a broader molecular weight distribution of the final polymer product.
The classical Smith–Ewart Theory assumes that emulsion polymerization occurs inside the polymer particles and suggests the relationships between the rate of polymerization (
Rp), the number of particles per volume (
Nc), and the average number of radicals per particle (
) [
24]. The role of BPAS should be evaluated by these three parameters to understand its influence on the course of the emulsion polymerization of styrene. According to the classical theory, emulsion polymerization is divided into three stages. Interval I stage starts with radicals entering micelles to form growing particles, and is followed by Interval II stage. During Interval II, no new particle is formed; therefore,
Nc is a constant. The
Rp in a conventional emulsion polymerization during Interval II is given by Equation (3):
where
kp is the rate constant for propagation,
Cp is the concentration of monomer in the particle during Interval II, and
NA is Avogadro’s number.
Nc can be calculated from the number-average diameter of polymer particle (
dp) during Interval II by Equation (4):
where
m0 is the initial mass of the monomer, ρ is the density of polystyrene (1062 g/L) ,
x is the monomer conversion, and
wp is the polymer mass fraction of latex particles during Interval II. If the sample was obtained at the end of Interval II, then x should equal
wp. The parameter vtotal is the total volume of the emulsion (0.18 L in this system). The value of
Rp can be calculated from the slopes of the curves of the monomer conversion versus reaction time and the initial molar concentration of the monomer. If
dp is a narrow particle size distribution (PSD) and is collected at approximately the end of Interval II, then the observed average number of radicals per particle (
) is derived using Equation (5):
The theoretical value of the average number of radicals per particle (
) can be calculated by a semi-empirical expression in zero-one emulsion polymerizations, in which termination is not rate-determining [
25]. The adsorption of the chain-transferred radical from a polymer particle and the entry of a radical into a particle are considered to be balanced, and radical termination in the aqueous phase can be neglected. When the desorption of monomer-derived radicals occurs, with the associated rate constant monomeric radical desorption from the polymer particles (
kdM), the semi-empirical expression for calculating
, given in Equation (6), provides an approximate relationship between
and
kdM using the ratio of radical production and the rate constants of bimolecular termination in the polymer particles as a parameter (
):
This equation converges to a limiting value of
= 0.5, when
m <<
, and
<< 1. Here,
is the rate of radical production in the aqueous phase, and
f is the initiator efficiency. The Maxwell–Morrison Model predicts
f well for styrene emulsions, with the efficiency term
[
26], where
(= 4 × 10
9 L/M/s) and
(estimated as
) are the termination and propagation rate coefficients for aqueous phase radicals, respectively,
Cw is the monomer concentration in the aqueous phase (taken as 4.3 × 10
−3 M ) [
27], and
z is the critical degree of polymerization for irreversible entry of a radical into a particle (
z = 2 fits most data well) [
28]. The parameter
is the rate constant of initiator decomposition,
is the initiator concentration,
is the volume of a polymer particle,
is the termination rate constant in the polymer particles, and
is the rate constant for monomeric radical desorption from the polymer particles, given by Equation (9):
where
ktr,M is the rate constant for chain transfer to monomer;
mdM is the partition coefficient of a monomer between particles and aqueous phase (chosen to be the same as that for a styrene monomer = [
styrene]
p/[
styrene]
aq);
Dw is the diffusion coefficient of a monomer radical in the aqueous phase; and δ is the ratio of the water-side mass transfer resistance and the overall mass transfer resistance for a radical desorbed from the polymer particles. These values can be estimated with Equation (10):
where
,
,
,
, and
are the Boltzmann constant, the reaction temperature, the viscosity inside a polymer particle, the diffusion coefficient of a radical in a polymer particle, and the number-average particle diameter, respectively.
is calculated for a polymeric radical based on viscosity and is difficult to use, because it is unlikely that the internal viscosity is measured and because viscosity will change with conversion. If monomeric radicals can be assumed to generally diffuse as fast as the monomer, then through most of the reaction,
(diffusion coefficients of monomer) is not much less than
. Because
is large, δ is approximately unity. Therefore, the above calculation for
can replace that of
. Scheren [
29] estimated the change in diffusion coefficients of monomer (
) with the weight fraction polymer (
wp) in an emulsion polymerization from measured diffusion coefficients of species of similar size and structure to styrene, to give the following expression for the diffusion of monomers:
= 3.373 × 10
−7 – 5.2894 × 10
−7wp − 1.2645 × 10
−7wp2 + 3.3429 × 10
−7wp3 (in dm
2·s
−1, up to
wp = 0.82). Therefore, the Equation (10) calculation for
replaces that of
Dp and is estimated using Equation (11):
The numerical values of the used constants are collected in
Table 2.
Table 2.
Numerical values of constants used in the calculation for .
Table 2.
Numerical values of constants used in the calculation for .
| Constant | Unit | Value | Reference |
|---|
| L/mol/s | 480 | [30] |
| L/mol/s | 7 × 107 | [31] |
| mol/s | 5.6 | [24] |
| s−1 | 5.8 × 10−6 | [31] |
| or | - | 1,629 | [31] |
| or | dm2/s | 1.50 × 10−7 | [32] |
| g/L | 1,062 | [24] |
| g/L | 905 | [24] |
| / | - | 0.000116 | [30] |
According to the rate coefficient for radical desorption from the particles, the rate of desorption coefficient
is given by Equation (12) when the transfer reaction to the chain transfer agents dominates [
19]:
where
,
and
are the concentrations of CTA, the monomer in polymer particles, and the rate for a radical leaving group (the R group mentioned in the introduction section) added to the styrene coefficient (this case is RS•, where R is the butyl group), respectively. BPAS itself has a similar structure to styrene, and it must be sparingly soluble in the aqueous phase, based on the efficient transport of BPAS through the aqueous phase from monomer droplets throughout the polymerization reactions. Therefore, it might be assumed that RS• can still efficiently desorb from the latex particle, and because the first monomer addition step is fast, the addition of the adduct radical RSM• to styrene (
) might be the important step competing with desorption. If it is assumed that the first step is sufficiently fast, because of very reactive sulfur-centered radicals, then the approximate value of the competition term would be
. The value of
has not been measured, but typical values for this reaction can be assumed similar to that of a monomeric radical,
i.e., with values of approximately
=
. Thus, the
value in Equation (12) requires that the appropriate
value is close to
.
is the diffusion coefficient of a CTA radical in the water phase (chosen to be the same as that for a styrene monomer radical in the water phase), and
is the partition coefficient of a CTA radical between particles and the aqueous phase (also chosen to be the same as that for a styrene monomer = [
styrene]
p/[
styrene]
aq). In this case, the average number of radicals per particle
can be expressed by the following equation when radical desorption is substantial:
This equation also requires that α
w << 1. Note that later (
Table 3), it is shown that α
w << 1.
Table 3.
Global results.
| Run | Rp a (10−4 mol/L/s) | dp (nm) | Nc b (1018/L) | c | (-) | (s−1) | m (-) | | f |
|---|
| 1 | 6.77 | 76.7 | 1.33 | 0.000116 d | 5.38 × 10−6 | 0.131e | 1.49 × 10−5 | 0.114 | 0.091 |
| 2 | 2.69 | 74.1 | 1.47 | 0.0009 | 1.42 × 10−5 | 0.271 | 1.15 × 10−4 | 0.041 | 0.106 |
| 3 | 3.65 | 58.3 | 3.03 | 0.0016 | 3.37 × 10−6 | 0.780 | 2.01 × 10−4 | 0.027 | 0.047 |
| 4 | 5.53 | 46.2 | 6.08 | 0.0013 | 8.34 × 10−7 | 1.008 | 1.61 × 10−4 | 0.020 | 0.029 |
| 5 | 7.68 | 40.2 | 9.23 | 0.0012 | 3.62 × 10−7 | 1.229 | 1.46 × 10−4 | 0.018 | 0.022 |
| 6 | 14.5 | 32.7 | 17.1 | 0.0016 | 1.05 × 10−7 | 2.478 | 1.91× 10−4 | 0.018 | 0.011 |
| 7 | 5.11 | 55.4 | 3.53 | 0.0015 | 4.09 × 10−6 | 0.809 | 1.88 × 10−4 | 0.032 | 0.053 |
| 8 | 5.62 | 52.7 | 4.09 | 0.0016 | 3.34 × 10−6 | 1.015 | 1.99 × 10−4 | 0.028 | 0.051 |
| 9 | 6.49 | 66.3 | 2.06 | 0.00047 | 2.24 × 10−6 | 0.176 | 5.92 × 10−5 | 0.071 | 0.065 |
| 10 | 5.13 | 65.8 | 2.11 | 0.0007 | 2.14 × 10−6 | 0.267 | 8.88 × 10−5 | 0.054 | 0.053 |
| 11 | 2.62 | 64.8 | 2.20 | 0.0013 | 1.96 × 10−6 | 0.512 | 1.65 × 10−4 | 0.037 | 0.026 |
| 12 | 2.33 | 61.3 | 2.60 | 0.002 | 1.40 × 10−6 | 0.881 | 2.53 × 10−4 | 0.020 | 0.027 |
| 13 | 1.50 | 60.1 | 2.35 | 0.0033 | 1.24 × 10−6 | 1.513 | 4.16 × 10−4 | 0.014 | 0.020 |
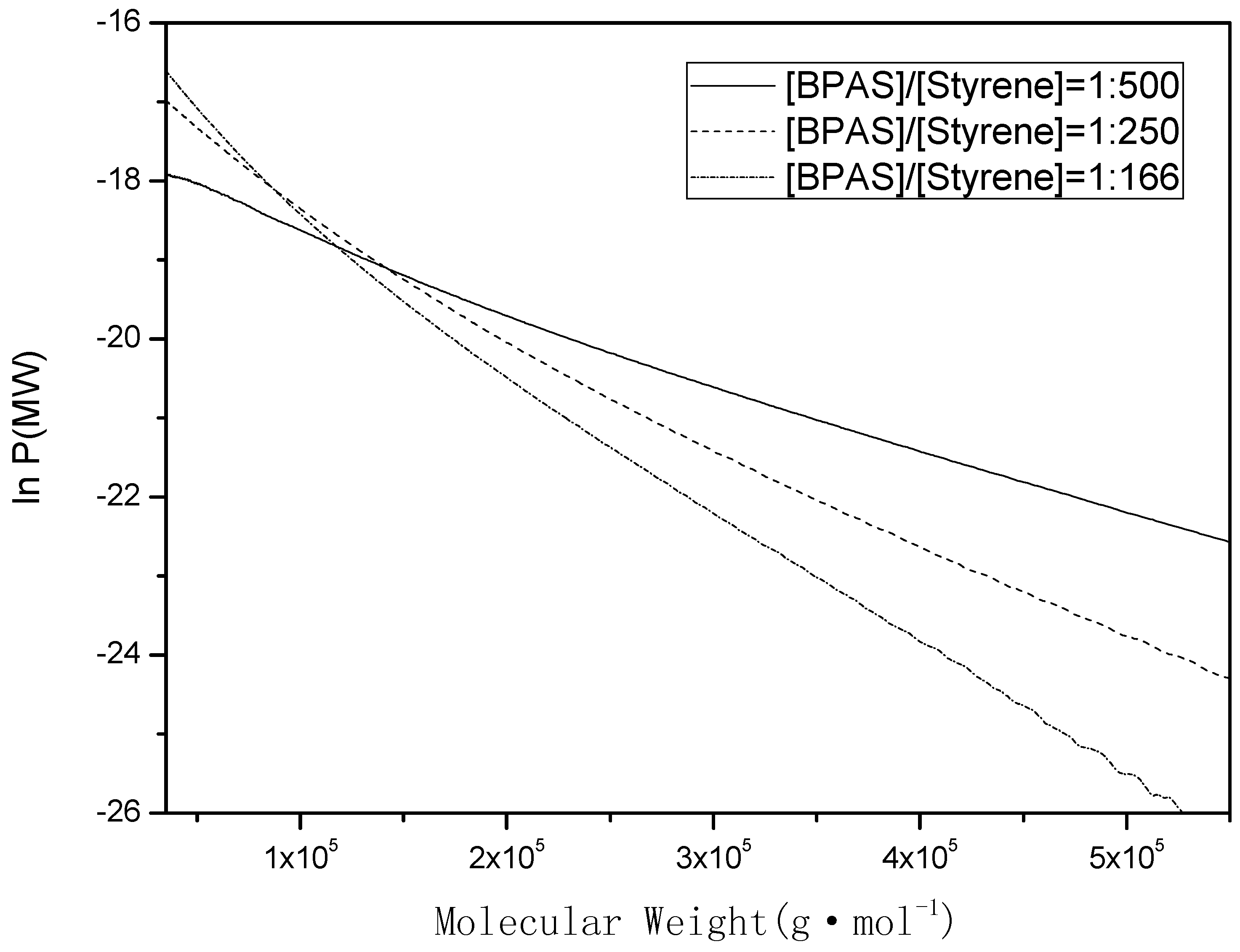
The values of
can be calculated by molecular weight distribution (MWD) analysis [
33]. The instantaneous number molecular weight distribution
Pinst(
MW) can be thought of as the MWD of the polymer that is formed over an infinitesimally small time interval. Instantaneous number MWDs collected over finite time intervals are more properly “pseudo-instantaneous” number MWDs. Clay and Gilbert developed a model describing the instantaneous number MWD for zero-one (Equation (14)) and pseudo-bulk (Equation (15)) emulsion polymerization systems. The instantaneous number MWD can be modeled by a single exponential for zero-one and pseudo-bulk systems, as follows:
where
MW is the polymer molecular weight, ρ is the pseudo-first-order entry rate coefficient per polymer particle, and
is the monomer molecular weight.
If the transfer event is the dominant chain-stopping event (
i.e.,
>> ρ,
,
), then Equations (14) and (15) simplify to the following form:
Therefore, a plot of vs. MW should yield a straight line with a slope equal to (). Thus, from the instantaneous number MWDs, which were measured by GPC traces, it is possible to determine the values of . If samples were obtained at the beginning of interval II, preferably over a small conversion interval (10%–15%), then the values of could be used in Equations (12) and (18).
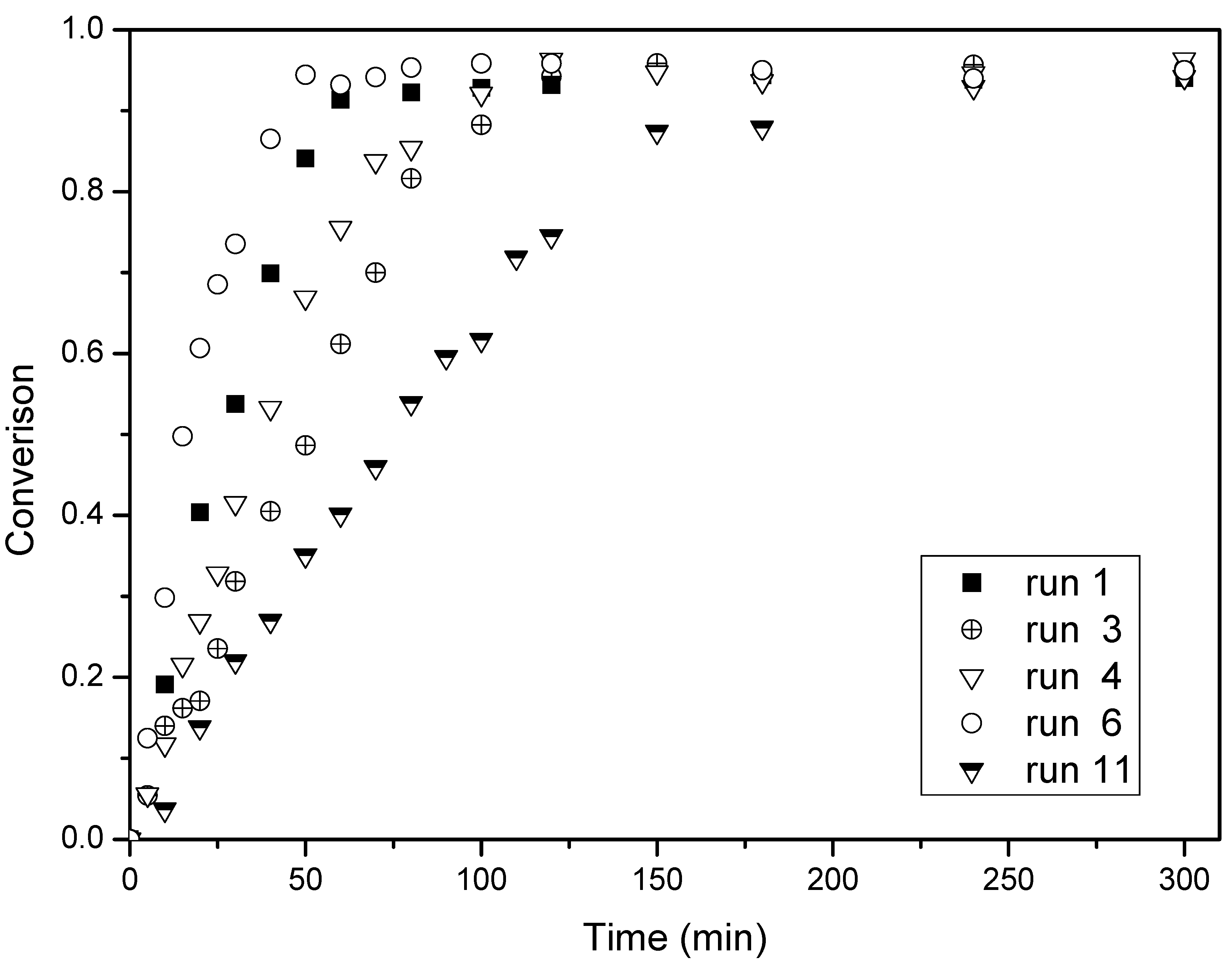
Figure 1.
Conversion-time histories for the butyl(2-phenylallyl)sulfane (BPAS) mediated emulsion polymerization. Reaction temperature: 70 °C.
Figure 1.
Conversion-time histories for the butyl(2-phenylallyl)sulfane (BPAS) mediated emulsion polymerization. Reaction temperature: 70 °C.
Figure 2.
Typical number molecular weight distribution plotted as lnP(MW) vs. MW. The region used was to determine the slope, which was subsequently used to determine the initial values of and , the polystyrene samples were obtained at the beginning of interval II (10%–15% monomer conversion).
Figure 2.
Typical number molecular weight distribution plotted as lnP(MW) vs. MW. The region used was to determine the slope, which was subsequently used to determine the initial values of and , the polystyrene samples were obtained at the beginning of interval II (10%–15% monomer conversion).
3.2. Influence of Surfactant and Initiator Concentration with BPAS on the Rate of Polymerization
Figure 1 depicts the typical monomer conversion as a function of reaction time in the presence of BPAS with a varying amount of surfactant and initiator, and
Table 3 lists the values of
Rp which were measured by the slope of every run in
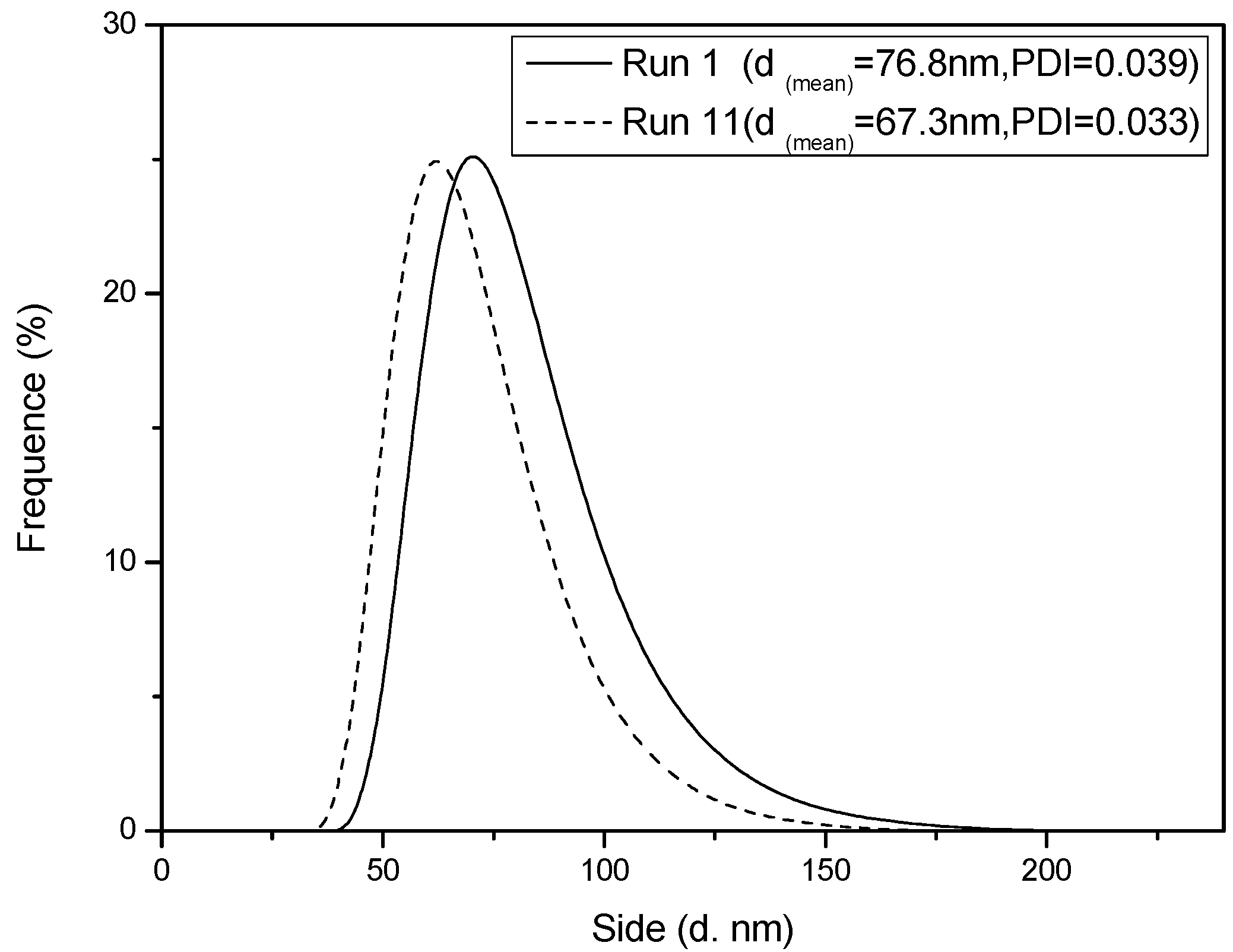
Figure 1. Meanwhile, the SEM photographs of PS spheres and the number-average diameter distribution of a latex particle with and without BPAS are shown in
Figure 3 and
Figure 4, respectively. These figures show that a higher concentration of surfactant or initiator increases
Rp when the amount of BPAS is constant (runs 2–8). As the surfactant or initiator concentration increases, more polymer particles are formed, resulting in higher rates of polymerization. As shown in
Figure 3 and
Figure 4, the particle size distribution (PSD) is narrow and monodispersed, indicating that the treatment of the kinetics is valid because many of the equations are highly sensitive to particle size. A log–log plot of either
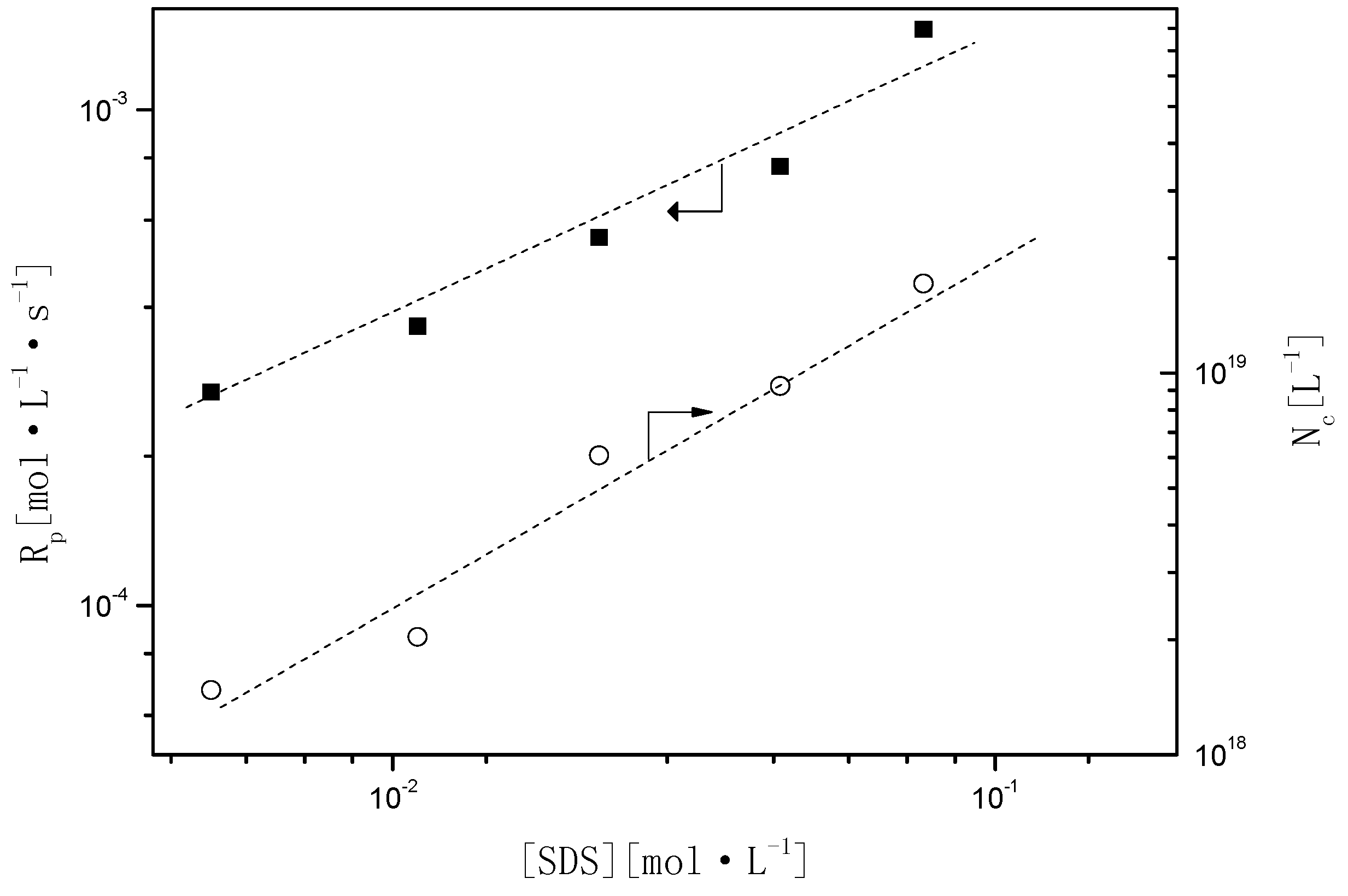
Rp or
Nc and the concentration of the surfactant or initiator should be constructed to evaluate the influences of the amounts of SDS or KPS on the
Rp and
Nc in the presence of BPAS. As shown in the results in
Figure 5, the relationship of
Rp and
Nc with the concentration of SDS was estimated as
and
, respectively.
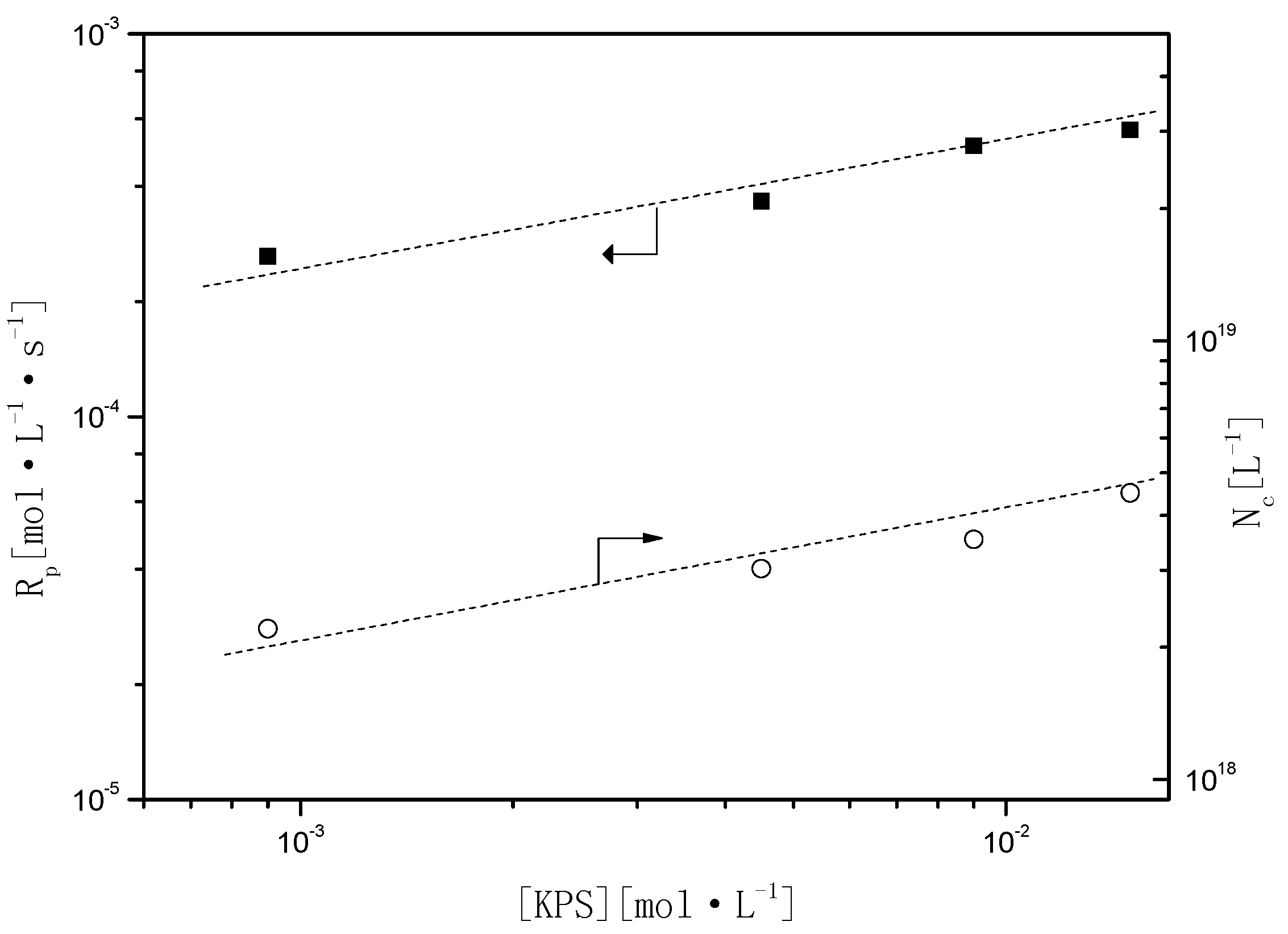
Figure 6 shows the effect of the concentration of KPS on
Rp and
Nc (
and
). According to Smith–Ewart’s Theory, relationships among
Rp,
Nc, [
SDS], and [
KPS] should be
,
,
and
. Nomura
et al. [
19] demonstrated that as the radical desorption from the particles increased, aided by chain transfer agents, the emulsifier dependence exponent for the number of polymer particles formed increased from 0.6 to 1.0, and the initiator dependence exponent decreased from 0.4 to 0. In addition, the sum of these exponents always equals unity, which is shown in Equation (17):
where
and
are the initial surfactant and initiator concentrations, respectively. The value of z satisfies the inequality 0.6 ≤
z ≤ 1.0 and increases with an increase in radical desorption from the particles. The obtained results are in good agreement with the sum of the dependence exponents. Because styrene is a hydrophobic monomer, the mechanism for particle formation is normally expected via the entry of micelles. The major contribution to the radical desorption is most likely from the departure of the leaving group on the BPAS from the particles, which terminate with radicals in either the water phase or through re-entering particles that terminate with growing polymer chains. When runs are compared at the same BPAS concentration and with a lower initiator concentration, the average diameter from run 11 was higher than that in run 3. This alludes to the fact that the nucleation mechanisms are different for both sets of experiments at low and high initiator concentrations. The evidence suggests that the exiting process increases the nucleation rate, but if the aqueous phase radical concentration is high, which is manifested by a high initiator concentration, then the increased entry rate dominates over re-entry with the possibility of termination of the exited radicals in the aqueous phase. Thus, the fate of desorbed radicals is termination in water. This becomes the dominant process at this high initiator concentration. At a low initiator concentration, the leaving group may have a lower rate of entry, which would result in a longer nucleation period, and larger polymer particles. Consequently, the dependence exponents among
Rp,
Nc, [
SDS], and [
KPS] are different from the predictions by the classical theory.
Figure 3.
Scanning electron microscope (SEM) photographs of PS spheres prepared by emulsion polymerization at the end of the reaction: (a) run1 and (b) run 11.
Figure 3.
Scanning electron microscope (SEM) photographs of PS spheres prepared by emulsion polymerization at the end of the reaction: (a) run1 and (b) run 11.
Figure 4.
Number-average diameter distribution of a latex particle during Interval II at 60% monomer conversion.
Figure 4.
Number-average diameter distribution of a latex particle during Interval II at 60% monomer conversion.
Figure 5.
Relationship of Rp and Nc as a function of the surfactant (sodium dodecyl sulfate (SDS)).
Figure 5.
Relationship of Rp and Nc as a function of the surfactant (sodium dodecyl sulfate (SDS)).
Figure 6.
Relationship of Rp and Nc as a function of the initiator (potassium peroxodisulfate (KPS)).
Figure 6.
Relationship of Rp and Nc as a function of the initiator (potassium peroxodisulfate (KPS)).
3.3. Analysis of the Rate of Emulsion Polymerization
To further evaluate the desorption of radicals from the particles in the presence of BPAS in the emulsion polymerization of styrene, the relationships among
Rp,
Nc, and
should be considered because
and Nc directly control
Rp. From the presented results, it is evident that the presence of BPAS affects the course of the emulsion polymerization (
Figure 1). The rates of polymerization in the presence of BPAS are lower than without BPAS. Additionally, the particle number can be affected by the exit of the leaving group on the BPAS from polymer particles. The rate of polymerization and the particle number are directly related to the average number of radicals per particle. The values of the observed average number of radicals per particle
) were calculated from Equation (5). The theoretical values of the average number of radicals (
) were calculated from Equation (6) for run 1, considering only the desorption of radicals formed by transfer to the monomer. Meanwhile, the
values for runs 2–13 were calculated from Equation (13), when the transfer reaction to chain transfer agents dominates. The similar
and
values (
Table 3) indicate that the emulsion polymerization of styrene in the presence and absence of BPAS can be described using the classical theoretical emulsion polymerization framework. This also implies that BPAS as CTA has a significant kinetic influence inside latex particles. Complete re-entry and minimal re-escape is the primary fate of desorbed radicals, as previously mentioned.
Styrene is classified as a Smith–Ewart Case 1 monomer. Case 1 occurs when
<< 1 and only a small fraction of polymer particles will contain a radical because the probability of radical desorption is higher than radical entry. Typically, Case 1 behavior is observed for small particles, as demonstrated in
Table 3.
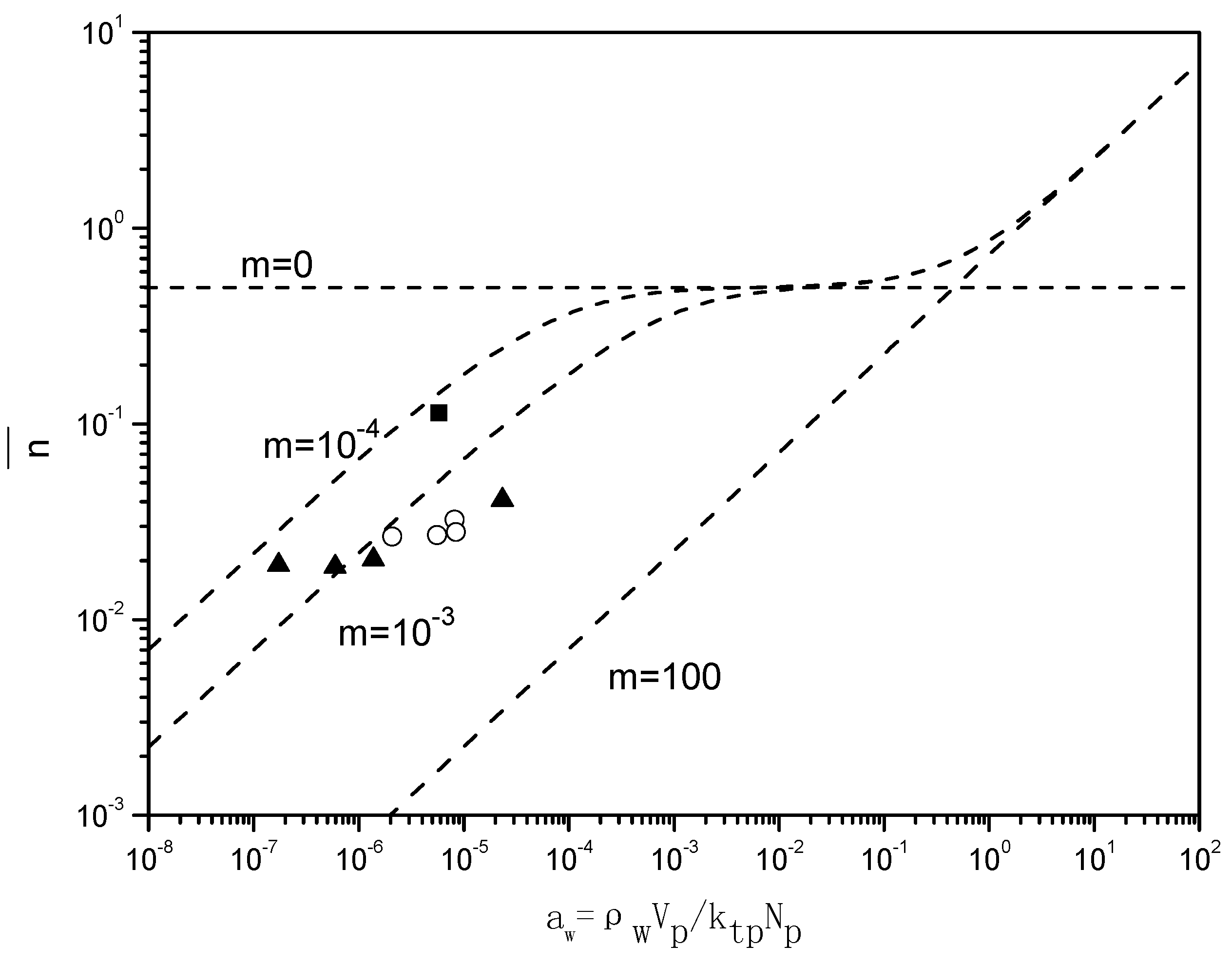
Figure 7 presents the “Ugelstad-plots” that compare the
with the theoretical lines for
m = 0,
m = 10
−4,
m = 10
−3 and
m = 10
2. For the emulsion polymerizations in the absence of BPAS, the ratio of radical desorption and radical termination, m given in Equation (7), equals 10
−4, which implies a small contribution of radical desorption.
When BPAS is added to the reaction system with a ratio of 1:250 of BPAS to styrene, the values of decrease 2–5 times, indicating that the desorption of the leaving group radicals from BPAS results in a decrease in . The results are those for a situation in which the polymerization displays a very pronounced Case 1 behavior; consequently, displays a strong dependency on the particle size. For the given experimental conditions, m = 10−3, which implies an increase in the radical desorption from the polymer particles. The particle size dependency of originates from the fact that the rate coefficient of radical desorption, is inversely proportional to the particle size squared, given in Equation (12). These low values for in the presence of BPAS explain the large difference between the rates of polymerization with and without BPAS.
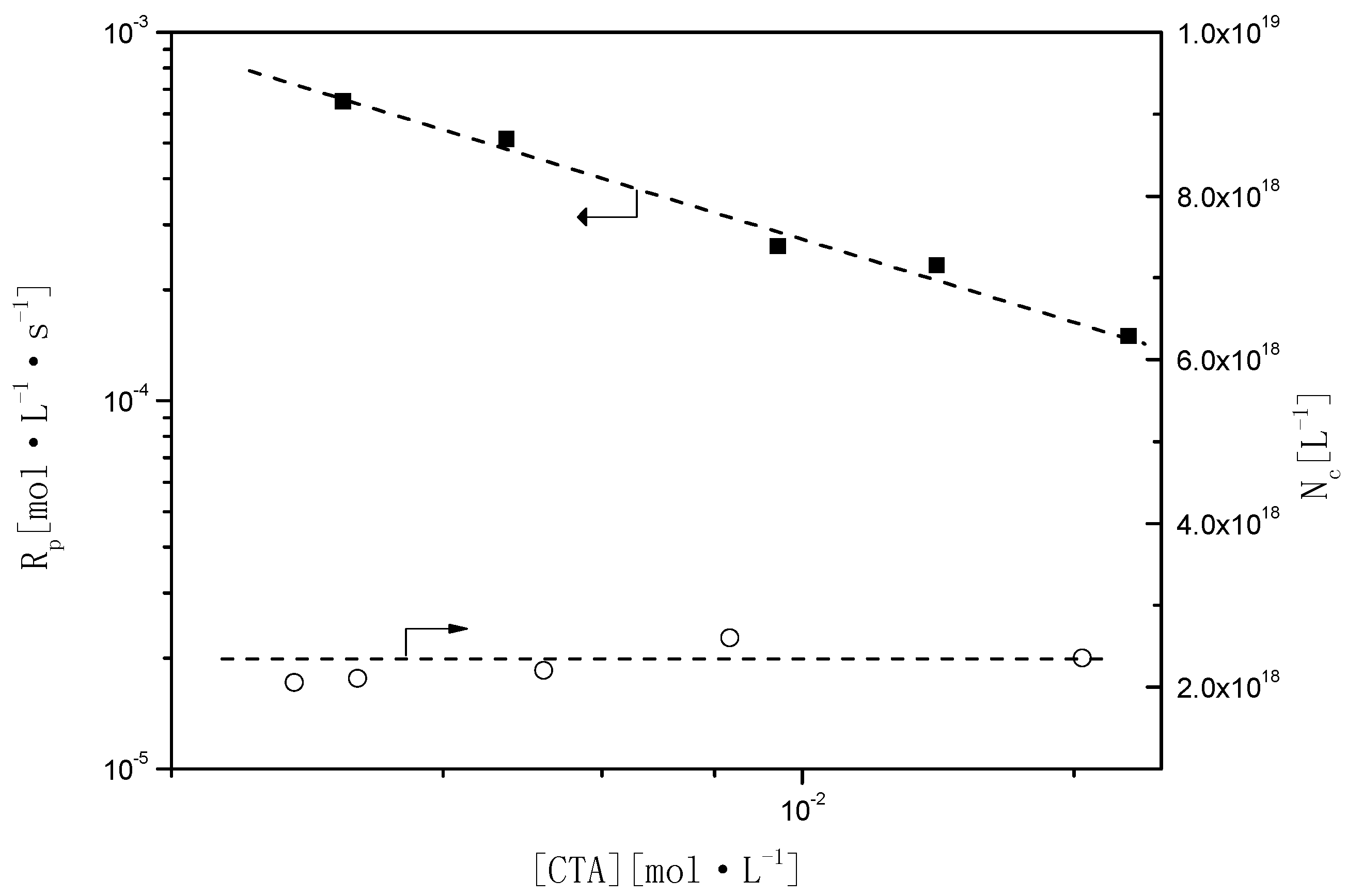
In styrene emulsion polymerization,
is strongly influenced by the presence of BPAS (
Table 3). As shown in
Figure 8,
Rp decreases upon increasing the concentration of BPAS. The relationship of
Rp with the amounts of BPAS was estimated as
, and there was no effect on Nc (
Table 3, runs 9–13) with a constant concentration of surfactant and initiator. As BPAS is consumed during the reaction, it is refreshed from monomer droplets during Interval II. Thus, BPAS remains towards the end of the reaction, which would lead to lower molecular weights. These results are illustrated in the following section.
Figure 9 also presents the relationship between
and
with a varying concentration of BPAS. A decrease of
as the concentration of BPAS increase implies that the desorption of radicals dramatically increases with a high BPAS concentration. The increased desorption originates from the fact that the rate coefficient of radical desorption
, is proportional to the concentration of the CTA. As the BPAS concentration increases, the chain-transferred radical concentration inside the particle increases.
Figure 7.
Relationship between the average number of radicals per particle and
(runs 1–8 and 11 in
Table 3. The values for
were obtained from
). The experimental values of
were calculated by Equation (7). ■ run 1; ○ varying initiator concentration, [
BPAS]/[
styrene] = 1:250; ▲ varying surfactant concentration, [
BPAS]/[
styrene] = 1:250. (---) Theoretical lines were generated by Equation (6) for
m = 0,
m = 10
−4,
m = 10
−3 and
m = 10
2.
Figure 7.
Relationship between the average number of radicals per particle and
(runs 1–8 and 11 in
Table 3. The values for
were obtained from
). The experimental values of
were calculated by Equation (7). ■ run 1; ○ varying initiator concentration, [
BPAS]/[
styrene] = 1:250; ▲ varying surfactant concentration, [
BPAS]/[
styrene] = 1:250. (---) Theoretical lines were generated by Equation (6) for
m = 0,
m = 10
−4,
m = 10
−3 and
m = 10
2.
Figure 8.
Relationship of Rp and Nc as a function of BPAS concentration.
Figure 8.
Relationship of Rp and Nc as a function of BPAS concentration.
Figure 9.
Relationship between the average number of radicals per particle and
(runs 9–13, in
Table 3. The values for
were obtained from
) The experimental values of
were calculated by Equation (7). ■ run 1; ○ varying BPAS concentration. (---) Theoretical lines were generated by Equation (6) for
m = 0,
m = 10
−4,
m = 10
−3 and
m = 10
2.
Figure 9.
Relationship between the average number of radicals per particle and
(runs 9–13, in
Table 3. The values for
were obtained from
) The experimental values of
were calculated by Equation (7). ■ run 1; ○ varying BPAS concentration. (---) Theoretical lines were generated by Equation (6) for
m = 0,
m = 10
−4,
m = 10
−3 and
m = 10
2.
3.4. Influence of the BPAS Concentration on the Number-Average Molecular Weight () of Polystyrene
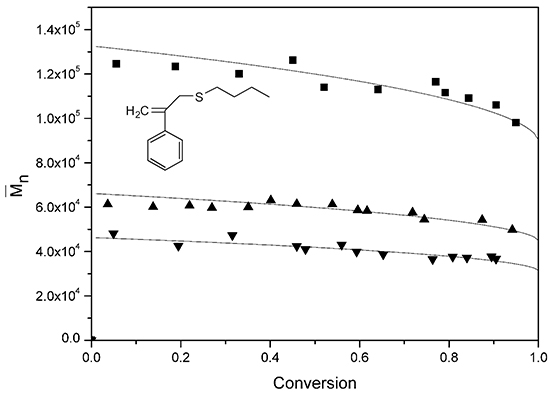
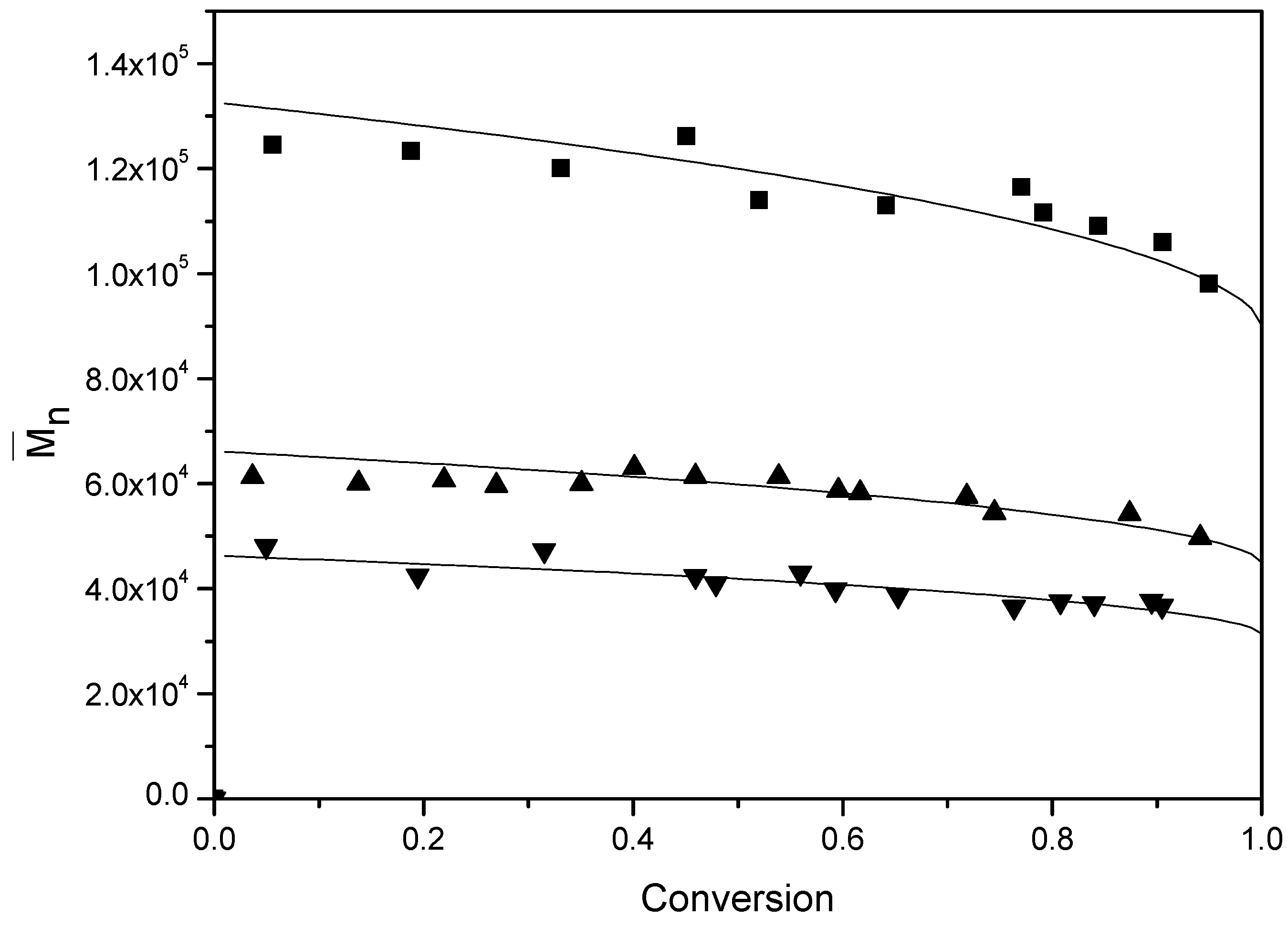
Figure 10 presents the number-average molecular weight (
) for various BPAS concentrations at a constant concentration of initiator and surfactant.
Table 4 lists the results of
at the final conversion. It can be seen that an increase of the BPAS concentrations leads to a decrease of
, and in the runs containing BPAS,
remains relatively constant over the entire conversion range. The concentrations of initiator and surfactant did not affect
when the amount of BPAS was the same (runs 2–8). The evolution of the molecular weight distributions throughout run 11 (
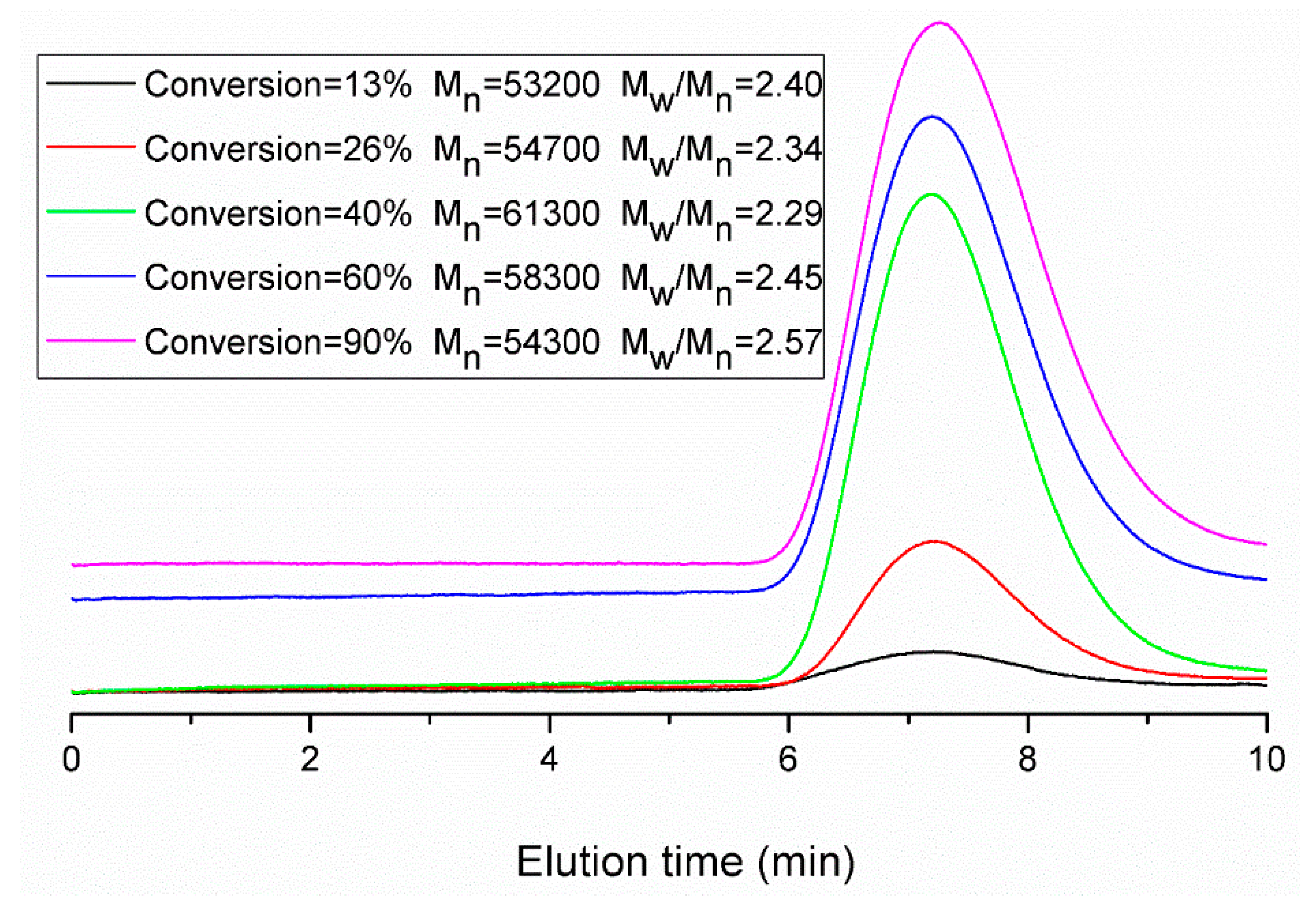
Figure 11) becomes broader at the end of the polymerization. This is primarily due to the BPAS consumed during the reaction and then refreshed from monomer droplets during interval II. Thus, BPAS remained towards the end of the reaction, leading to lower molecular weight polymer. The probability of the chain transfer reaction by macromonomers will increase. This reaction may form graft polymers or copolymers. This also may result in a broader molecular weight distribution.
The equation derived by Tobolsky [34] was used to predict the
of polystyrene by using CTAs. The equation is as follows:
where [
M]
p/[
T]
p is the initial molar ratio between monomer and CTA in the polymer particles and
is a fractional conversion. This equation considers the change of the concentrations of monomer and CTA compared with the Mayo Equation.
Figure 10.
Evolution of for various concentrations of BPAS. ■ run 10; ▲ run 11; ▼ run 12; the solid curve is the theoretical curve calculated from Equation (18).
Figure 10.
Evolution of for various concentrations of BPAS. ■ run 10; ▲ run 11; ▼ run 12; the solid curve is the theoretical curve calculated from Equation (18).
Table 4.
The results of the number-average molecular weight in the final conversion, and the initial values of the ratio between CTAs and the monomer in a polymer particle.
Table 4.
The results of the number-average molecular weight in the final conversion, and the initial values of the ratio between CTAs and the monomer in a polymer particle.
| Run | [Styrene]/[CTA] | a | | |
|---|
| 1 | - | - | 7.05 × 105 | - |
| 2 | 250 | 0.0013 | 4.89 × 104 | - |
| 3 | 250 | 0.0023 | 5.31 × 104 | - |
| 4 | 250 | 0.0019 | 4.79 × 104 | - |
| 5 | 250 | 0.0018 | 4.96 × 104 | - |
| 6 | 250 | 0.0023 | 4.76 × 104 | - |
| 7 | 250 | 0.0022 | 4.78 × 104 | - |
| 8 | 250 | 0.0023 | 4.73 × 104 | 0.68 |
| 9 | 750 | 0.0007 | 1.36 × 105 | - |
| 10 | 500 | 0.0011 | 9.81 × 104 | - |
| 11 | 250 | 0.0019 | 4.98 × 104 | - |
| 12 | 166 | 0.0029 | 3.67 × 104 | - |
| 13 | 100 | 0.0049 | 2.15 × 104 | - |
| 14 | 500 | 0.0012 | 8.11 × 104 | - |
| 15 | 250 | 0.0019 | 5.24 × 104 | - |
Figure 11.
Evolution of molecular weight distributions during run 11.
Figure 11.
Evolution of molecular weight distributions during run 11.
Figure 10 also presents a comparison of the effect of BPAS on
predicted by Equation (18) with that observed experimentally. Styrene solution polymerization experiments with BPAS were reported by Rizzardo and provided valuable raw data for
= 0.68 at 60 °C. It is evident that the experimentally obtained
and theoretical outcomes using 0.68 for
are in good agreement over the entire conversion range in
Figure 10. The
Ctr of irreversible AFCT agents is defined in the typical manner
); thus, the transfer rate coefficient (
) of BPAS can be estimated as 326 L/mol/s at 70 °C for styrene (
= 480 L/mol/s at 70 °C for styrene). The agreement between the theoretical values for
and the experimentally obtained values implies that the ratio of monomer and CTA remains generally constant throughout the reaction, and the transport is most likely efficient. This can also be achieved in Interval II because [
M]
p is constant; therefore, if a small amount of CTA is consumed, then [
M]
p/[
T]
p will also be generally constant until Interval III starts. At that stage, a significant amount of polymer is already formed; thus, the effects of small deviations at higher conversions will be difficult to observe, but a broader MWD at the end of the polymerization can be seen.
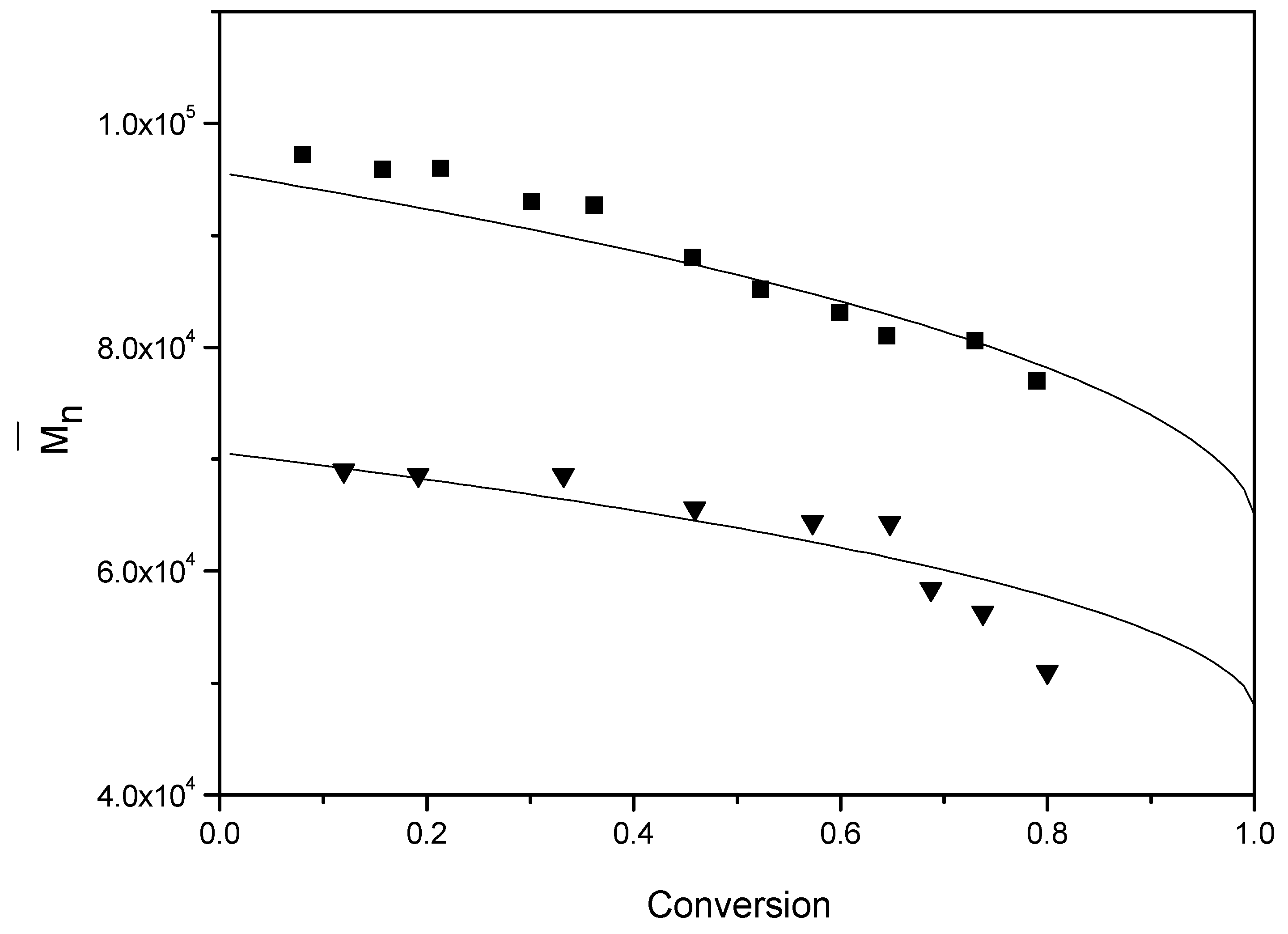
Because some synthetic rubbers are typically produced at low temperatures, the emulsion polymerization of styrene was also performed at room temperature using a redox initiation system to investigate the efficiency of BPAS for molecular weight control (runs 14 and 15). According to the results shown in
Figure 12 and
Table 4, the evolution of
and the final obtained
for polystyrene were the same as the runs at a relatively high temperature. However, the predictions become too high at approximately 70% conversion in
Figure 12. This is around the glass transition, where propagation might become diffusion controlled. This would lead to lower molecular weights than predicted, as observed. The same final
values were obtained between low and high temperature polymerizations, indicating that the efficiency of BPAS for molecular weight control is relatively independent of reaction temperature. Thus, BPAS shows potential for application as a molecular weight regulator in synthetic rubber production.
Figure 12.
Evolution of for various concentrations of BPAS at room temperature. ■ run 14; ▲ run 15; the solid curve is the theoretical curve calculated from Equation (18).
Figure 12.
Evolution of for various concentrations of BPAS at room temperature. ■ run 14; ▲ run 15; the solid curve is the theoretical curve calculated from Equation (18).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
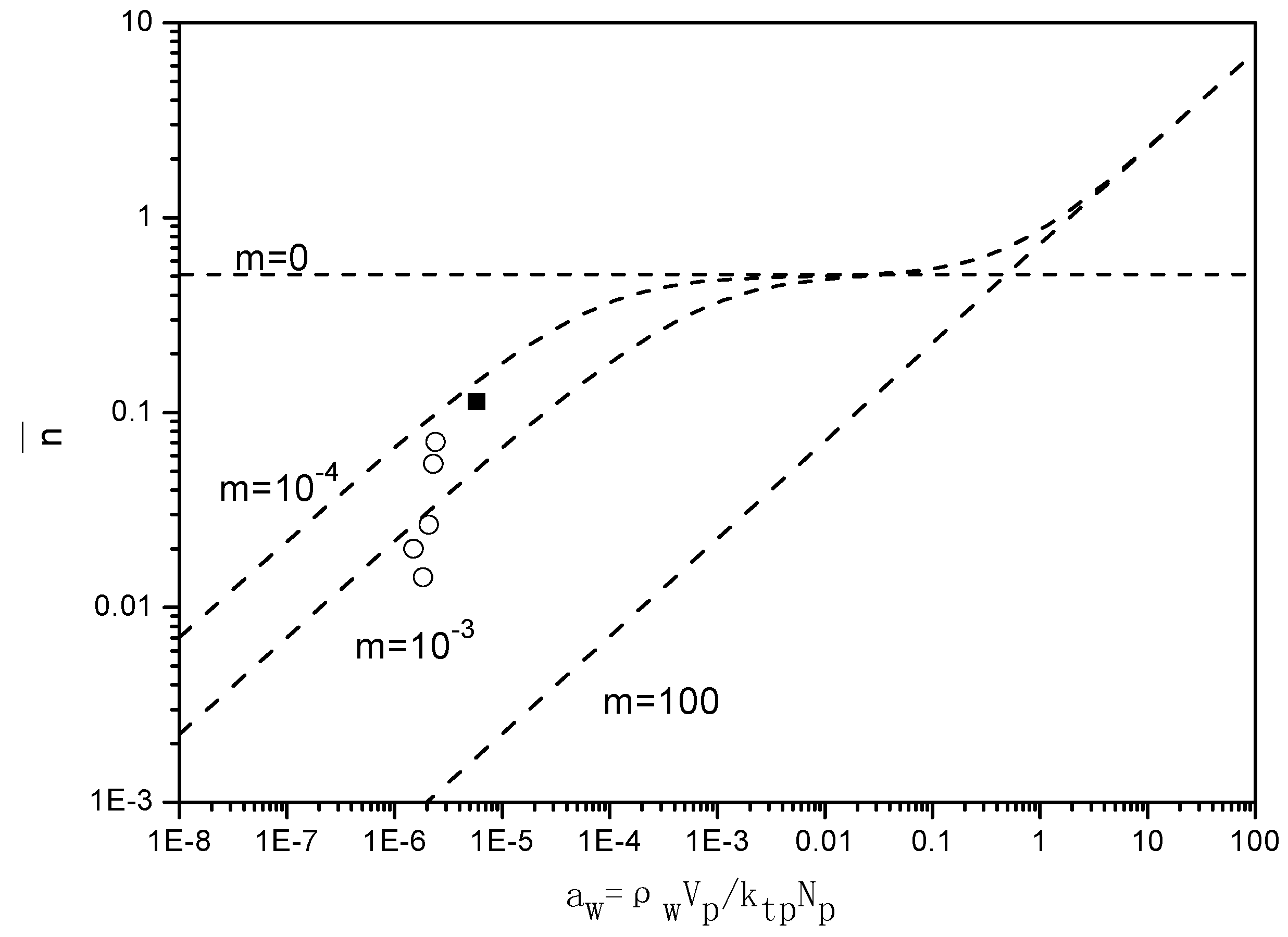{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}












