1. Introduction
Chitin and cellulose represent two of the most abundant polysaccharides in nature, where their functions are to give strength and structural support to crustacean shells and plants, respectively. Both are homopolymers, built up by 1-4-β-linked (d)-N-Acetyl-glucosamine and (d)-glucose, respectively. The lack of solubility of these polymers is fundamental for their functions in nature but for industrial applications, it has limited their practical uses. To circumvent the solubility issue and expand their uses, both have been subjected to a multitude of further chemical manipulations in order to obtain water solubility and other useful properties. Once in derivatized and soluble forms, both polymers have found plentiful uses in different areas.
The processes for isolation of chitin and chitosan involve a number of variable steps and different manufacturers use shells from different species and have their own set of chemical conditions. Typically, shells from shrimp and crab are used. The protocols used in the isolation processes involve different concentrations of acids and alkali and the processes operate at different temperatures and reaction times. A consequence of these variations in process parameters is that the isolated chitosans obtain different properties and purity profiles leading to difficulties in evaluating data from biological studies in which different chitosans have been used. The chitosans produced all have in common that their typical degrees of deacetylation vary from about 70% to 95%, they have a “blockwise” acetylation pattern, and they are soluble in water as long as pH is kept below 6.5. However, when pH is raised they all precipitate out from solution. The differences between these chitosans are seen in e.g., purity, polymer chain length, solubility and appearance. Taken together, the lack of solubility at physiological conditions, i.e., pH 7.4, and the poor consistency between batches, have been and still are a hurdle for development of pharmaceutical products from the “blockwise” type acetylated chitosans and this has limited their practical use in medicine.
Another type of chitosans has a random acetylation pattern and a higher degree of acetylation [
1,
2,
3,
4]. Today, this kind of chitosans is not commercially available, at least not in larger quantities, but methods for their preparation are found in the literature. There are essentially two ways of preparing them, either by deacetylation from chitin, or by reacetylation with acetic anhydride of a highly deacetylated chitosan. These “random” type acetylated chitosans show some different properties compared to their “blockwise” deacetylated relatives. From a drug development perspective, some of these differences are attractive, e.g., they are soluble at physiological conditions, do not precipitate
in vivo, and their biodegradation is substantially faster.
Both types of chitosans, “blockwise” and “randomly” deacetylated, possess obvious features which makes them highly attractive from a drug development perspective. They are nontoxic, biodegradable and mucoadhesive. In addition, chitosan has an inherent antimicrobial property, can be made in different physical forms and is relatively inexpensive. If it had not been for the issues of solubility, purity and batch inconsistencies, all regulatory show stoppers, chitosan would be an ideal polymer for pharmaceutical development. Nevertheless, a large number of products based on chitosan chemistry are under development and a few have made it all the way to the markets.
Of the physical forms possible, chitosan hydrogels have probably gained the largest interest and they are being developed for numerous applications [
5]. One way of describing the different types of hydrogels is to define them from their structure, are they covalently cross-linked or are they not? There is an almost endless number of different variations in which different gelling mechanisms have been used, e.g., thermo gelling, photo induced gelling and chemical cross linking in combinations with chitosans of different degrees of deacetylation, different molecular weights and in combination with a large number of cross linkers, other polymers and pharmaceutically acceptable excipients. For an overview, see e.g., [
6].
In regenerative medicine, chitosan hydrogels have found many uses e.g., for tissue repair, as support material for growing cells or used as transport and depot vehicles for living cells [
7,
8,
9], in some cases further doped with growth factors and drugs [
10]. Other uses are, e.g., to give a temporary tissue support in cataract surgery or controlled release of various types of drugs and biomolecules [
11,
12] and in an interesting combination with Fe
3O
4 affording a magnetic self-healing hydrogel [
13,
14,
15]. The properties of chitosan, physical and biological, have made it a promising candidate for delivery of pharmaceutically active components and as a delivery vehicle for, e.g., vaccines [
16,
17], gene fragments [
18] and micro-RNA [
19]. Drug formulations based on chitosan technology are today under development for many different purposes, e.g., as vaccine carriers [
20], drug releasing hydrogels [
21], membranes, gauze and more. Some formulations are designed to give a sustained release over time whereas release from others is more instant. When hydrogels of chitosan are used it has been found that covalent cross-linking is preferred since gels without cross-links have a tendency to dissolve. Another advantage of using covalent cross-linking is that the release rate from the gel can be altered by using different degrees of cross-linking [
5].
Irrespective for what type of medical use the gels are made, they must comply with the regulatory standards and tight specifications set up by the pharmaceutical industry, in order to be used as commercial products. Formulations for oral and topical administration have fewer requirements with regard to purity demands than formulations intended for parenteral use. Other product properties desired for hydrogels are e.g., good stability, injectability through fine needles and the possibility of a terminal sterilization process, e.g., autoclaving.
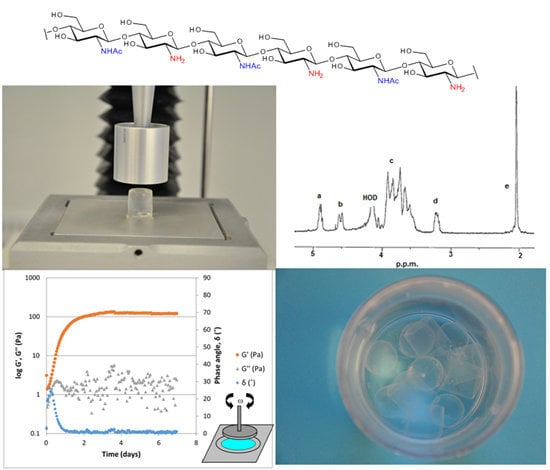
Viscogel AB, a Swedish company active in the pharmaceutical area, has developed a proprietary process for making randomly acetylated chitosan. The process is GMP (good manufacturing practice) compliant, and delivers chitosans (Viscosan™) meeting the high quality standards set by the pharmaceutical industry. In addition to delivering a highly purified material, especially with respect to endotoxins and proteins, the process achieves batch to batch consistency and produces chitosans which are soluble even at physiological pH 7.4. An advantage of having the ability to operate at physiological, higher pH without inducing precipitation is the concomitant increased reactivity of the amino function on C-2 of the glucosamine moiety. A more reactive amine leads to new and more efficient types of chemistries when making derivatives from these materials. A relevant example that illustrates the benefit of having an increased reactivity on the glucosamine amino group in combination with the solubility properties of a randomly deacetylated chitosan at neutral pH is the cross linking reaction by 3,4-Diethoxy-3-cyclobutene-1,2-dione (diethyl squarate, DES) [
22]. Even at a very low cross linker concentration, the reaction with the chitosan (1%) in solution proceeds smoothly and provides after consumption of the cross linker a rigid viscoelastic hydrogel. The gel formed is remarkably rigid which indicates a high number of functional crosslinks and elastic segments in the gel lattice. In fact, the gel is rigid enough to allow for further mechanical processing into particles of defined sizes. The gels formed, sometimes referred to as “crushed gels”, consist of soft hydrogel particles containing 99% water. The particles typically have an average size of 10–250 µm and do not aggregate. In this form, the gels are injectable, even through very fine needles, e.g., 30 gauge and can be sterilized by autoclaving. When the average particle size is below 50 µm, it is no longer possible to experience the particulate nature of the crushed hydrogels; instead they are experienced as smooth creams leaving only a slight cooling sensation when applied to skin. The cooling sensation likely originates from the huge surface area exposed by the processed particles, which allows for a relatively fast evaporation of water.
The aim of this study is to characterize hydrogels made from randomly deacetylated chitosan and to stress the importance of having well characterized raw materials fulfilling the requirements set by the pharmaceutical industry. The overall aim of the project is to provide supporting data for further development of chitosan controlled-release products.
2. Experimental Section
2.1. Materials
Three chitosan batches (Viscosan™, Viscogel AB, Solna, Sweden) of variable molecular weight (low, medium, high) and comparable fraction of acetylation,
FA (~0.5) were studied (refer to
Table 1 for characterization data).
Table 1.
Properties of the chitosan samples.
Table 1.
Properties of the chitosan samples.
| Batch No. | FA | [η] (mL/g) | Molecular Weight (from [η]) | Relative Viscosity | | | PI (/) |
|---|
| V1D2 | 0.47 | 520 | 161,000 | 34 | 156,000 | 92,500 | 1.70 |
| V2D2 | 0.49 | 950 | 310,000 | 226 | 335,000 | 184,600 | 1.82 |
| V3D2 | 0.50 | 1620 | 554,000 | 3000 | 686,000 | 397,100 | 1.73 |
The listed chemicals were applied in the experimental work:
Acetic acid (Scharlau, Barcelona, Spain), deuterium chloride (Acros Organics, Thermo Fisher Scientic, Waltham, MA, USA), deuterium oxide (Sigma Aldrich, St. Louis, MO, USA), 3,4-Diethoxy-3-cyclobutene-1,2-dione (Acros Organics, Thermo Fisher Scientific), hydrochloric acid (Scharlau, Barcelona, Spain), MES buffer (2-(N-morpholino)ethanesulfonic acid, Sigma Aldrich), methyl paraben (Sigma Aldrich), propyl paraben (Sigma Aldrich), PBS buffer (Phosphate-buffered saline buffer, Gibco®, Thermo Fisher Scientific), pilocarpine (Sigma Aldrich), purified water (Baxter, Deerfield, IL, USA), Ringer’s Acetate (B. Braun Medical, Melsungen, Germany), sodium acetate (Fluka, Sigma Aldrich), sodium chloride (Scharlau, Barcelona, Spain), sodium hydrogen carbonate (Thermo Fisher Scientific), sodium hydroxide (Fluka, Sigma Aldrich), Teflon Based Oil TF2 (Weldtite, Barton-on-Humber, UK), paraffin oil (Merck, Darmstadt, Germany), ethanol (Kemetyl, Haninge, Sweden).
2.2. Characterization of Chitosan
2.2.1. NMR Characterization of Monomer Composition
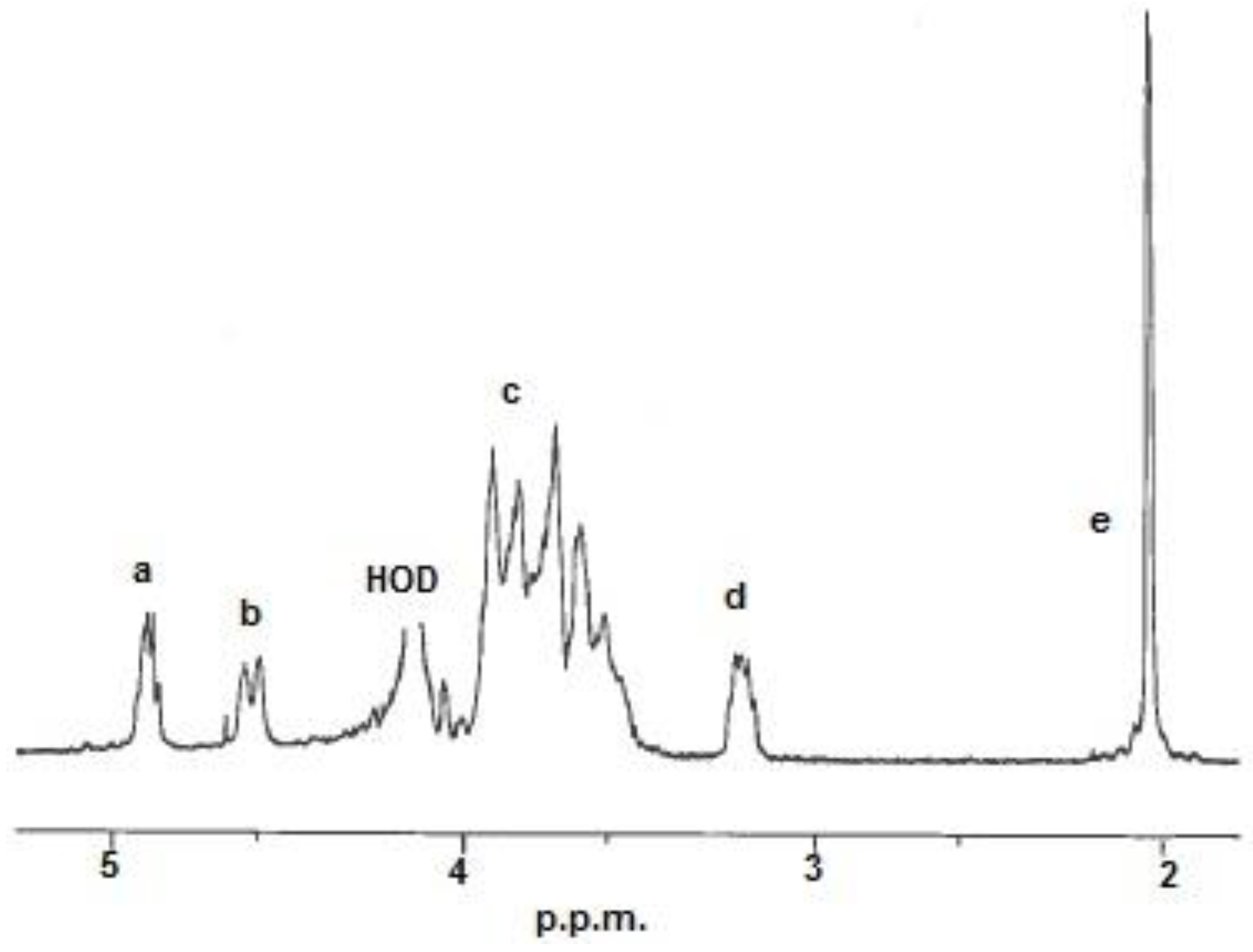
The monomer composition,
i.e., fraction of acetylation (
FA) was determined by
1H-NMR (Nuclear Magnetic Resonance) spectroscopy. The
FA was calculated from the integrals of the peaks by using the equation below [
23]:
The integrals (
Ia to
Ie) were determined from the resonances shown in
Figure 1.
The calculated
FA-values and properties of the chitosan samples are listed in
Table 1.
Figure 1.
1H-NMR spectrum of a chitosan. Resonance: (a) H-1 in D-units; (b) H-1 in A-units; (c) H-2/6 in A-units and H-3/6 in D-units; (d) H-2 in D-units and (e) 3H from the acetyl group. HOD = water.
Figure 1.
1H-NMR spectrum of a chitosan. Resonance: (a) H-1 in D-units; (b) H-1 in A-units; (c) H-2/6 in A-units and H-3/6 in D-units; (d) H-2 in D-units and (e) 3H from the acetyl group. HOD = water.
2.2.2. Intrinsic Viscosity Measurements
The intrinsic viscosities [η] were determined as previously described [
24].
For chitosans the solvent used was 0.02 M NaAc/HAc + 0.1 M NaCl, pH = 4.5. Ionic strength, I = 0.1 M. Temperature: 20 °C. The calculated [η]-values are given in
Table 1.
The molecular weight (
Mw) was calculated from the intrinsic viscosity by use of the Mark-Houwink-Sakurada-equation ([η] =
K ×
Ma) with a = 0.92 and
K = 8.43 × 10
−3 [
25], see
Table 1.
2.2.3. SEC-MALLS Characterization of Average Mw and Mw-Distribution
By use of Size Exclusion Chromatography-Multi Angle Laser Light Scattering (SEC-MALLS) the number and weight average molecular weights were determined according to [
26]. The weight average molecular weight,
, and the number average molecular weight,
, together with the polydispersity index,
/
, are given in
Table 1.
2.2.4. Dynamic Viscosity Measurements
The relative viscosity was determined with a Brookfield RVDV I+ (Brookfield Engineering Inc., Middleboro, MA, USA) equipped with spindle # 18 and a small sample adapter. Samples were prepared as 1% (w/w) in 1% acetic acid and equilibrated at 20 °C prior to measurement.
2.3. Dissolution of Chitosan
Chitosan was suspended in water to a concentration of approximately 1.1% (w/w) and 1 mL 2 M HCl per gram of chitosan was added. The pH of the solution was adjusted to 6.6 with 1 M NaOH once all of the chitosan had dissolved. Water was added to obtain a final chitosan concentration of 1% (w/w).
2.4. Preparation of a Typical ViscoGel
Three percent (molar equivalents relative the amount of chitosan monomers) 3,4-Diethoxy-3-cyclobutene-1,2-dione (DES) (in a 10% (v/v) solution in ethanol) was slowly added to the chitosan solution and stirred for 1 h at room temperature, transferred to a sealed gelling chamber and placed in a heating cabinet at 40 °C for more than 10 days.
2.5. Preparation of ViscoGel for the Swelling Studies
2.5.1. Preparation of a Hypotonic Chitosan Solution (A)
Chitosan (FA 0.51, 490 mPa·s, 500 mg) dry powder was dissolved in water (45 mL) and 2 M HCl (500 μL). The chitosan solution was titrated to pH 6.6 with 1 M NaOH and the volume of the solution adjusted to 50 mL with purified water. This gave an ionic strength of 30 mM.
2.5.2. Preparation of an Isotonic Chitosan Solution (B)
Chitosan (FA 0.51, 490 mPa·s, 500 mg) dry powder was dissolved in water (45 mL) and 2 M HCl (500 μL). The chitosan solution was titrated to pH 6.6 with 1 M NaOH and sodium chloride (321.5 mg) added prior to adjusting the volume to 50 mL with purified water resulting in an ionic strength of 140 mM.
2.5.3. Preparation of the ViscoGel Blocks
A 24-well tissue culture plate (Sarstedt AG, Nümbrecht, Germany) was used as a mold. To prevent the ViscoGel from adhering, the mold was covered in a thin layer of Teflon oil. 122 µL of 10% (v/v) DES in ethanol was then added to solutions A and B, respectively, and the flasks were left to stir at room temperature for 20 min. One milliliter of solution A was transferred to each of 12 wells in the 24-well plate and 1 mL of solution B was transferred to each of the remaining 12 wells. The plate was covered with a lid, sealed to prevent water evaporation and placed in a heating cabinet (40 °C) for two weeks.
After maturation, each block was very carefully removed. The gel blocks had the shape of a cylinder with a diameter of 20 mm and a height of about 3 mm.
2.5.4. Preparation of Buffer Solutions and Cell Strainers
Ringer’s acetate (pH 7.4) was poured into two 100 mL beakers. Then pH was adjusted to 5.0 and 4.0, respectively, using a few drops of 2 M HCl.
Three different PBS buffers of physiologic ion strength were prepared in the same way to give three portions of 100 mL PBS of pH 6.0, 7.0 and 8.0.
Using two 6-well plates (Sarstedt AG, Nümbrecht, Germany), two wells were filled with each buffer type (pH 4, 5, 6, 7 and 8). To each well, one cell strainer containing one block of ViscoGel was added, one block with hypotonic ViscoGel to each type of buffer, and one block of isotonic ViscoGel to each type of buffer. The cell strainers with ViscoGel were weighed before immersion in 10 mL of the buffer, and they were then taken out of the buffer at regular intervals. Excess buffer was wiped off and the cell strainers with ViscoGel were weighed. The results were plotted in
Figure 2 and
Figure 3.
Figure 2.
Weight change over time for blocks made from hypotonic ViscoGel™.
Figure 2.
Weight change over time for blocks made from hypotonic ViscoGel™.
Figure 3.
Weight change over time for blocks made from isotonic ViscoGel™.
Figure 3.
Weight change over time for blocks made from isotonic ViscoGel™.
2.6. Characterization of ViscoGel
2.6.2. Small Strain Oscillatory Measurements: Gelling Experiments
Gelling experiments were performed on StressTech Rheometer (Rheologica, Lund, Sweden) for ViscoGels of 15 different compositions, one example shown (
Figure 4). The measuring geometry used was plate-plate 40 mm with 1 mm gap. The settings used were strain 0.001, frequency 1 Hz, discontinuous oscillation mode and five integration periods. All experiments were performed at 40 °C and pH 6.6 and the duration of the measurements was seven days. After the gelation measurement, a frequency sweep was performed on the formed gel with an increasing frequency between 0.1 and 10 Hz at a strain of 0.001. For all measurements, paraffin oil was deposited around the sample in order to avoid evaporation of water during the measurement. Paraffin oil does not interfere with the measurements or the gelling process and was chosen as the best option based on optimization tests.
Figure 4.
Typical gelling curve for a ViscoGel™, 1.0% (w/w) V2D2 at a 3% theoretical crosslinking (DES relative to primary amino groups) obtained through small strain oscillatory measurement (strain 1 × 10−3, frequency 1 Hz, 1 mm gap, 40 mm plate-plate geometry).
Figure 4.
Typical gelling curve for a ViscoGel™, 1.0% (w/w) V2D2 at a 3% theoretical crosslinking (DES relative to primary amino groups) obtained through small strain oscillatory measurement (strain 1 × 10−3, frequency 1 Hz, 1 mm gap, 40 mm plate-plate geometry).
2.7. Pilocarpine Loaded ViscoGel
Pilocarpine (240 mg, 1.15 mmol) was dissolved in 4.0 mL of purified water and added dropwise to a stirred solution of chitosan (8 g, 1.5% (w/w), FA 0.50, viscosity 430 mPa·s). Twenty-seven microliters of a solution of DES (10% (v/v) in ethanol (corresponding to 3% theoretical cross-linking) was added and the stirring continued for 1 h at room temperature. The mixture was placed in a closed flask and transferred to a heating cabinet (40 °C).
After two weeks at 40 °C, the ViscoGel formed was mechanically processed into smaller fragments (average size 200 µm) and the release of pilocarpine from the ViscoGel was followed by HPLC (high performance liquid chromatography).
3. Results and Discussion
3.1. Characterization of Chitosan
Three chitosan batches were selected for this study as specified in
Table 1.
The molecular weights of the three chitosan samples ranged from 161 via 310 to 554 kDa. Except for the low molecular weight sample, the chitosans had molecular weights in the higher range of commercial chitosan samples. The fraction of acetylation (FA) was around 0.5 for all of the batches.
All the chitosan samples showed molecular weight distributions with polydispersity indexes (PI) between 1.7 and 2, close to the expected “Kuhn-distribution” with a PI of 2 that is the “most probable distribution” resulting from a random degradation of a long chain [
27].
3.2. ViscoGel Swelling at Different Ionic Strength and pH
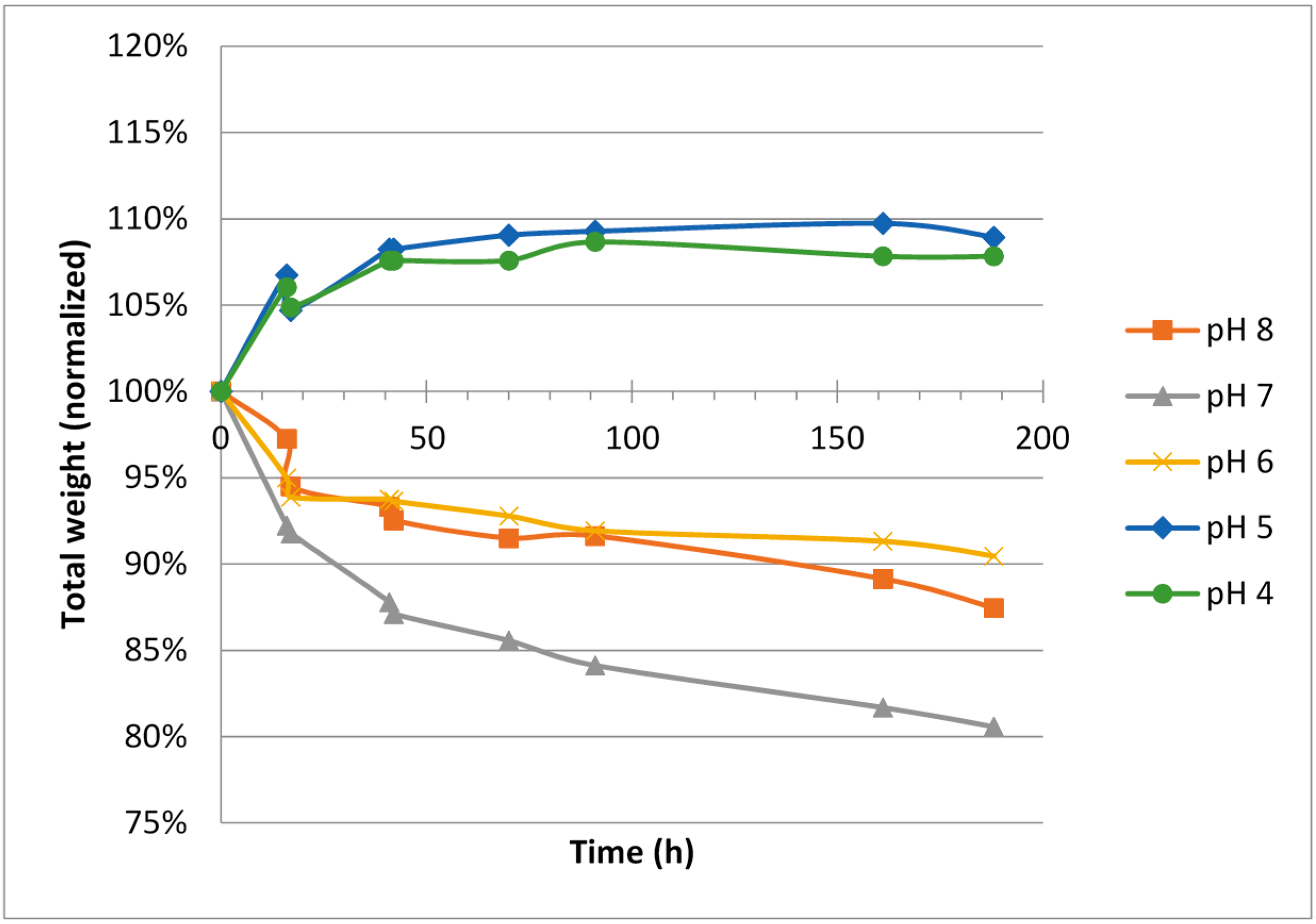
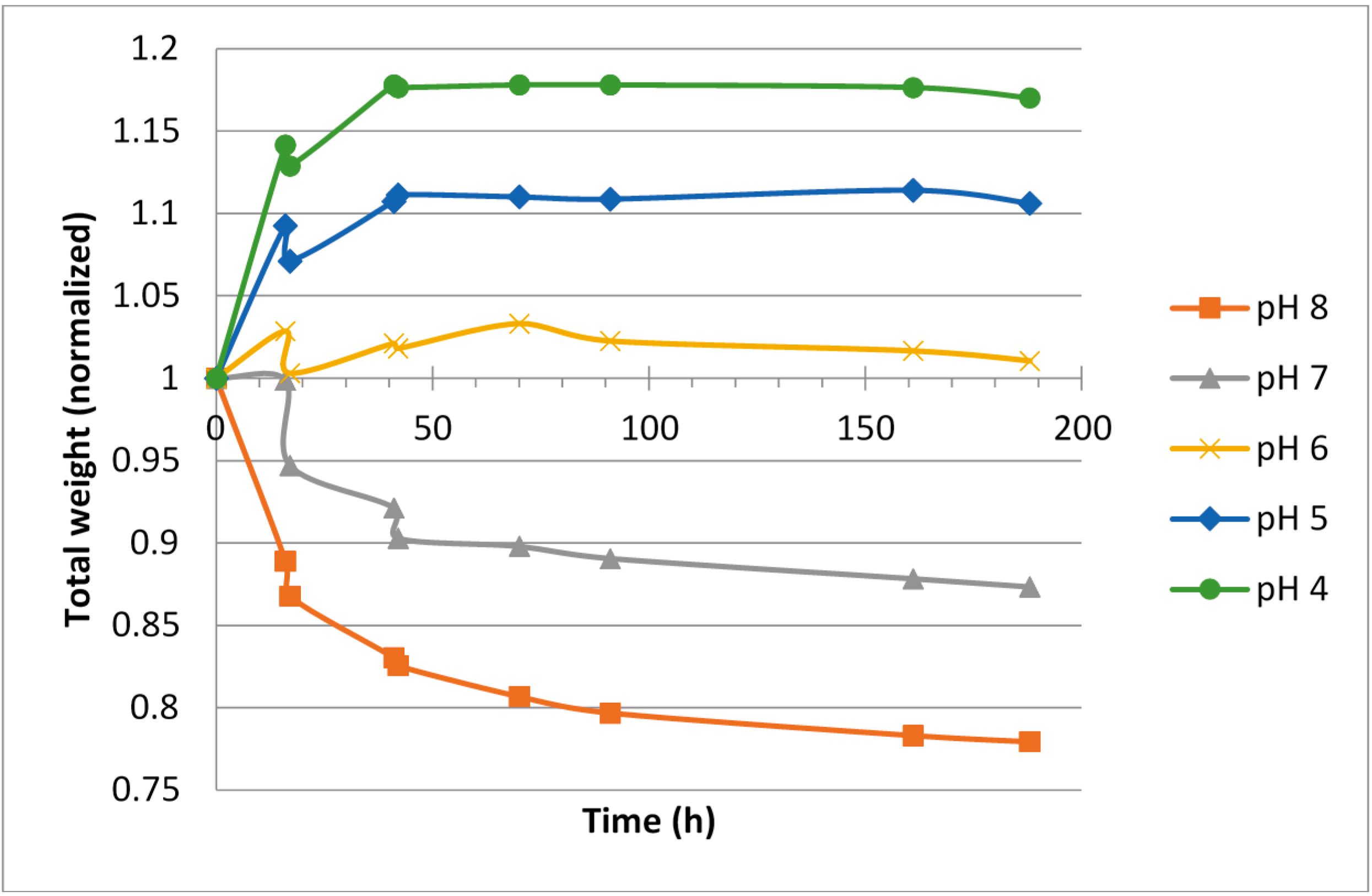
For hypotonic ViscoGels, an expansion (5%–10% weight increase) was observed for the gels when soaked in buffers having low pH, 4 and 5, respectively. A corresponding shrinkage (5%–15% weight decrease) was observed for the ViscoGels when soaked in buffers having pH 6–8 (
Figure 2).
The difference in salinity between the surrounding buffer, which had an isotonic ionic strength, and the ViscoGels, which were hypotonic, would theoretically cause the ViscoGels to release water and shrink in order to increase their salt concentrations. However, on the other hand, it is reasonable to believe that the free ions constituting the difference in ionic strength would reach an even distribution between the gel and the buffer in a relative short time. It is therefore more reasonable to consider the fixed charges, i.e., those on the chitosan molecule to explain most of the observed results. At lower pH, the chitosan chains are more positively charged and thus repel each other more forcefully causing the network of chitosan chains to expand and thereby leaving more room for water to increase volume and mass.
The isotonic ViscoGels had the same ionic strength as the surrounding buffer solutions, and thus were not expected to change in weight when considering the ionic strength differences (
Figure 3). However, the effects of pH as seen for the hypotonic ViscoGels remained, again pointing towards the behavior being linked mainly to the fixed charges in the gel network. A lowering of the pH resulted in swelling of the gels and an increase in pH resulted in shrinkage of the gels. Since there was no difference in ionic strength, to counteract the effect of pH, the swelling was slightly larger for these gels.
Irrespective of initial ionic strength in the ViscoGel, the change in relative gel volume was rather small and all gels had almost reached their new equilibrium weights after 50–70 h in the respective buffer.
The results showed that the hypotonic ViscoGel was slightly less inclined to swell at lower pH. This was expected since water left the gel to compensate for the lower salinity in the ViscoGel compared with the buffer. This effect countered the swelling effect of the pH at least to some extent. In general, the hypotonic ViscoGel seemed less inclined to change its volume, both when it comes to swelling at low pH and shrinking at high pH.
3.3. Characterization of ViscoGel
3.3.1. Gelling Kinetic Experiments
Strain sweeps were performed on all ViscoGel samples analyzed on StressTech (Rheologica, Lund, Sweden). The results showed (data not included) a linear viscoelastic region up to strains above 0.50, which implies that the chosen experimental strain of 0.001 was well within this region for all samples tested.
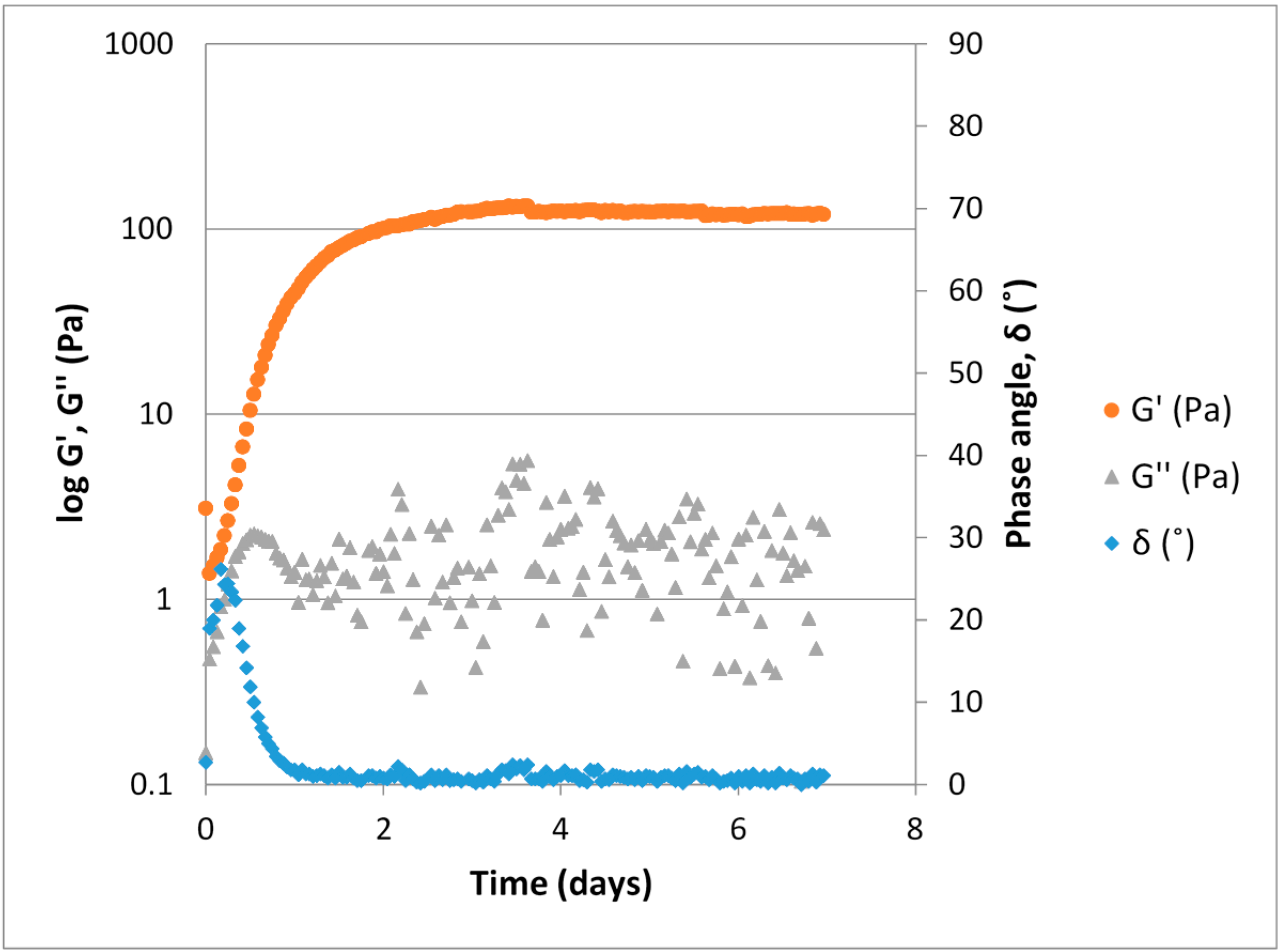
The ViscoGel solutions were produced as described above and a typical example illustrating their maturation process is shown in
Figure 4. During gel formation the sample goes from displaying predominantly viscous properties (liquid like) to predominantly elastic properties (solid like). The elastic modulus, G', clearly increases as the number of crosslinks increase, while the measured values for the viscous modulus (G'') are lower than the G'. A phase angle below 45° indicates that elastic properties are predominant and that a phase transition from liquid to gel has occurred. After approximately 3.5 days the gel strength plateaus and remains at a constant level for the remainder of the measurement time.
Figure 5 displays a typical frequency sweep for ViscoGel after cross-linking. G' > G'' over the entire range and G' being almost frequency independent. ViscoGel is therefore a true gel within this frequency range.
Figure 5.
Frequency sweep for a 1.0% (w/w) V3D2 gel at 3% theoretical crosslinking. The diagram displays the G' and G'' with increasing frequency 0.1–10 Hz.
Figure 5.
Frequency sweep for a 1.0% (w/w) V3D2 gel at 3% theoretical crosslinking. The diagram displays the G' and G'' with increasing frequency 0.1–10 Hz.
3.3.2. ViscoGel Stability
The long-term storage experiment was conducted in order to investigate the storage stability of bulk ViscoGels. The experiments duration was 12 months and the stability of the gels were controlled by monthly compression measurements on the Texture Analyzer, measuring the Young’s modulus, stress at failure and deformation at failure for different types of ViscoGel (with respect to both Viscosan molecular weight and the degree of crosslinking). In total, five different gel types were produced.
3.3.3. Effect of Concentration of Crosslinker
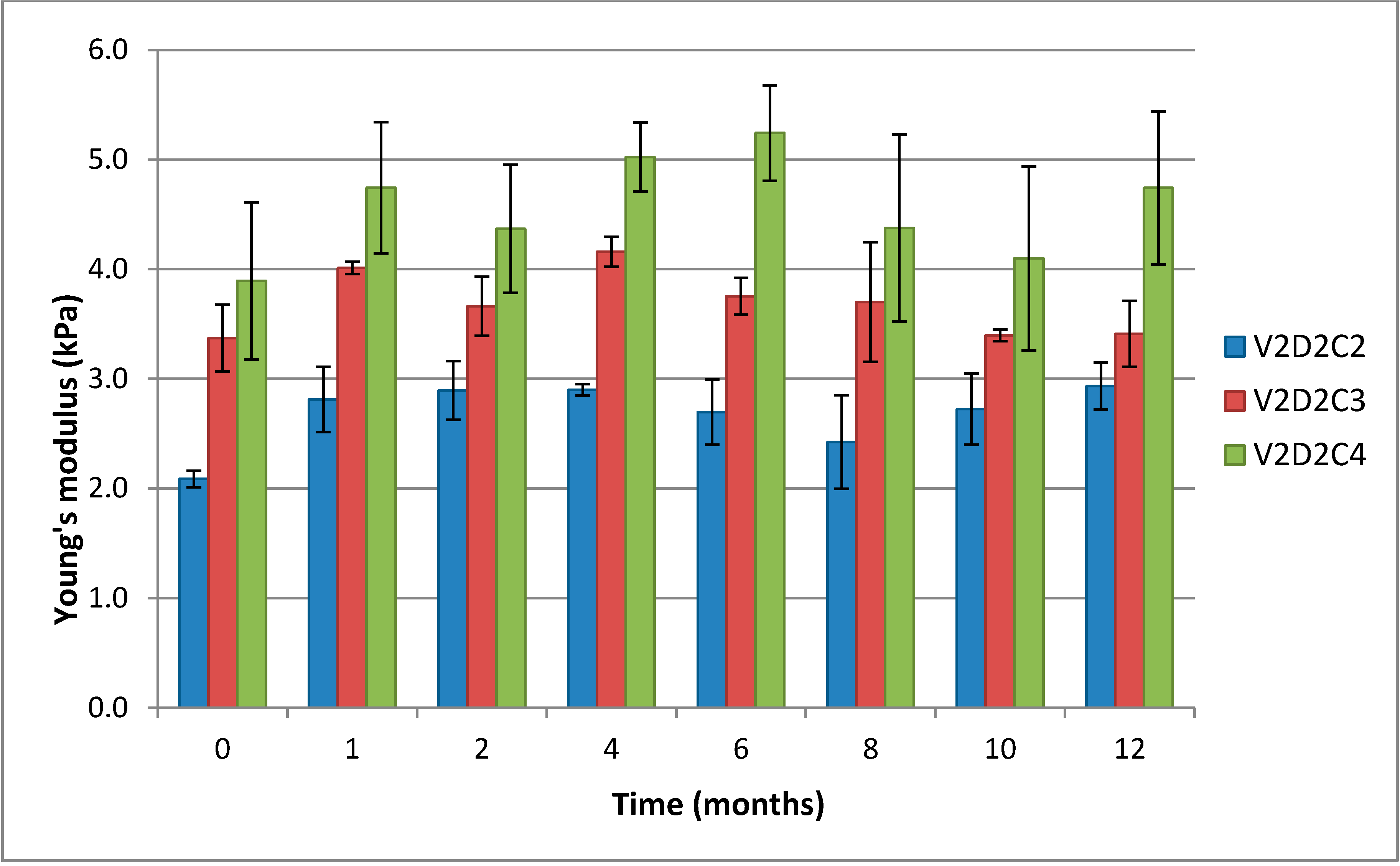
It is clearly seen that the Young’s modulus increases with increasing concentration of DES (
Figure 6). Young’s modulus is a measure of the “stiffness” (mechanical response) of a material, the materials ability to recapture its original shape after deformation and reflects the number and length of elastic segments. The diagram also shows that in the initial phase of the storage experiment, particularly from start to month 1, there is a weak, though non-significant, trend towards Young’s modulus increasing. This can be explained by additional functional crosslinks being formed over time, resulting in a tightening of the gel lattice and the gels becoming more solid-like.
Large scale deformation measurements (results not included) on gels with different degrees of crosslinking showed a trend towards failure at lower relative deformation with increasing degree of crosslinking. This is to be expected as a result of shortening the elastic segments. Stress at failure for these gels did not exhibit any clear trend. It was confirmed that the gels are stable over a 12 month period.
Figure 6.
Comparison of the Young’s modulus over 12 months for gels of 1% (w/w) chitosan V2D2 and variable concentration of crosslinker. C2, C3 and C4 represent 2%, 3% and 4% theoretical cross-linking, respectively.
Figure 6.
Comparison of the Young’s modulus over 12 months for gels of 1% (w/w) chitosan V2D2 and variable concentration of crosslinker. C2, C3 and C4 represent 2%, 3% and 4% theoretical cross-linking, respectively.
3.3.4. Effect of the Chitosan Molecular Weight
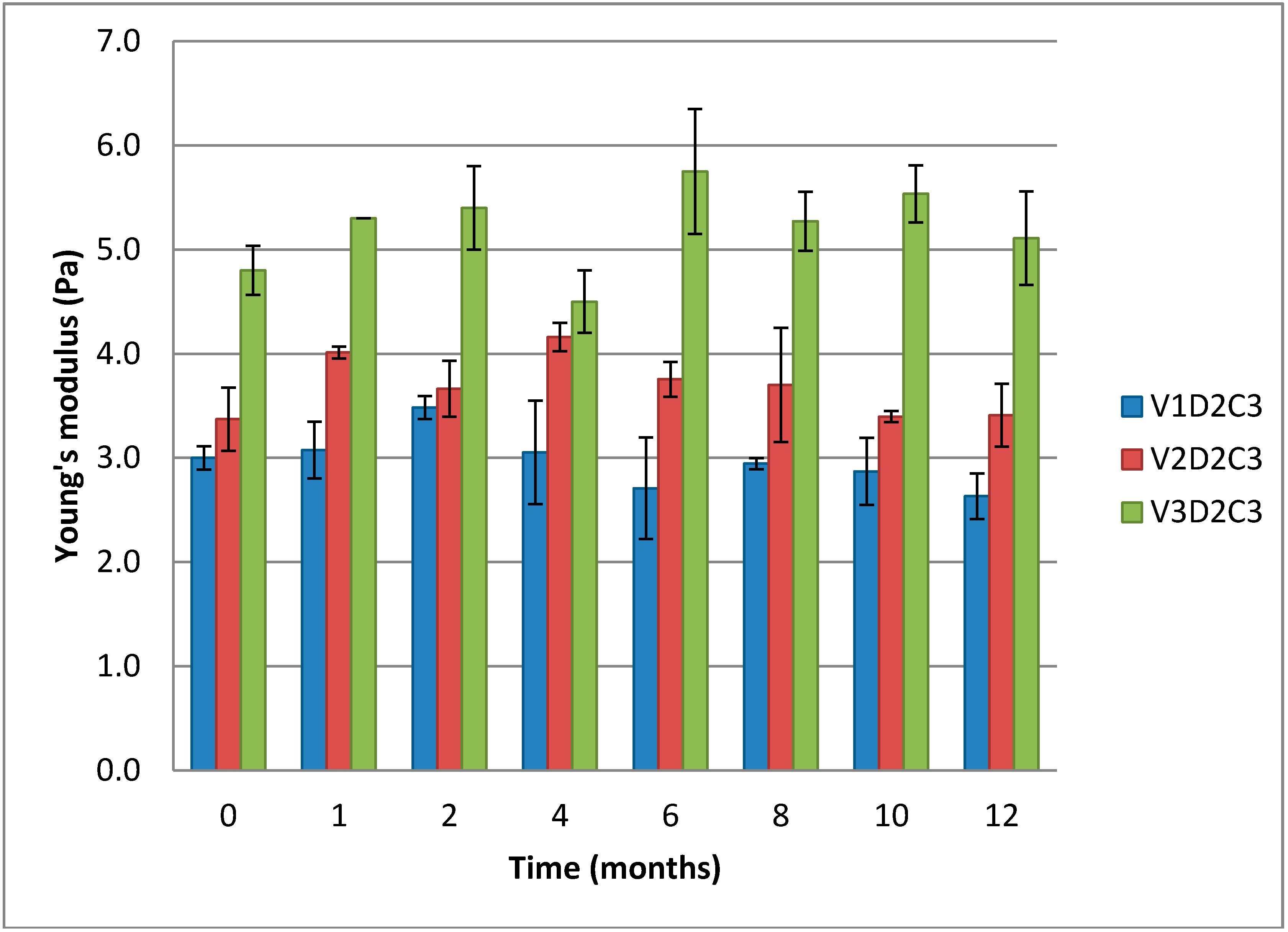
Figure 7 below displays Young’s modulus for three different gels with increasing molecular weight. Longer chains of chitosan in the gels make formation of more functional crosslinks possible and give a lower sol- and loose end fraction of chains. That means that a higher fraction of the chains is contributing to the gel lattice and the crosslinking is more effective. This again leads to a higher number of elastic segments. As seen in
Figure 6, there is a small trend towards an increase in Young’s modulus in the initial stage observed for all gels.
There are some variations in the measured stress at failure for the gels of variable chitosan molecular weight, as shown in
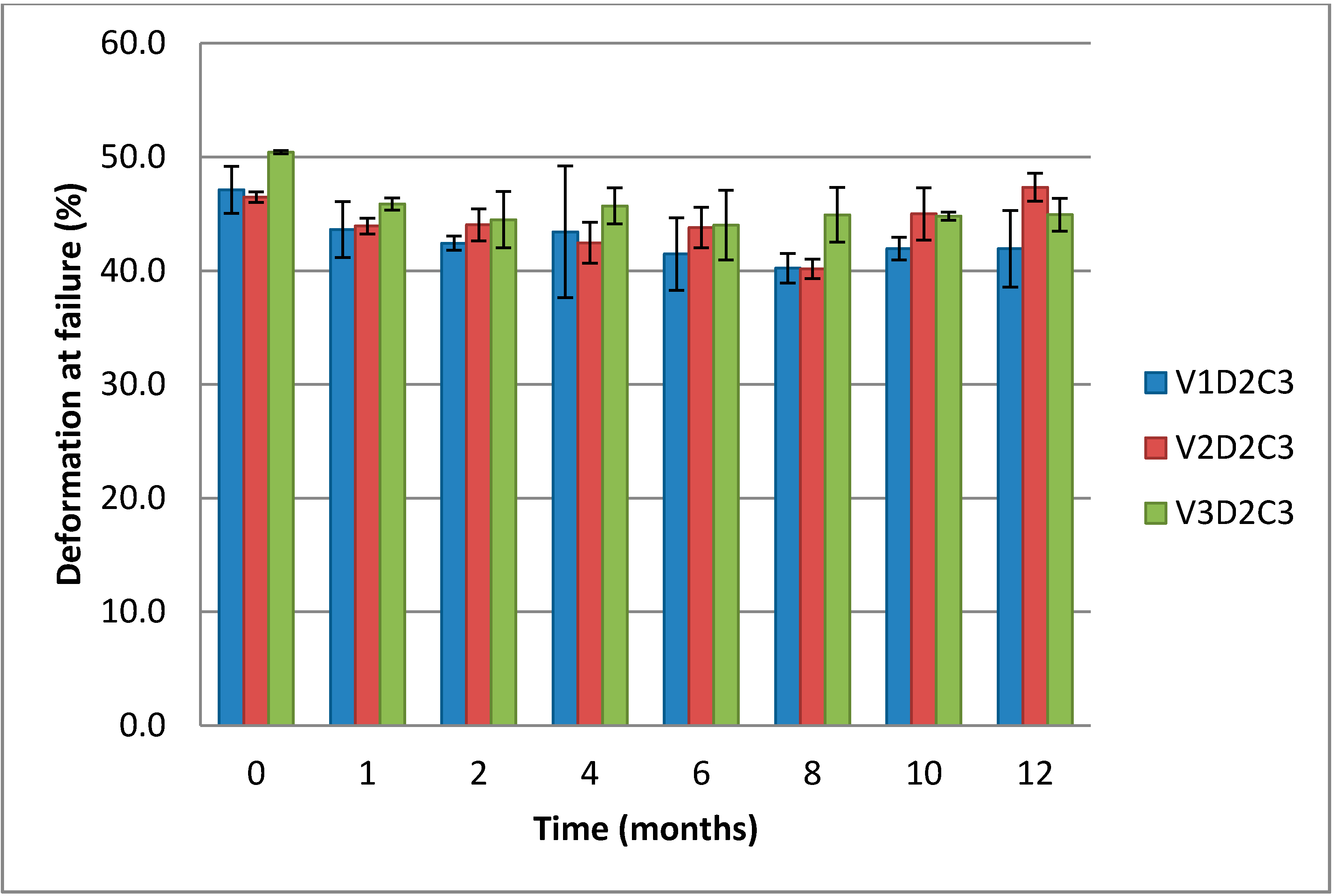
Figure 8. For the gels of low and medium molecular weight chitosan (V1D2C3 and V2D2C3) the differences are small, but the gels of highest molecular weight chitosan (V3D2C3) require a significantly higher force to break. Chitosan of longer chain lengths produces gels that are more rigid as a higher number of functional crosslinks per chain can be formed. Hence, when longer chitosan chains are available, a more ideal gel network can form. In the case of shorter chains, there will be more loose end segments present not contributing to the lattice resulting in a less ideal gel lattice.
When it comes to the deformation at failure, there are only small, non-significant differences between the measured values for the three gel types of varying molecular weight chitosan (
Figure 9).
Figure 7.
Comparison of Young’s modulus over 12 months for cylindrical gels of 1% (w/w) chitosan of variable molecular weight at 3% theoretical DES crosslinking.
Figure 7.
Comparison of Young’s modulus over 12 months for cylindrical gels of 1% (w/w) chitosan of variable molecular weight at 3% theoretical DES crosslinking.
Figure 8.
Comparison of stress at failure over 12 months for cylindrical gels of 1% (w/w) chitosan of variable molecular weight at 3% theoretical DES crosslinking.
Figure 8.
Comparison of stress at failure over 12 months for cylindrical gels of 1% (w/w) chitosan of variable molecular weight at 3% theoretical DES crosslinking.
Figure 9.
Comparison of deformation at failure over 12 months for cylindrical gels of (1% w/w) chitosan of variable molecular weight at 3% theoretical DES crosslinking.
Figure 9.
Comparison of deformation at failure over 12 months for cylindrical gels of (1% w/w) chitosan of variable molecular weight at 3% theoretical DES crosslinking.
3.4. Release of Pilocarpine from Drug-Loaded ViscoGel
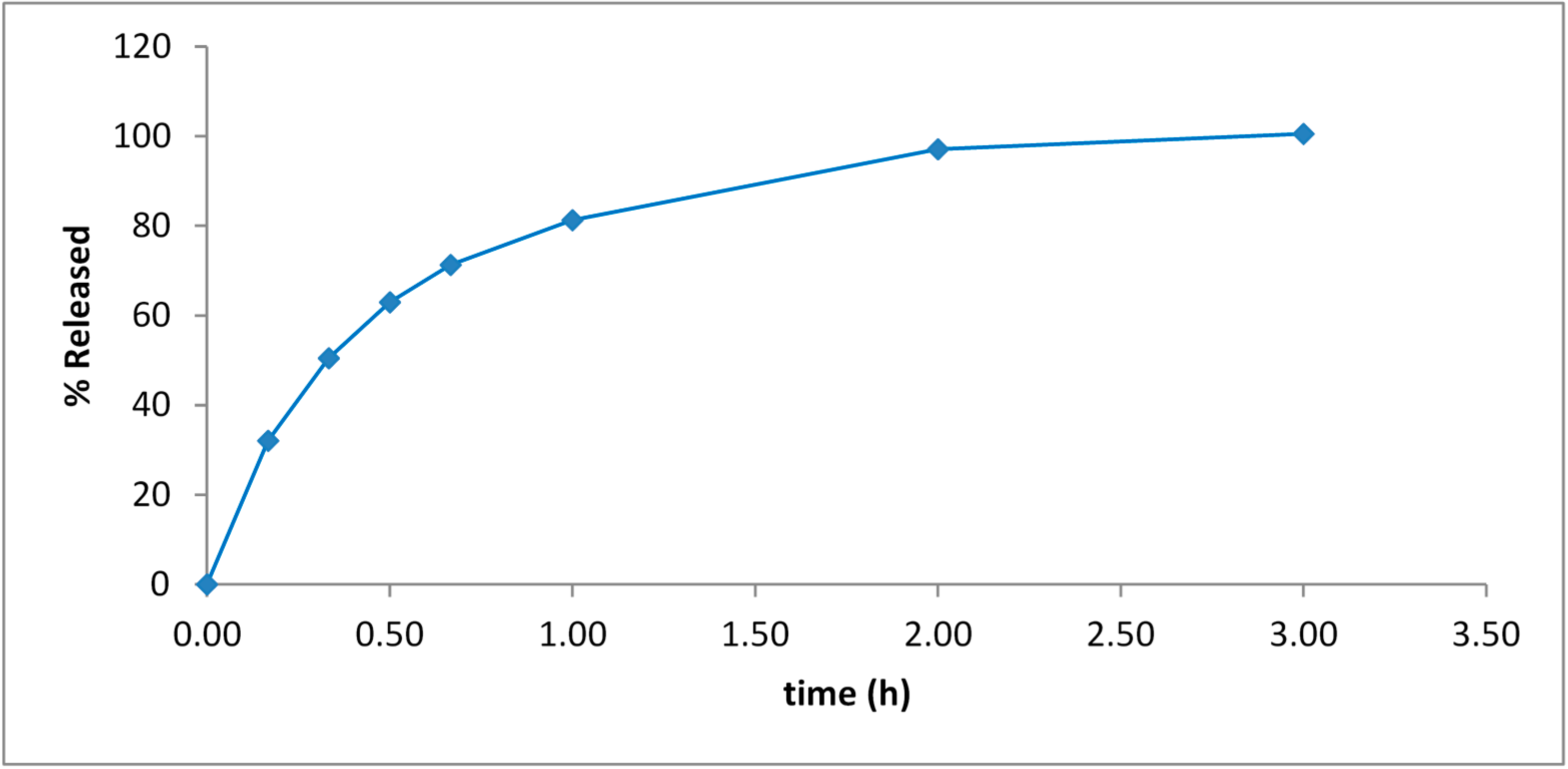
ViscoGel is a versatile system for drug delivery applications. An example of such a drug delivery system is described in
Section 2.7. Pilocarpine is a drug used to treat dry mouth, chronic open-angle glaucoma and acute angle-closure glaucoma and the release of pilocarpine from ViscoGel is shown in
Figure 10. The pilocarpine was fully released from the ViscoGel in 2–3 h with no burst effect.
Figure 10.
Release of Pilocarpine from ViscoGel™ in PBS at 37 °C.
Figure 10.
Release of Pilocarpine from ViscoGel™ in PBS at 37 °C.
3.5. General Observations
The data presented above, together with unpublished results, show that ViscoGels can be produced from chitosans of different fractions of acetylation and molecular weight. The process for their preparation is mild and robust, in a water solution at near neutral pH (6.6) and at a slightly elevated temperature (40 °C), and the gels formed possess useful physical properties. We also found that the DES is fully consumed in less than 22 h, even when five times the normal amount (15%) of DES is used, i.e., DES forms at least one bond with the polymer rather quickly. (The reaction was analyzed by HPLC at 253 nm (LoD 5.8 ng/L). Results not included). The viscoelastic properties of the ViscoGels allow for further mechanical processing into particles. When comparing the different systems, some general tendencies can be observed. When increasing the molecular weight of the chitosan, increased gel strength is observed. Increasing the degree of crosslinking for systems made of the same chitosan augments the gel stiffness. The gelation curves for the different systems show a steady increase in both the elasticity modulus and the viscosity during the time of measurement and they reached equilibrium values after approximately 5–6 days.
Like other chitosans, chitosans with high FA possess all the attractive properties but there are some differences. In addition to being soluble at physiological conditions, they are slightly less anti-microbial and degrade substantially faster than chitosans having a lower FA. Interestingly, when converted into the form of a ViscoGel, some of these properties are altered and one of the most striking changes is that of the antimicrobial properties. ViscoGel in particulate form does not, in contrast to a chitosan solution of the same concentration, exert strong antimicrobial properties (in-house report). A likely explanation for this observation is that the polymer chains in the ViscoGel are locked up in a three dimensional network and cannot, like chitosan, wrap the bacteria in a chitosan cage, eventually leading to bacterial death. This lack of antimicrobial properties combined with a positive charged surface of the particles at physiological conditions makes ViscoGel particles useful as vehicles for transportation of living cells and microorganisms. The cells and microorganisms, when mixed with the particles, will bind tightly to the surface of the particles, without losing their viability, and can then be injected. This makes ViscoGel particles potentially useful in e.g., vaccination with live virus, bacteria-vaccines and in transplantation of cells.
4. Conclusions
In contrast to super-swelling gels [
28,
29], the swelling of ViscoGel is modest. The magnitude of the swelling/shrinking of ViscoGel in different buffers was ±20%, even after 91 h exposure to the buffers in the range of pH 4–8. A hypotonic (standard) ViscoGel changes its volume slightly less than the isotonic ViscoGel, but no major swelling/shrinking occurs in either case.
The ViscoGel formulations, independent upon Mw and degree of cross-linking, exhibited excellent stability over the one year period both with respect to the elastic modulus and force/deformation at failure. As expected, the strongest gels were obtained using a high concentration of crosslinker and chitosans with high molecular weight.
The ViscoGel technology is versatile and examples of more advanced uses are drug delivery of small molecules and biologicals. The drug, which has been incorporated in the three dimensional chitosan network, is released from the ViscoGel particles through diffusion as seen in the example with Pilocarpine, or via a mixed mechanism—diffusion and erosion by enzymatic degradation—when studied in an in vivo system. The above presented data support the possible use of ViscoGels in medicine, e.g., as a carrier of living organisms or as a sustained drug release vehicle.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









