
Study on the Effect of Silanization and Improvement in the Tensile Behavior of Graphene-Chitosan-Composite

Abstract
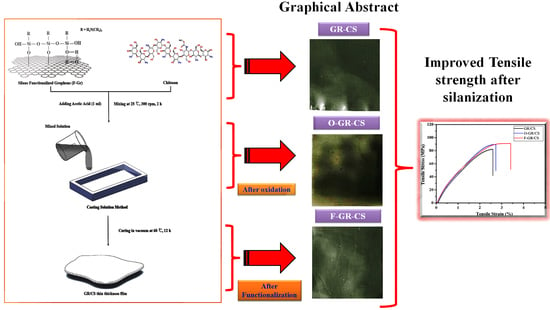
:
1. Introduction
2. Experimental Section
2.1. Reagents
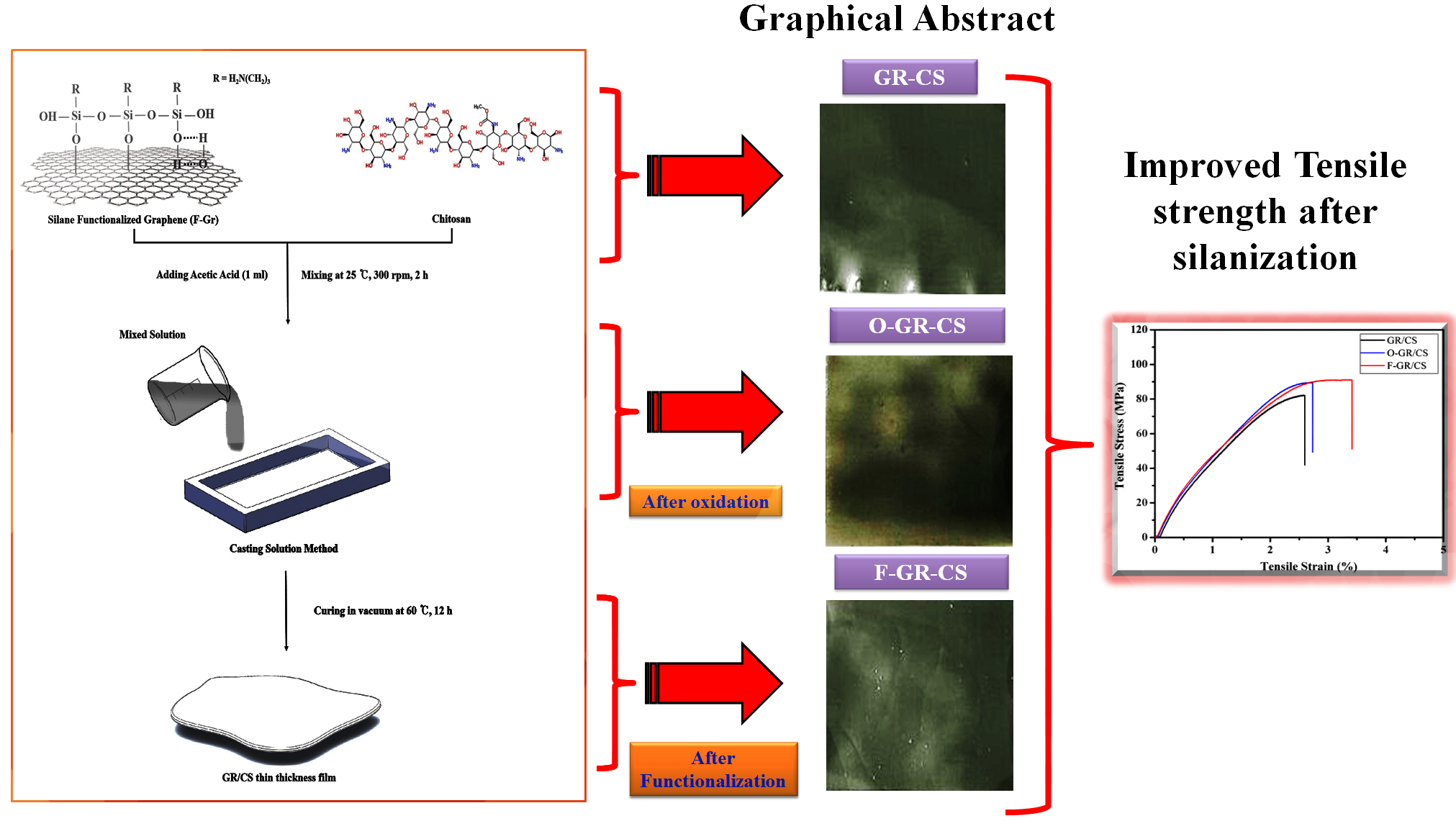
2.2. Oxidation of Graphene
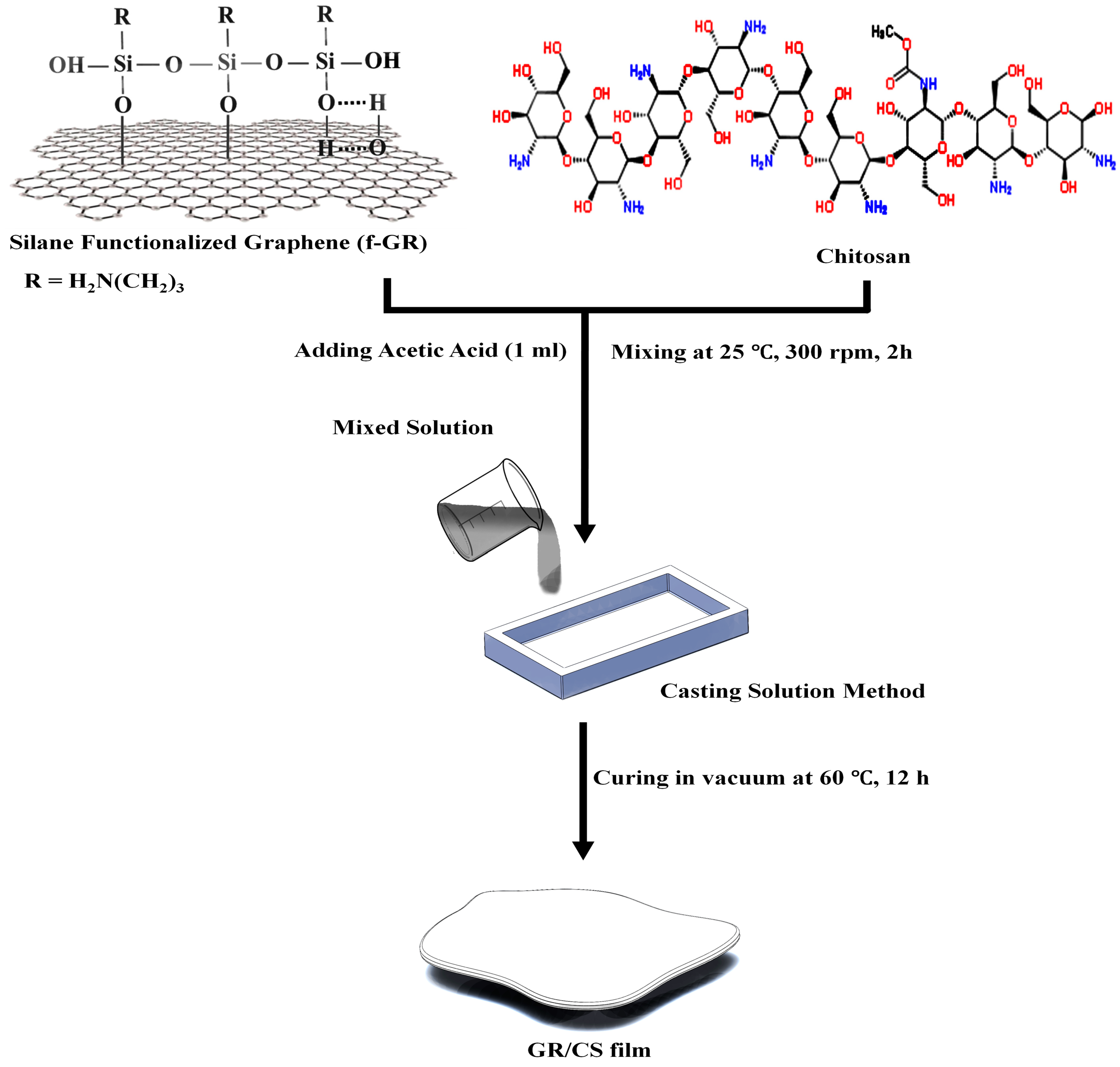
2.3. Silanization of Graphene with 3-Aminopropyltriethoxysilane (3-APTES)

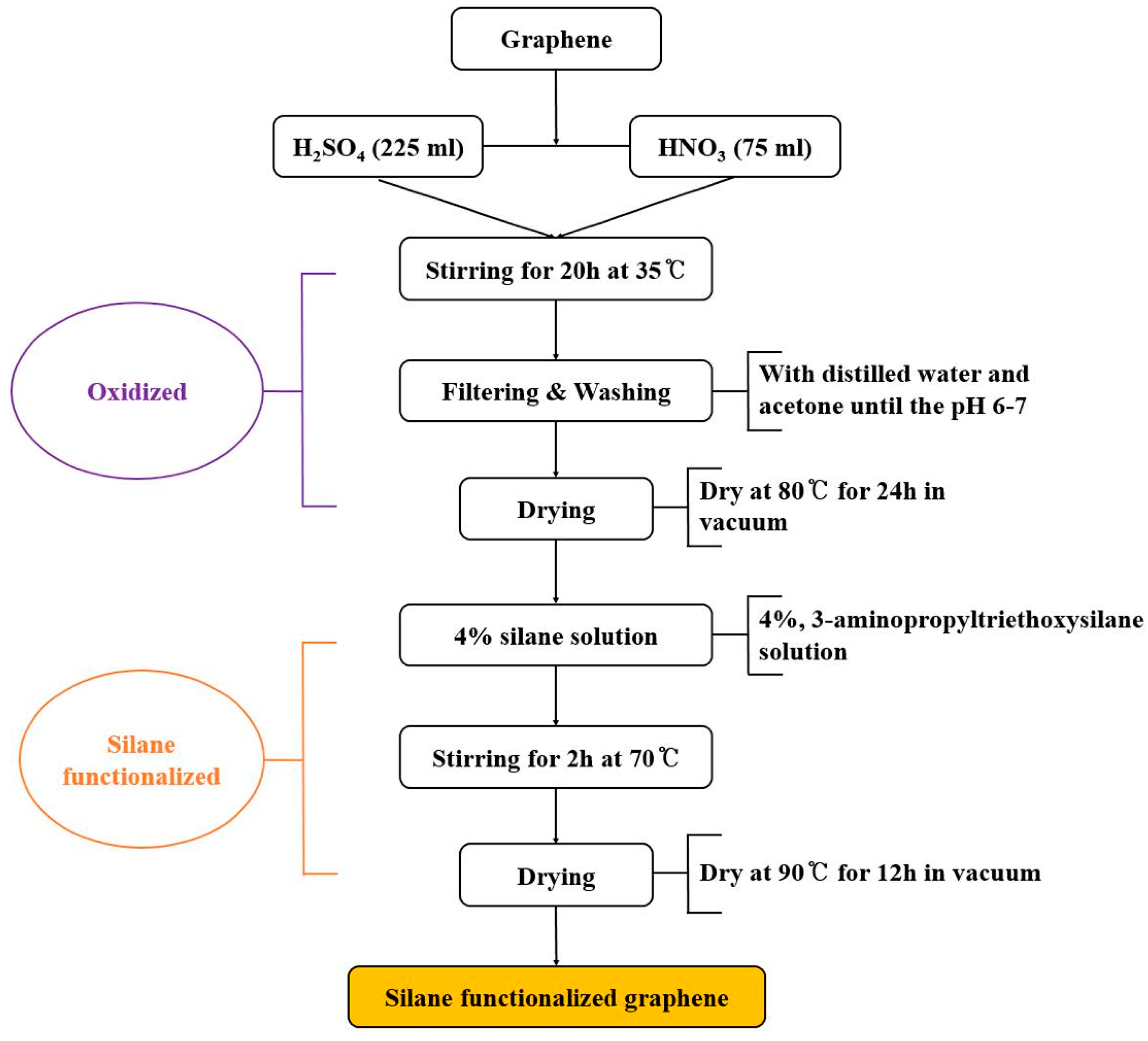
2.4. Preparation of Chitosan/Graphene Nanocomposite Films

3. Characterization
4. Results and Discussions
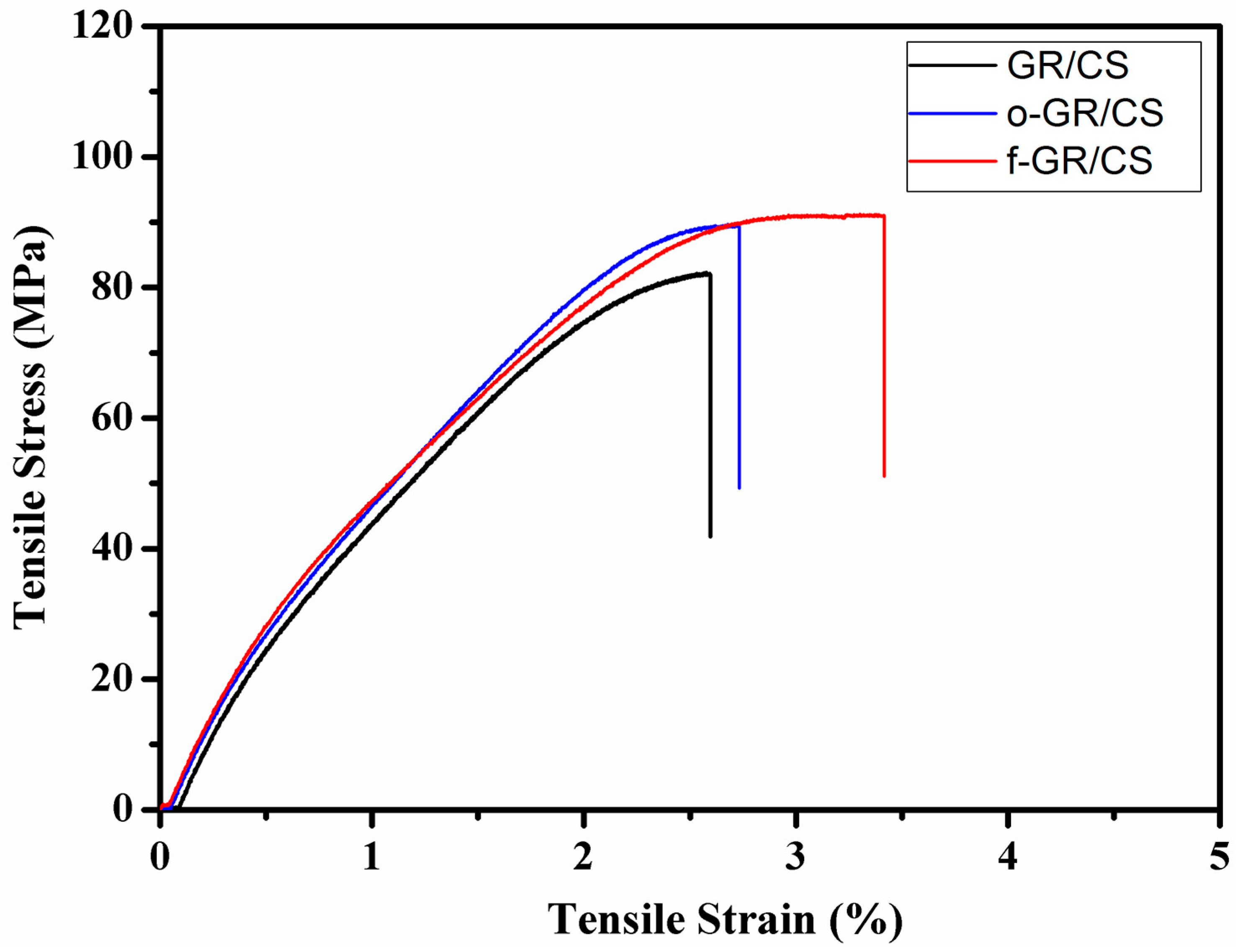
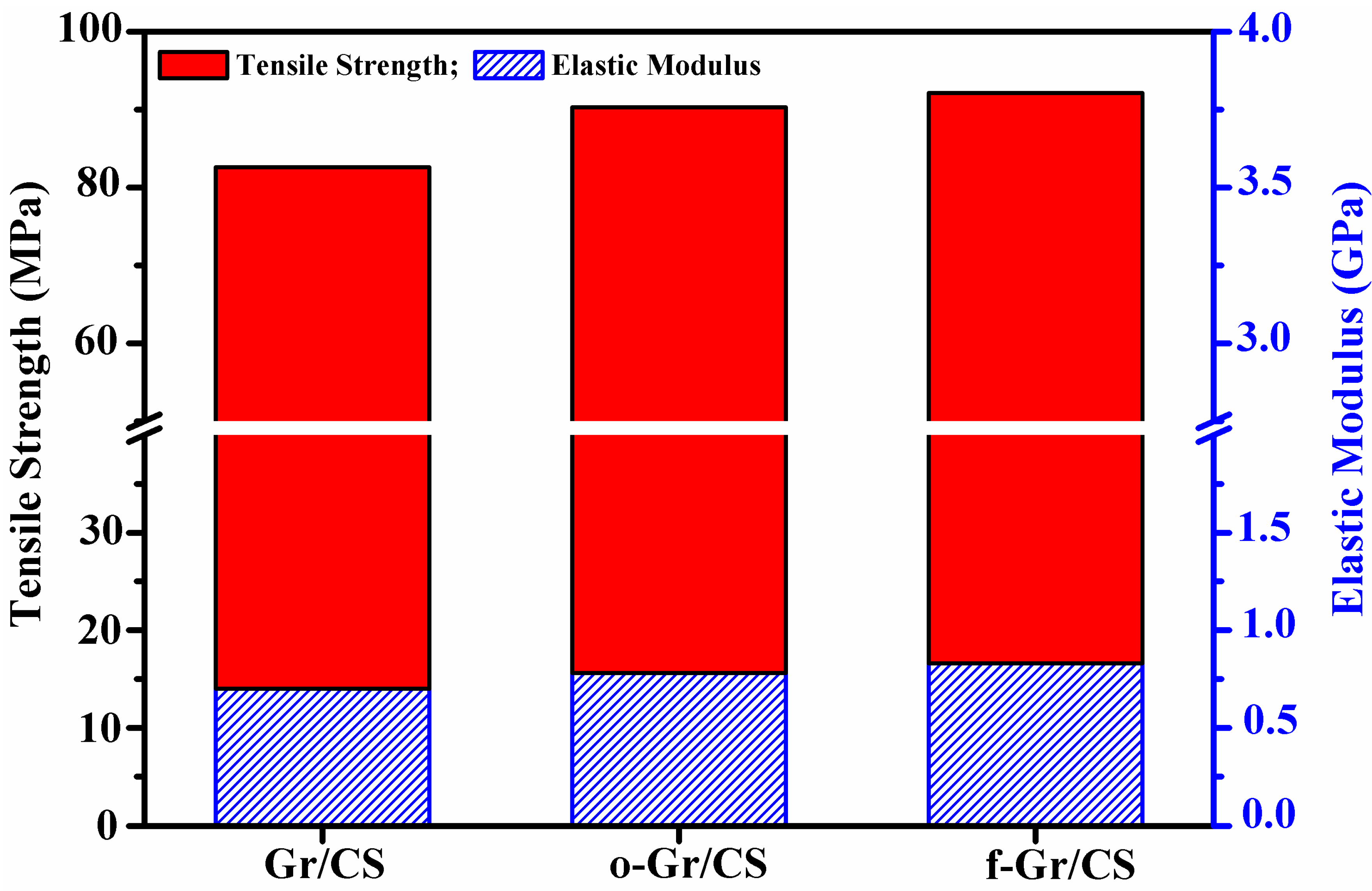
4.1. Tensile Characteristics of Graphene/Chitosan Nanocomposite Films



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample type | Young’s modulus (GPa) | Ultimate tensile strength (MPa) | Elongation at break (%) |
|---|---|---|---|
| GR/CS | 0.70 (±0.021) | 81.73 (±1.43) | 2.50 (±0.32) |
| o-GR/CS | 0.78 (±0.017) | 89.43 (±1.22) | 2.55 (±0.29) |
| f-GR/CS | 0.83 (±0.016) | 91.01 (±1.19) | 3.37 (±0.31) |
| Sample: GR/CS | Tensile Stress (Mpa) | Young’s Modulus (Gpa) | Strain% |
| No. 1 | 84.3212 | 0.7521 | 1.6675 |
| No. 2 | 81.1147 | 0.6817 | 3.3212 |
| No. 3 | 76.0059 | 0.6632 | 3.3342 |
| No. 4 | 82.061 | 0.6335 | 2.4225 |
| No. 5 | 85.1472 | 0.7695 | 1.7751 |
| MEAN | 81.73 | 0.7 | 2.5041 |
| SD | 3.213877695 | 0.052260961 | 0.720377337 |
| SE | 1.437289799 | 0.023371812 | 0.322162539 |
| Sample o-GR/CS | Tensile Stress (Mpa) | Young’s Modulus (Gpa) | Strain% |
| No. 1 | 86.1145 | 0.7721 | 1.9871 |
| No. 2 | 87.1152 | 0.7879 | 2.6664 |
| No. 3 | 92.0775 | 0.8441 | 1.8891 |
| No. 4 | 88.7138 | 0.7675 | 2.5484 |
| No. 5 | 93.1145 | 0.7345 | 3.6875 |
| MEAN | 89.4271 | 0.78122 | 2.5557 |
| SD | 2.736732949 | 0.035924053 | 0.642018775 |
| SE | 1.223904182 | 0.016065725 | 0.287119525 |
| Sample f-GR/CS | Tensile Stress (Mpa) | Young’s Modulus (Gpa) | Strain% |
| No. 1 | 89.1475 | 0.8723 | 2.7784 |
| No. 2 | 91.05 | 0.8615 | 2.9785 |
| No. 3 | 95.7785 | 0.7824 | 4.2978 |
| No. 4 | 87.9612 | 0.8351 | 2.6958 |
| No. 5 | 91.1158 | 0.7972 | 4.0975 |
| MEAN | 91.0106 | 0.8297 | 3.3696 |
| SD | 2.665163101 | 0.035067649 | 0.685255681 |
| SE | 1.191897173 | 0.015682729 | 0.306455657 |
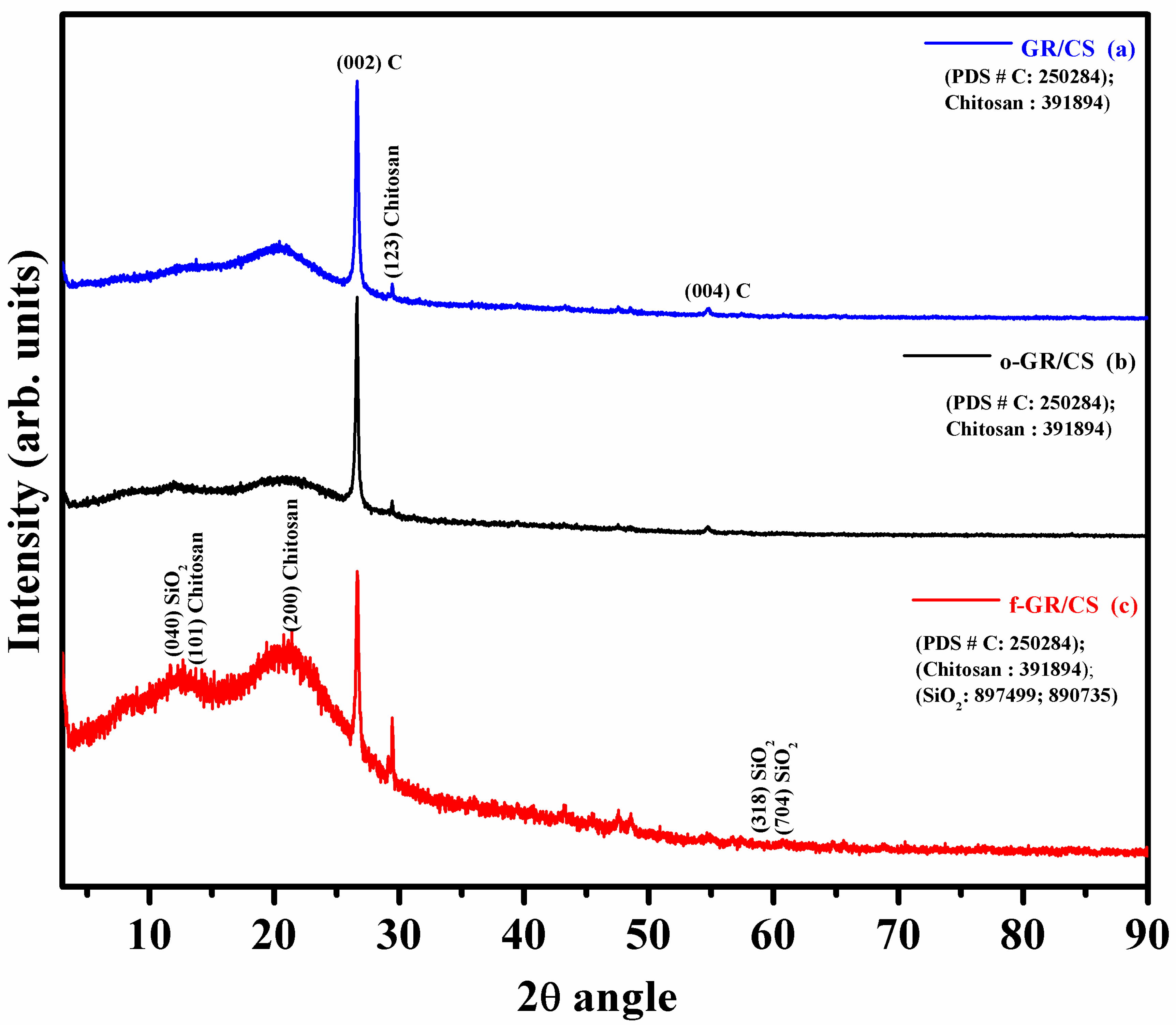
4.2. X-ray Diffraction

| Sample Name | 2θ Value | hkl | System | Lattice | JCPDF No. | Mineral Phase |
|---|---|---|---|---|---|---|
| (a) GR/-CS | 26.618 | 002 | Hexagonal | Primitive | 25-0284 | Graphite |
| 29.500 | 123 | Orthorhombic | NA | 39-1894 | Chitosan | |
| 54.73 | 004 | Hexagonal | Primitive | 25-0284 | Graphite | |
| (b) o-GR/CS | 26.609 | 002 | Hexagonal | Primitive | 25-0284 | Graphite |
| 29.414 | 123 | Orthorhombic | NA | 39-1894 | Chitosan | |
| 54.721 | 004 | Hexagonal | Primitive | 25-0284 | Graphite | |
| (c) f-GR/CS | 11.667 | 040 | Monoclinic | End centered | 89-7499 | SiO2 |
| 13.602 | 101 | Orthorhombic | NA | 39-1894 | Chitosan | |
| 21.100 | 200 | Orthorhombic | NA | 39-1894 | Chitosan | |
| 26.637 | 002 | Hexagonal | Primitive | 25-0284 | Graphite | |
| 29.453 | 123 | Orthorhombic | NA | 39-1894 | Chitosan | |
| 57.403 | 318 | Rhombohedral | Rhombo-central | 89-0735 | SiO2 | |
| 60.700 | 704 | Rhombohedral | Rhombo-central | 89-0735 | SiO2 |
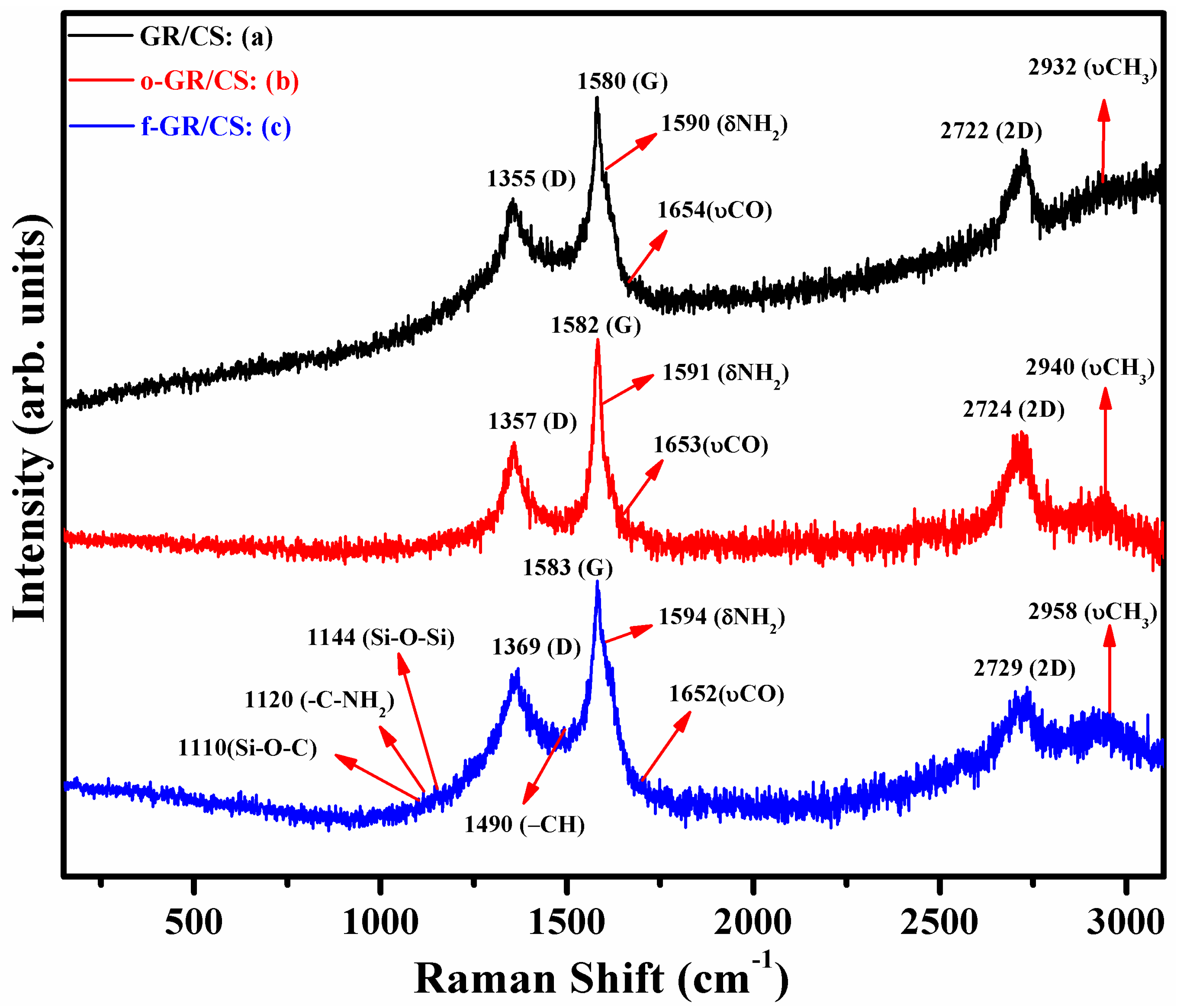
4.3. Raman Spectroscopy

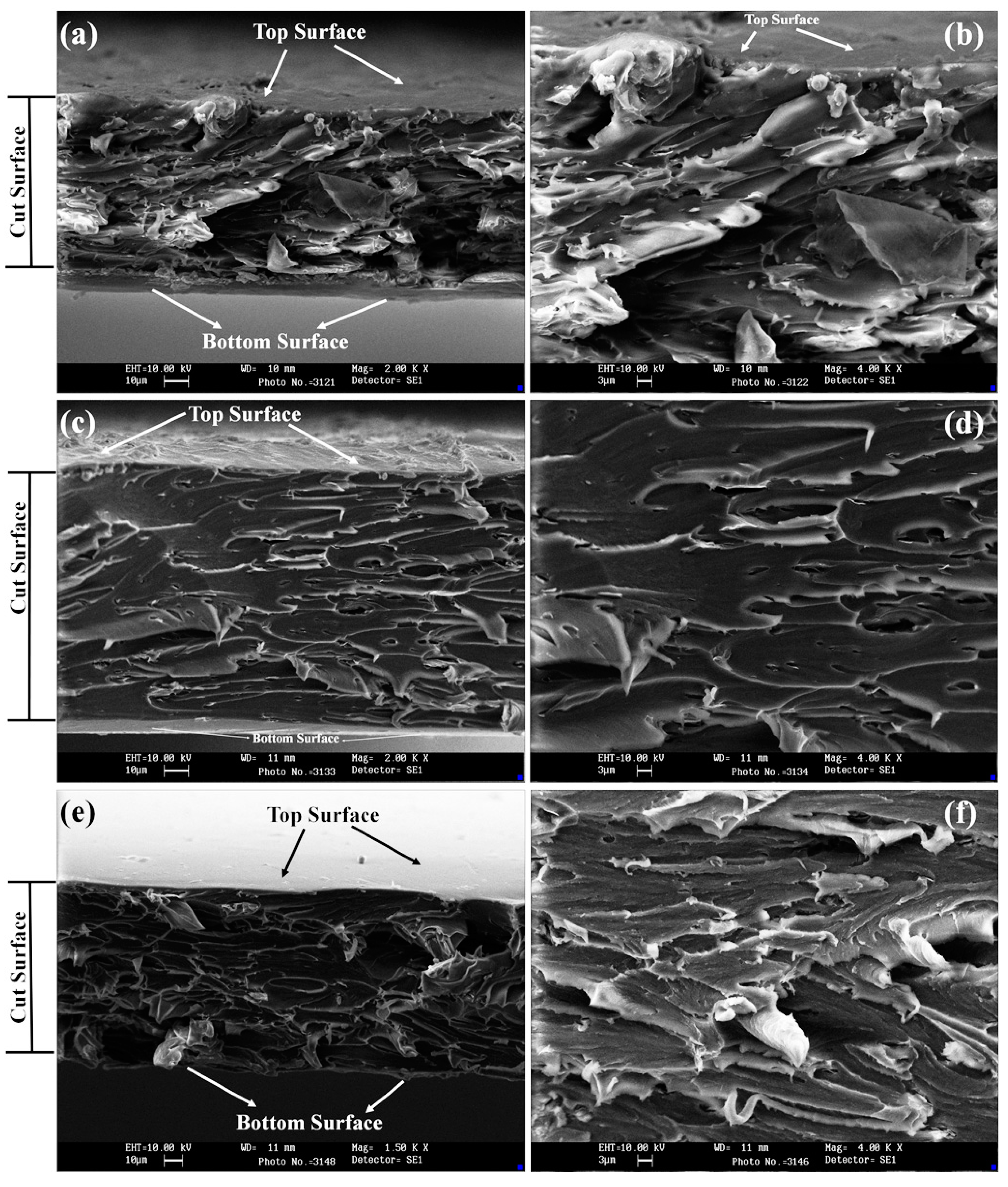
4.4. Scanning Electron Microscope (SEM) Analysis


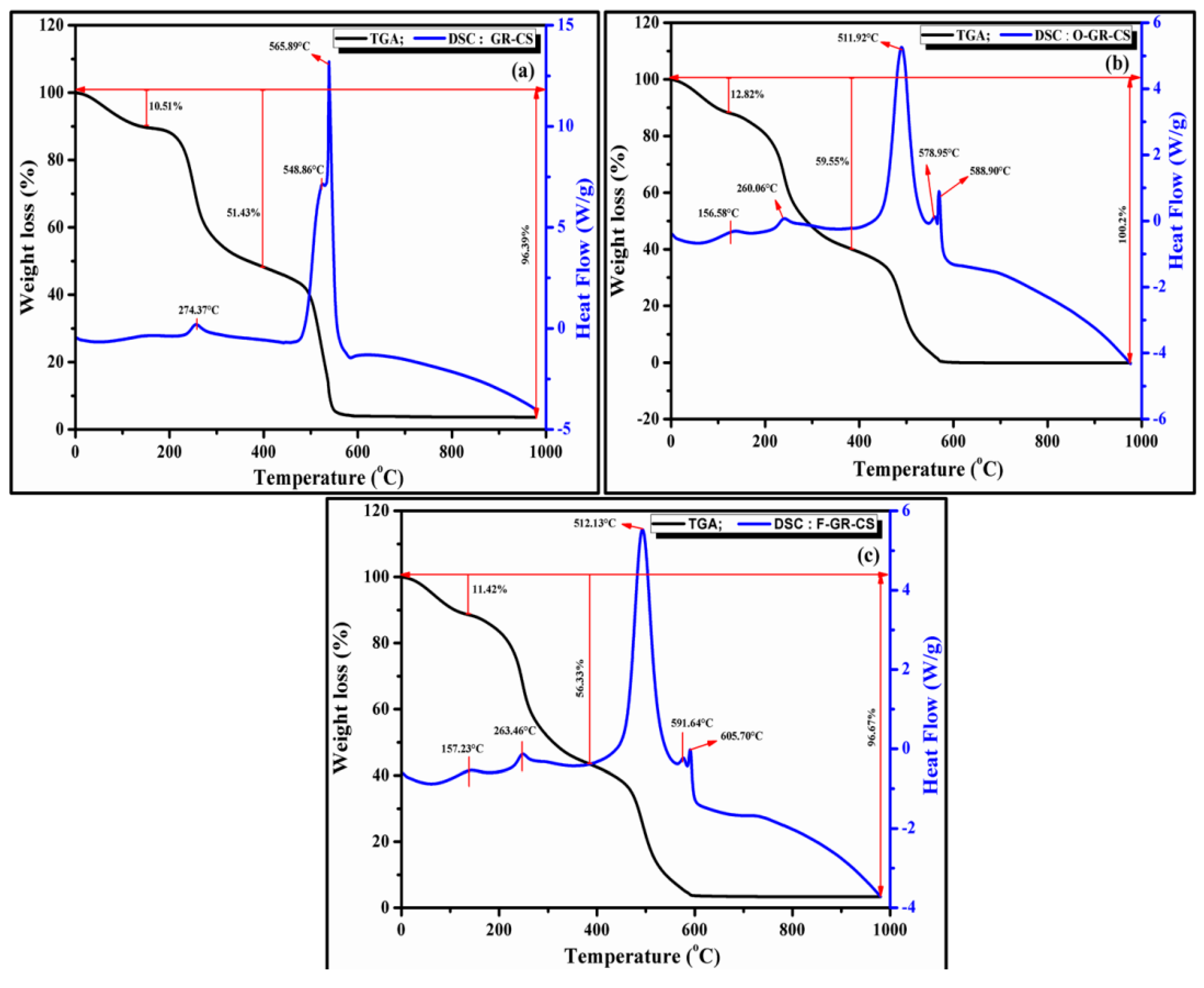
4.5. Thermal Analysis

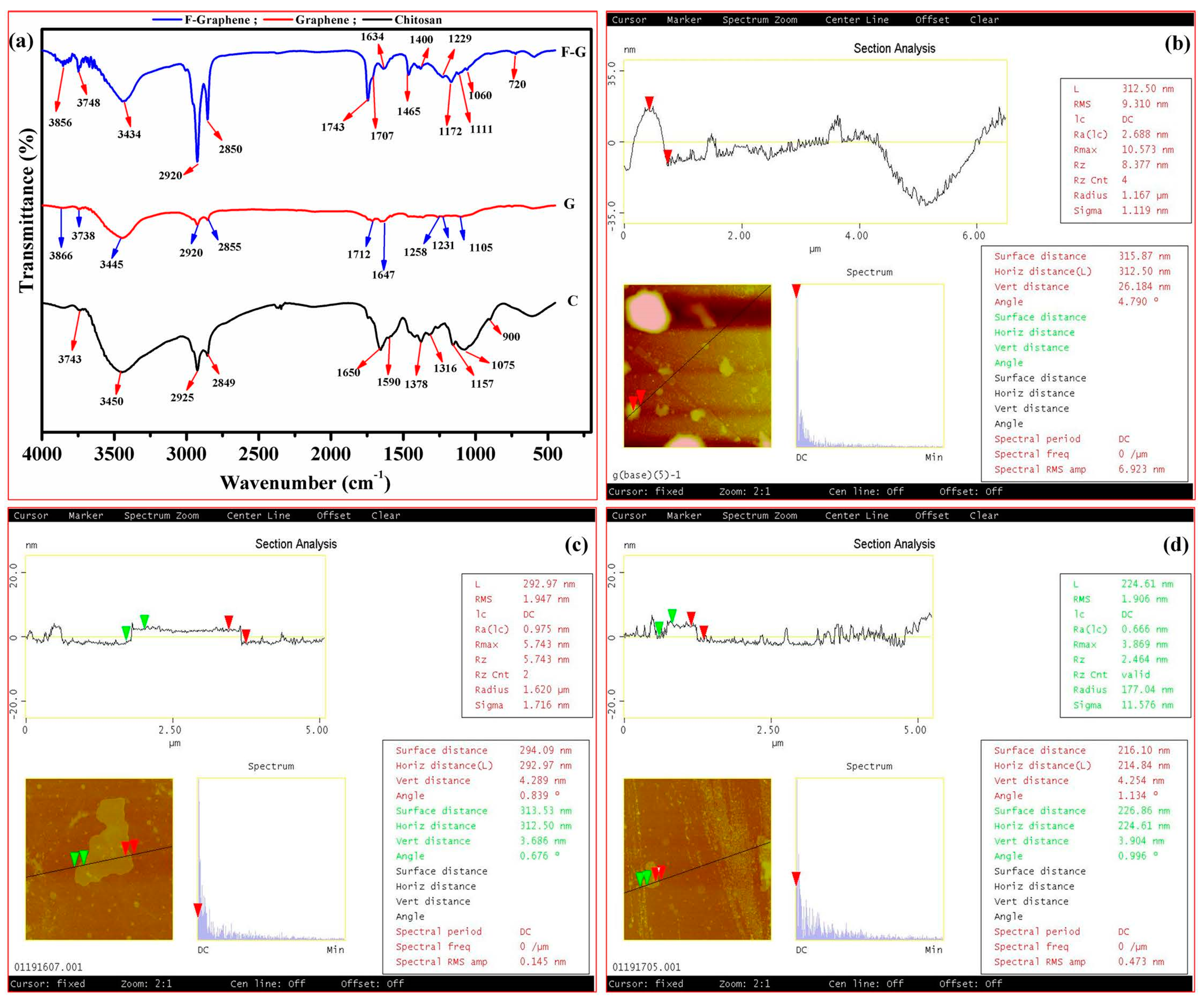
4.6. FTIR Spectroscopy

| Chemical | Peak position (cm−1) | Assignment | Reference |
|---|---|---|---|
| Chitosan | 900 | Stretching of C–O from: C–O–H; symmetric and asymmetric stretch from C–O–C bridge | [46] |
| 1075 | Saccharide structure of chitosan and C–O stretching | [47,48] | |
| 1157 | Saccharide structure of chitosan and anti-symmetric stretching from C–O–C bridge | [47,48] | |
| 1316 | Acetyl Groups | [49] | |
| 1378 | Amide III/C–H bending and stretching | [50] | |
| 1590 | Amine (NH2) band bending vibration | [51] | |
| 1650 | Carboxamide O=C–NHR | [51] | |
| 2849 | (CH3) anti-symmetric and symmetric stretching vibrations | [48] | |
| 2925 | (CH3) anti-symmetric and symmetric stretching vibrations | [48] | |
| 3450 | –OH/––NH2 stretching | [51] | |
| 3743 | –OH/–NH2 stretching | [51] | |
| Graphene | 1105 | C–O stretching | [52] |
| 1231 | C–OH stretching | [52] | |
| 1258 | C–O asymmetric stretching | [53] | |
| 1647 | C=C ring stretching | [54] | |
| 1712 | C=O stretching arising from carbonyl and carboxylic groups | [52] | |
| 2855 | –CH2 | [54] | |
| 2920 | –CH2 | [54] | |
| 3445 | –OH stretching | [52] | |
| 3738 | –OH stretching | [52] | |
| 3866 | –OH stretching | [52] | |
| f-Graphene | 720 | Si–O–Si | [55] |
| 1060 | Si–O–Si | [52] | |
| 1111 | Si–O–C | [52] | |
| 1172 | Si–O–Si | [55] | |
| 1229 | C–OH stretching | [52] | |
| 1400 | δ(O–H) basal plane CO–H | [53] | |
| 1465 | υ(C=C), δ(O–H), υ(C–O) | [53] | |
| 1634 | C=C ring stretching | [54] | |
| 1707 | C=O stretching from carbonyl and carboxylic groups | [52] | |
| 1743 | υ(C=O) | [53] | |
| 2850 | –CH2 | [54] | |
| 2920 | –CH2 | [54] | |
| 3434 | –OH stretching | [52] | |
| 3748 | –OH stretching | [52] | |
| 3856 | –OH stretching | [52] |
4.7. Atomic Force Microscopy
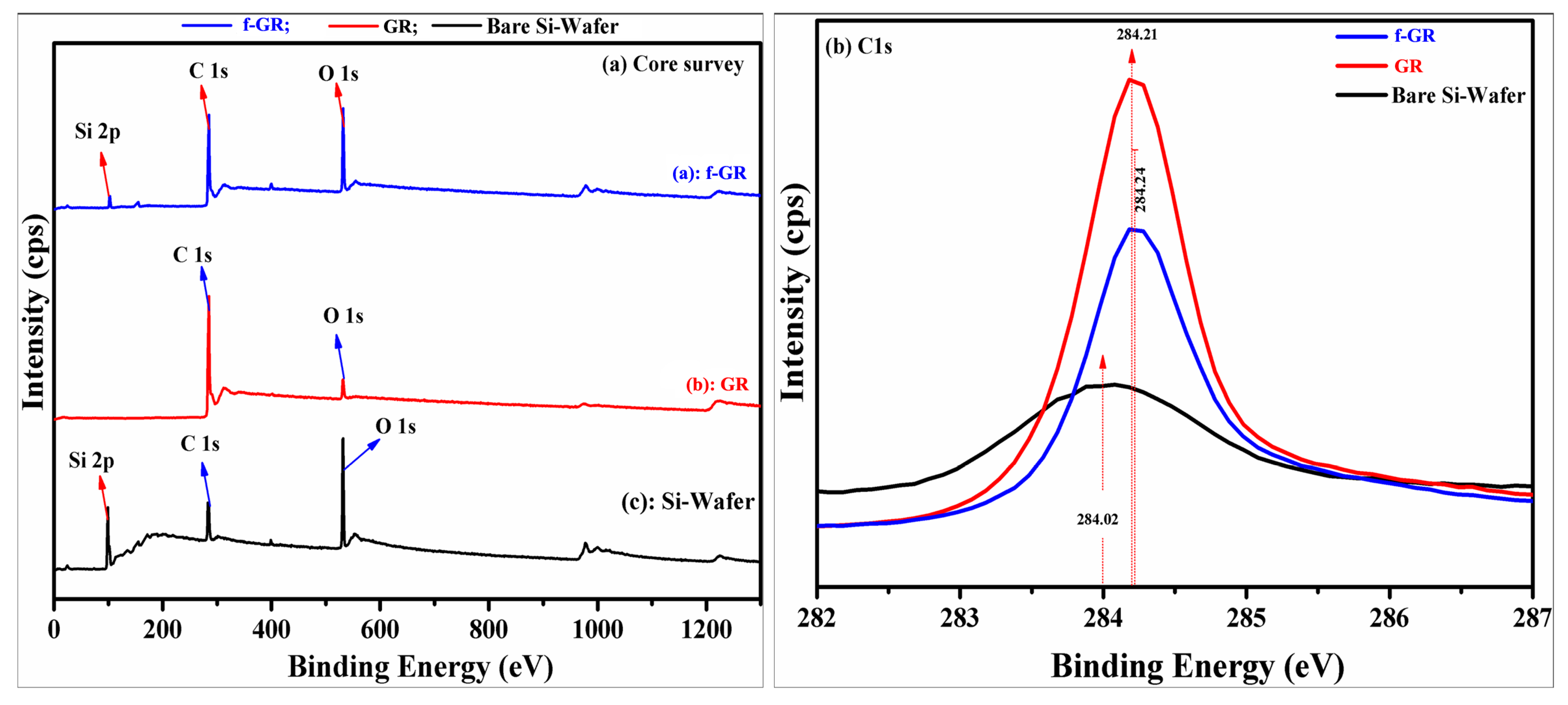
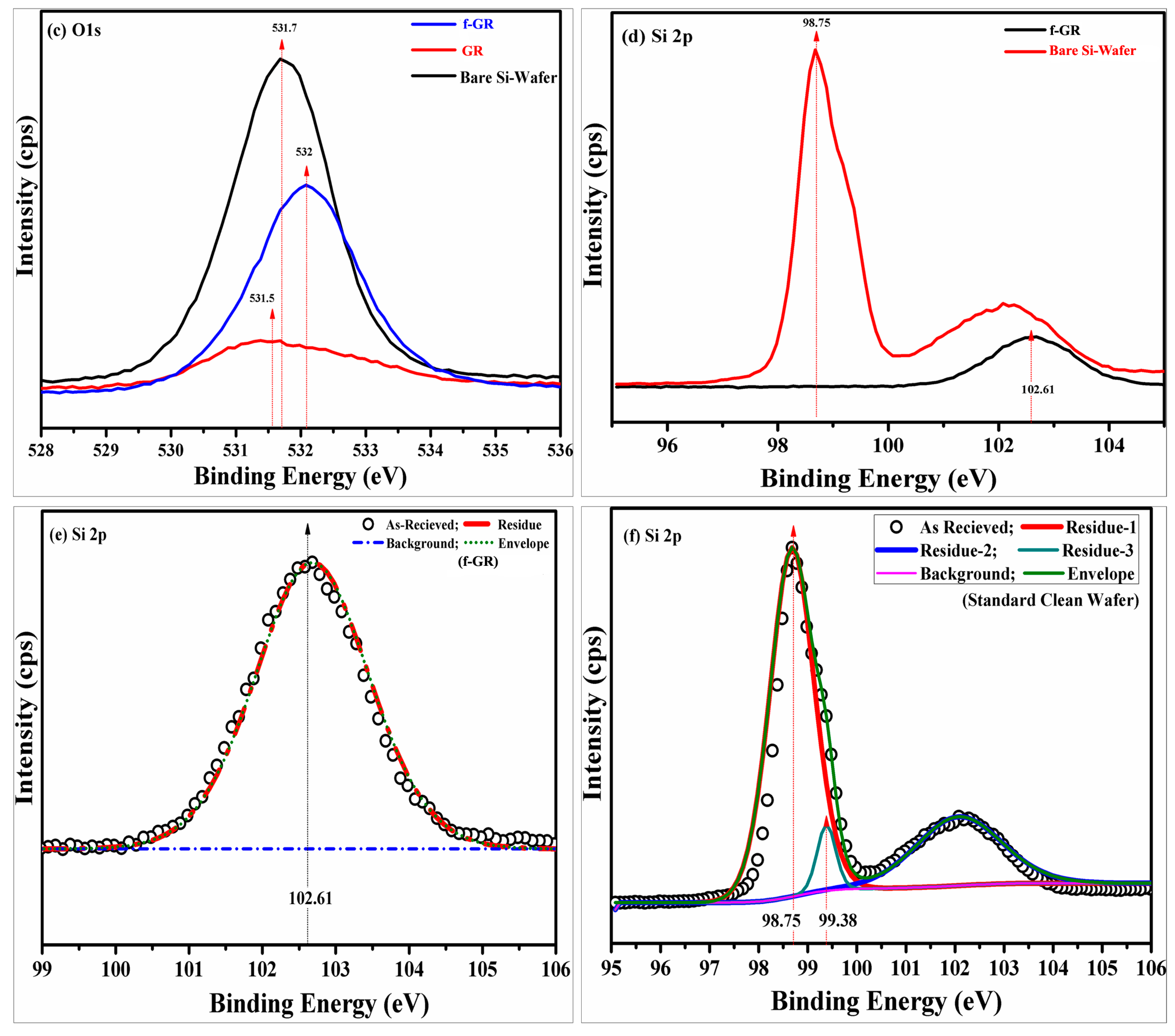
4.8. X-ray Photoelectron Spectroscopy


5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Maksimov, R.D.; Ivanova, T.; Kalnins, M.; Zicans, J. Mechanical properties of high-density polyethylene/chlorinated polyethylene blends. Mech. Compos. Mater. 2004, 40, 331–340. [Google Scholar] [CrossRef]
- Stevanovic, D.; Lowe, A.; Kalyanasundaram, S.; Jar, P.Y.B.; Alego, V.O. Chemical and mechanical properties of vinyl-ester/ABS blends. Polymer 2002, 43, 4503–4514. [Google Scholar] [CrossRef]
- Guzmán de Villoria, R.; Miravete, A.; Cuartero, J.; Chiminelli, A.; Tolosana, N. Mechanical properties of SWNT/epoxy composites using two different curing cycles. Compos. B Eng. 2006, 37, 273–277. [Google Scholar]
- Kinoshita, H.; Kaizu, K.; Fukuda, M.; Tokunaga, H.; Koga, K.; Ikeda, K. Development of green composite consist of woodchip, bamboo fibers and biodegradable adhesive. Compos. B Eng. 2009, 40, 607–612. [Google Scholar] [CrossRef]
- Zhang, X.; Do, M.D.; Bilyk, A. Chemical modification of wheat-protein-based natural polymers: Formation of polymer networks with alkoxysilanes to modify molecular motions and enhance the material performance. Biomacromolecules 2007, 8, 1881–1889. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.; Bristow, J.F.; Smith, P.J.; Payne, G.F. Enzymatic gelation of the natural polymer chitosan. Polymer 2000, 41, 2157–2168. [Google Scholar] [CrossRef]
- Muzzarelli, R.A.A.; Boudrant, J.; Meyer, D.; Manno, N.; DeMarchis, M.; Paoletti, M.G. Current views of fungal chitin/chitosan, human chitinases, food preservation, glucans, pectins and inulin: A tribute to Henri Braconnot, precursor of the carbohydrate polymers science, on the chitin bicentennial. Carbohydr. Polym. 2012, 87, 995–1012. [Google Scholar] [CrossRef]
- Fernandes, S.C.M.; Freire, C.S.R.; Silvestre, A.J.D.; Neto, C.P.; Gandini, A.; Berglund, L.A.; Salmén, L. Transparent chitosan films reinforced with a high content of nanofibrillated cellulose. Carbohydr. Polym. 2010, 81, 394–401. [Google Scholar] [CrossRef]
- Muzazarelli, R.A.A.; Greco, F.; Busilacchi, A.; Sollazzo, V.; Gigante, A. Chitosan, hyaluronan and chondroitin sulfate in tissue engineering for cartilage regeneration: A review. Carbohydr. Polym. 2012, 89, 723–739. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Chen, N.; Zhang, Q.; Wang, K.; Fu, Q.; Zhang, X. Preparation and properties of chitosan nanocomposites with nanofillers of different dimensions. Polym. Degrad. Stabil. 2009, 94, 124–131. [Google Scholar] [CrossRef]
- Venkatesan, J.; Kim, S.K. Chitosan composites for bone tissue engineering-an overview. Mar. Drugs 2010, 8, 2252–2266. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.R.; Jian, R.; Zheng, P.; Yu, J.; Ma, X. Preparation and properties of glycerol plasticized-starch (GPS)/cellulose nanoparticle (CN) composites. Carbohydr. Polym. 2010, 79, 301–305. [Google Scholar] [CrossRef]
- Cai, X.; Tong, H.; Shen, X.; Chen, W.; Yan, J.; Hu, J. Preparation and characterization of homogeneous chitosan–polylactic acid/hydroxyapatite nanocomposite for bone tissue engineering and evaluation of its mechanical properties. Acta Biomater. 2009, 5, 2693–2703. [Google Scholar] [CrossRef] [PubMed]
- Darder, M.; Colilla, M.; Ruiz-Hitzky, E. Biopolymer−clay nanocomposites based on chitosan intercalated in montmorillonite. Chem. Mater. 2003, 15, 3774–3780. [Google Scholar] [CrossRef]
- Li, G.; Jiang, Y.; Huang, K.; Ding, P.; Chen, J. Preparation and properties of magnetic Fe3O4–chitosan nanoparticles. J. Alloy Compd. 2008, 466, 451–456. [Google Scholar] [CrossRef]
- Wang, S.; Shen, L.; Zhang, W.; Tong, Y. Preparation and mechanical pproperties of chitosan/carbon nanotubes composites. Biomacromolecules 2005, 6, 3067–3072. [Google Scholar] [CrossRef] [PubMed]
- Azeez, A.A.; Rhee, K.Y.; Park, S.J.; Kim, H.J.; Jung, D.H. Application of cryomilling to enhance material properties of carbon nanotube reinforced chitosan nanocomposites. Compos. B Eng. 2013, 50, 127–134. [Google Scholar] [CrossRef]
- Marroquin, J.B.; Rhee, K.Y.; Park, S.J. Chitosan nanocomposite films: Enhanced electrical conductivity, thermal stability, and mechanical properties. Carbohydr. Polym. 2013, 92, 1783–1791. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, N.G.; Rana, S.; Cho, J.W.; Li, L.; Chan, S.H. Polymer nanocomposites based on functionalized carbon nanotubes. Prog. Poylm. Sci. 2010, 35, 837–867. [Google Scholar] [CrossRef]
- Kim, M.T.; Rhee, K.Y.; Lee, J.H.; Hui, D.; Lau, A.K.T. Property enhancement of a carbon fiber/epoxy composite by using carbon nanotubes. Compos. B Eng. 2011, 42, 1257–1261. [Google Scholar] [CrossRef]
- Liu, X.; He, X.Q.; Yang, Q.S.; Mai, Y.W. Overall behavior and microstructural deformation of R-CNT/polymer composites. Compos. B Eng. 2011, 42, 2123–2129. [Google Scholar] [CrossRef]
- Nayak, T.R.; Jian, L.; Phua, L.C.; Ho, H.K.; Ren, Y.; Pastorin, G. Thin films of functionalized multiwalled carbon nanotubes as suitable scaffold materials for stem cells proliferation and bone formation. ACS Nano 2010, 4, 7717–7725. [Google Scholar] [CrossRef] [PubMed]
- Malarkey, E.B.; Fisher, K.A.; Bekyarova, E.; Liu, W.; Haddon, R.C.; Parpura, V. Conductive single-walled carbon nanotube substrates modulate neuronal growth. Nano Lett. 2009, 9, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Murphy, R.; Coleman, J.N.; Cadek, M.; McCarthy, B.; Bent, M.; Drury, A.; Barklie, R.C.; Blau, W.J. High-yield, nondestructive purification and quantification method for multiwalled carbon nanotubes. J. Phys. Chem. B 2002, 106, 3087–3091. [Google Scholar] [CrossRef]
- Lau, C.; Cooney, M.J. Conductive macroporous composite chitosan−carbon nanotube scaffolds. Langmuir 2008, 24, 7004–7010. [Google Scholar] [CrossRef] [PubMed]
- Geim, A.K.; Novoselov, K.S. The rise of graphene. Nat. Mater. 2007, 6, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Rao, C.N.R.; Sood, A.K.; Subrahmanyam, K.S.; Govindaraj, A. Graphene: The new two-dimensional nanomaterial. Angew. Chem. Ed. 2009, 48, 7752–7777. [Google Scholar] [CrossRef]
- Stankovich, S.; Dikin, D.A.; Dommett, G.H.B.; Kohlhaas, K.M.; Zimney, E.J.; Stach, E.A.; Piner, R.D.; Nguyen, S.T.; Ruoff, R.S. Graphene-based composite materials. Nature 2006, 442, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Potts, J.R.; Dreyer, D.R.; Bielawski, C.W.; Ruoff, R.S. Graphene-based polymer nanocomposites. Polymer 2011, 52, 5–25. [Google Scholar] [CrossRef]
- Eda, G.; Chhowalla, M. Graphene-based composite thin films for electronics. Nano Lett. 2009, 9, 814–818. [Google Scholar] [CrossRef] [PubMed]
- Das, B.; Prasad, K.E.; Ramamurty, U.; Rao, C.N.R. Nano-indentation studies on polymer matrix composites reinforced by few-layer graphene. Nanotechnology 2009, 20. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K.E.; Das, B.; Maitra, U.; Ramamurty, U.; Rao, C.N.R. Extraordinary synergy in the mechanical properties of polymer matrix composites reinforced with 2 nanocarbons. Proc. Natl. Acad. Sci. USA 2009, 106, 13186–13189. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Ren, L.; Wang, R.; Zhang, C.; Yang, Y.; Liu, T. Water dispersible graphene noncovalently functionalized with tryptophan and its poly(vinyl alcohol) nanocomposite. Compos. B Eng. 2011, 42, 2130–2135. [Google Scholar] [CrossRef]
- Lee, J.H.; Marroquin, J.; Rhee, K.Y.; Park, S.J.; Hui, D. Cryomilling application of graphene to improve material properties of graphene/chitosan nanocomposites. Compos. B Eng. 2013, 45, 682–687. [Google Scholar] [CrossRef]
- Hummers, W.S., Jr.; Offeman, R.E. Preparation of graphitic oxide. J. Am. Chem. Soc. 1958, 80. [Google Scholar] [CrossRef]
- Kathi, J.; Rhee, K.Y. Surface modification of multi-walled carbon nanotubes using 3-aminopropyltriethoxysilane. J. Mater. Sci. 2008, 43, 33–37. [Google Scholar] [CrossRef]
- Ma, P.C.; Kim, J.K.; Tang, B.Z. Functionalization of carbon nanotubes using a silane. Carbon 2006, 44, 3232–3238. [Google Scholar] [CrossRef]
- Nebogatikova, N.A.; Antonova, I.V.; Volodin, V.A.; Prinz, V.Y. Functionalization of graphene and few-layer graphene with aqueous solution of hydrofluoric acid. Phys. E 2013, 52, 106–111. [Google Scholar] [CrossRef]
- Okpalugo, T.I.T.; Papakonstantinou, P.; Murphy, H.; Mclaughlin, J.; Brown, N.M.D. Oxidative functionalization of carbon nanotubes in atmospheric pressure filamentary dielectric barrier discharge (APDBD). Carbon 2005, 43, 2951–2959. [Google Scholar] [CrossRef]
- Saidi, W.A.; Norman, P. Probing single-walled carbon nanotube defect chemistry using resonance Raman spectroscopy. Carbon 2014, 67, 17–26. [Google Scholar] [CrossRef]
- Zając, A.; Hanuza, J.; Wandas, M.; Dymińska, L. Determination of N-acetylation degree in chitosan using Raman spectroscopy. Spectrochim. Acta Mol. Biomol. Spectros. 2015, 134, 114–120. [Google Scholar] [CrossRef]
- Sundar, S.; Mariappan, R.; Piraman, S. Synthesis and characterization of amine modified magnetite nanoparticles as carriers of curcumin-anticancer drug. Powder Technol. 2014, 266, 321–328. [Google Scholar] [CrossRef]
- Gui, M.M.; Yap, Y.X.; Chai, S.P.; Mohamed, A.R. Multi-walled carbon nanotubes modified with (3-aminopropyl)triethoxysilane for effective carbon dioxide adsorption. Int. J. Greenh. Gas Control 2013, 14, 65–73. [Google Scholar] [CrossRef]
- Hiraoui, M.; Guendouz, M.; Lorrain, N.; Moadhen, A.; Haji, L.; Oueslati, M. Spectroscopy studies of functionalized oxidized porous silicon surface for biosensing applications. Mater. Chem. Phys. 2011, 128, 151–156. [Google Scholar] [CrossRef]
- Gunda, N.S.K.; Singh, M.; Norman, L.; Kaur, K.; Mitra, S.K. Optimization and characterization of biomolecule immobilization on silicon substrates using (3-aminopropyl)triethoxysilane (APTES) and glutaraldehyde linker. Appl. Surf. Sci. 2014, 305, 522–530. [Google Scholar] [CrossRef]
- John, A.; Liu, F.; Majeed, H.; Zhong, F. Characterization of tara gum edible films incorporated with bulk chitosan and chitosan nanoparticles: A comparative study. Food Hydrocoll. 2015, 44, 309–319. [Google Scholar] [CrossRef]
- Boonsongrit, Y.; Mueller, B.W.; Mitrevej, A. Characterization of drug–chitosan interaction by 1H NMR, FTIR and isothermal titration calorimetry. Eur. J. Pharm. Biopharm. 2008, 69, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Rubentheren, V.; Ward, T.A.; Chee, C.Y.; Tang, C.K. Processing and analysis of chitosan nanocomposites reinforced with chitin whiskers and tannic acid as a crosslinker. Carbohydr. Polym. 2015, 115, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.B.V.; Varadaraj, M.C.; Gowda, L.R.; Tharanathan, R.N. Characterization of chito-oligosaccharides prepared by chitosanolysis with the aid of papain and Pronase, and their bactericidal action against Bacillus cereus and Escherichia coli. Biochem. J. 2005, 391, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.B.; Han, Z.; Zeng, X.; Yu, S.J.; Kennedy, J.F. Study on the degradation of chitosan by pulsed electric fields treatment. Innov. Food Sci. Emerg. 2010, 11, 587–591. [Google Scholar] [CrossRef]
- Kadir, M.F.Z.; Aspanut, Z.; Majid, S.R.; Arof, A.K. FTIR studies of plasticized poly(vinyl alcohol)–chitosan blend doped with NH4NO3 polymer electrolyte membrane. Spectrochim. Acta Mol. Biomol. Spectros. 2011, 78, 1068–1074. [Google Scholar] [CrossRef]
- Wan, Y.J.; Gong, L.X.; Tang, L.C.; Wu, L.B.; Jiang, J.X. Mechanical properties of epoxy composites filled with silane-functionalized graphene oxide. Compos. A Appl. Sci. Manuf. 2014, 64, 79–89. [Google Scholar] [CrossRef]
- Frogley, M.D.; Wang, C.; Cinque, G.; Barber, A.H. Polarised infrared microspectroscopy of edge-oriented graphene oxide papers. Vib. Spectrosc. 2014, 75, 178–183. [Google Scholar] [CrossRef] [Green Version]
- Dato, A.; Lee, Z.; Jeon, K.J.; Erni, R.; Radmilovic, V.; Richardsonc, T.J.; Frenklach, M. Clean and highly ordered graphene synthesized in the gas phase. Chem. Commun. 2009, 40, 6095–6097. [Google Scholar] [CrossRef]
- Zhou, Z.; Wang, S.; Lu, L.; Zhang, Y.; Zhang, Y. Functionalization of multi-wall carbon nanotubes with silane and its reinforcement on polypropylene composites. Compos. Sci. Technol. 2008, 68, 1727–1733. [Google Scholar] [CrossRef]
- Hong, Y.; Wang, Z.; Jin, X. Sulfuric acid intercalated graphite oxide for graphene preparation. Sci. Rep. 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Szwarckopf, H.E. XPS photoemission in carbonaceous materials: A “defect” peak beside the graphitic asymmetric peak. Carbon 2004, 42, 1713–1721. [Google Scholar] [CrossRef]
- Lesiak, B.; Stobinski, L.; Malolepszy, A.; Mazurkiewicz, M.; Kövér, L.; Tóth, J. Preparation of graphene oxide and characterisation using electron spectroscopy. J. Electron. Spectrosc. Relat. Phenom. 2014, 193, 92–99. [Google Scholar] [CrossRef]
- Hong, W.G.; Kim, B.H.; Lee, S.M.; Yu, H.Y.; Yun, Y.J.; Jun, Y.; Lee, J.B.; Kim, H.J. Agent-free synthesis of graphene oxide/transition metaloxide composites and its application for hydrogen storage. Int. J. Hydrog. Energy 2012, 37, 7594–7599. [Google Scholar] [CrossRef]
- Vitale, A.; Pollicino, A.; Bernardi, E.; Bongiovanni, R. Ultrathin perfluoropolyether coatings for silicon wafers: A XPS study. Prog. Org. Coat. 2015, 78, 480–487. [Google Scholar] [CrossRef]
- Sun, Y.N.; Feldman, A.; Farabaugh, E.N. X-ray photoelectron spectroscopy of O 1s and Si 2p lines in films of SiOx formed by electron beam evaporation. Thin Solid Films 1988, 157, 351–360. [Google Scholar] [CrossRef]
- Barr, T.L. An XPS study of Si as it occurs in adsorbents, catalysts, and thin films. Appl. Surf. Sci. 1983, 15, 1–35. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, D.S.; Dhand, V.; Rhee, K.Y.; Park, S.-J. Study on the Effect of Silanization and Improvement in the Tensile Behavior of Graphene-Chitosan-Composite. Polymers 2015, 7, 527-551. https://doi.org/10.3390/polym7030527
Kim DS, Dhand V, Rhee KY, Park S-J. Study on the Effect of Silanization and Improvement in the Tensile Behavior of Graphene-Chitosan-Composite. Polymers. 2015; 7(3):527-551. https://doi.org/10.3390/polym7030527
Chicago/Turabian StyleKim, Dae Sung, Vivek Dhand, Kyong Yop Rhee, and Soo-Jin Park. 2015. "Study on the Effect of Silanization and Improvement in the Tensile Behavior of Graphene-Chitosan-Composite" Polymers 7, no. 3: 527-551. https://doi.org/10.3390/polym7030527
APA StyleKim, D. S., Dhand, V., Rhee, K. Y., & Park, S. -J. (2015). Study on the Effect of Silanization and Improvement in the Tensile Behavior of Graphene-Chitosan-Composite. Polymers, 7(3), 527-551. https://doi.org/10.3390/polym7030527