
Synthesis of Highly Branched Polyolefins Using Phenyl Substituted α-Diimine Ni(II) Catalysts



Abstract
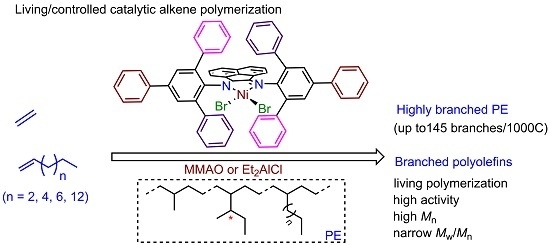
:
1. Introduction
2. Experimental Section
2.1. General Considerations and Materials
2.2. Instrumentation
2.3. Synthesis and Characterizations
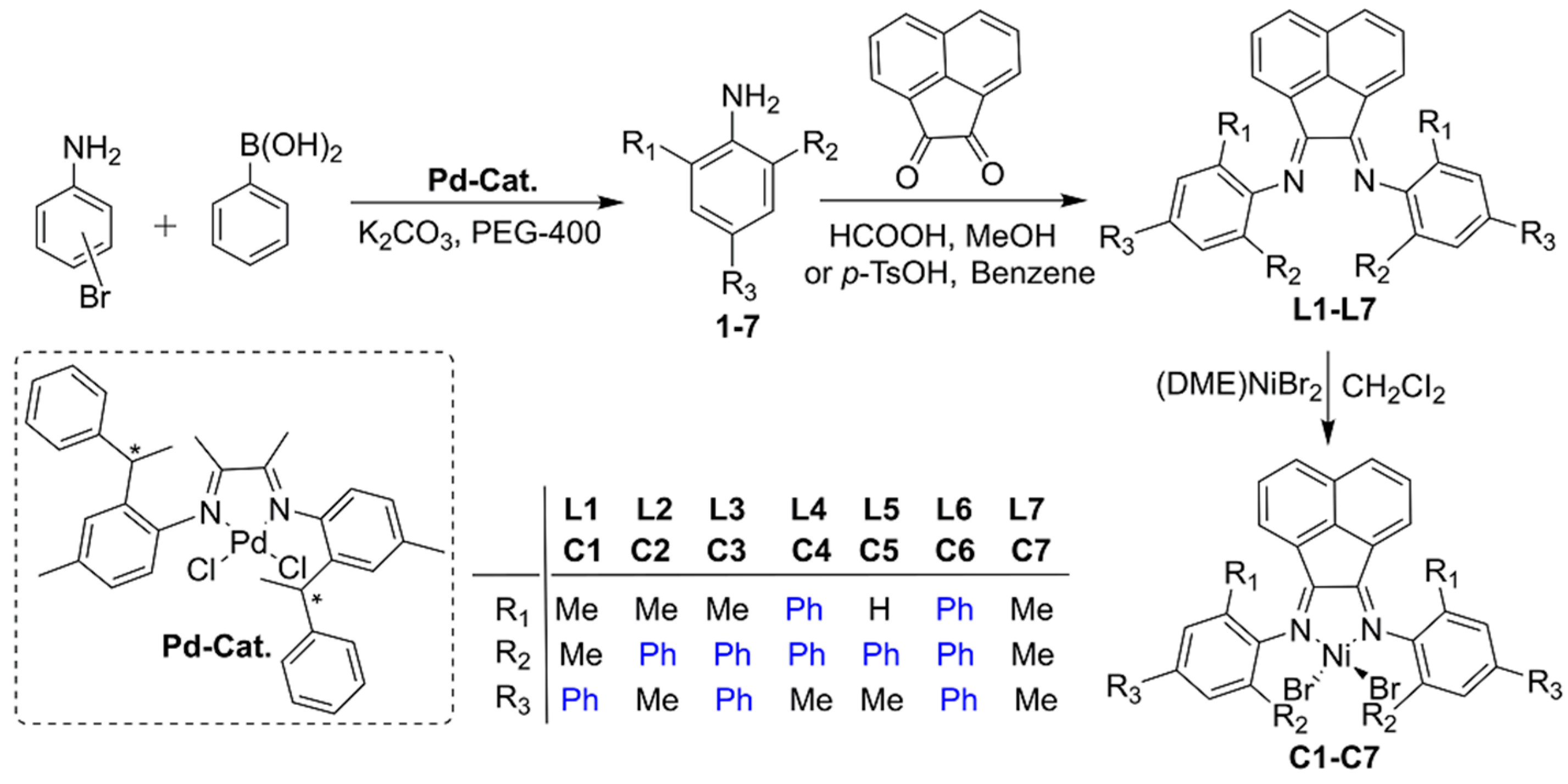
2.3.1. Synthesis of Aniline Derivatives 1–3
Synthesis of 2,6-Dimethyl-4-Phenylaniline 1
Synthesis of 2,4-Dimethyl-6-Phenylaniline 2
Synthesis of 2-Methyl-4,6-Diphenylaniline 3
2.3.2. Synthesis of Ligands L1–L3
Synthesis of bis[N,N′-(2,6-Dimethyl-4-Phenylphenyl)imino]Acenaphthene L1
Synthesis of bis[N,N′-(2,4-Dimethyl-6-Phenylphenyl)imino]Acenaphthene L2
Synthesis of bis[N,N′-(2-Methyl-4,6-Diphenylphenyl)imino]Acenaphthene L3
2.3.3. Synthesis of Complexes C1–C3 and C6
Synthesis of {bis[N,N′-(2,6-Dimethyl-4-Phenylphenyl)imino]Acenaphthene}Dibromonickel C1
Synthesis of {bis[N,N′-(2,4-Dimethyl-6-Phenylphenyl)imino]Acenaphthene}Dibromonickel C2
Synthesis of {bis[N,N′-(2-Methyl-4,6-Diphenylphenyl)imino]Acenaphthene}Dibromonickel C3
Synthesis of {bis[N,N′-(2,4,6-Triphenylphenyl)imino]Acenaphthene}Dibromonickel C6
2.4. X-ray Structure Determinations
2.5. Ethylene Polymerization
2.6. 1-Alkenes Polymerization
3. Results and Discussion
3.1. Synthesis and Characterization of the Organic Compounds and Their Complexes C1–C7
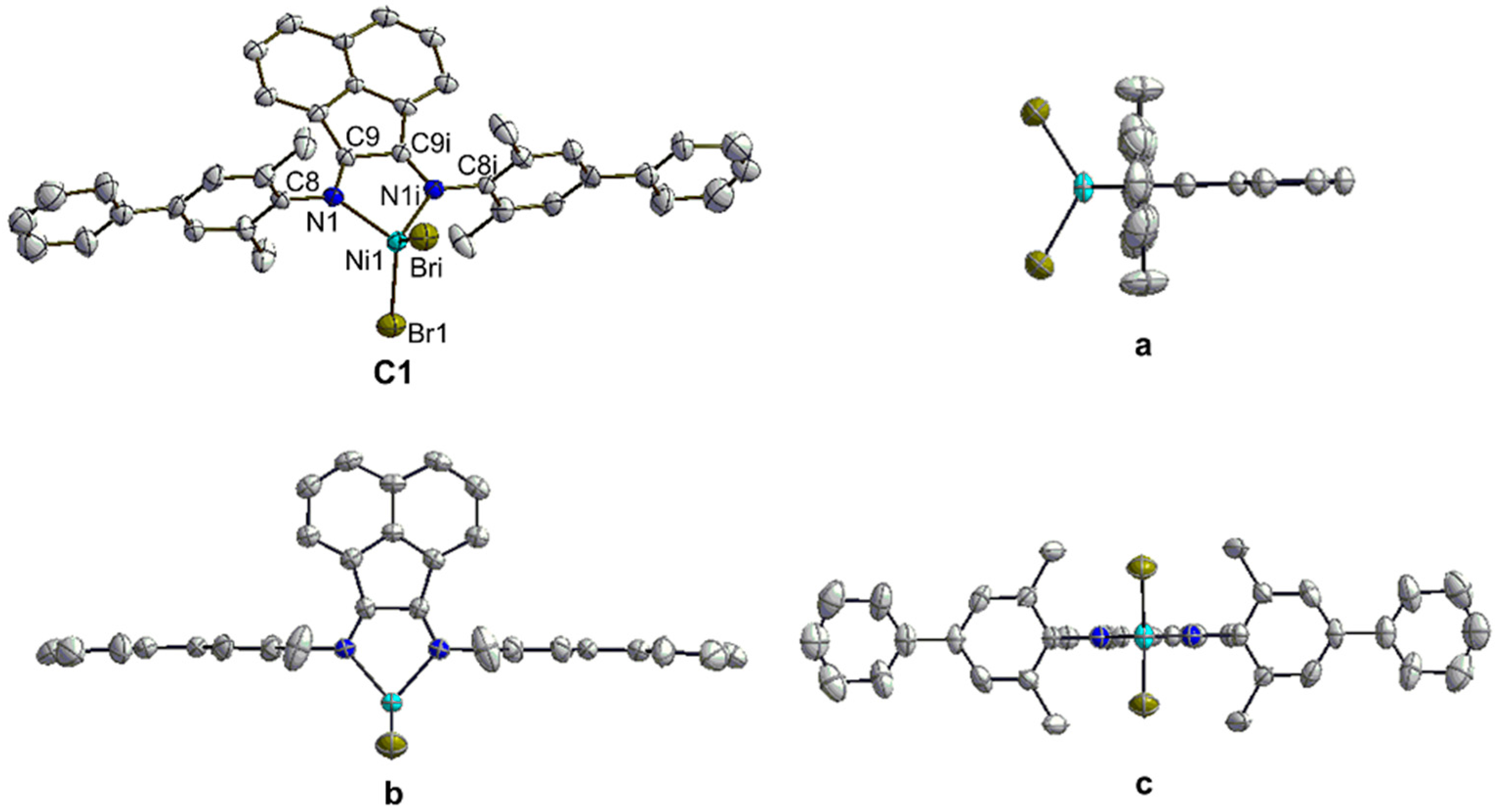
3.2. X-ray Crystallographic Studies
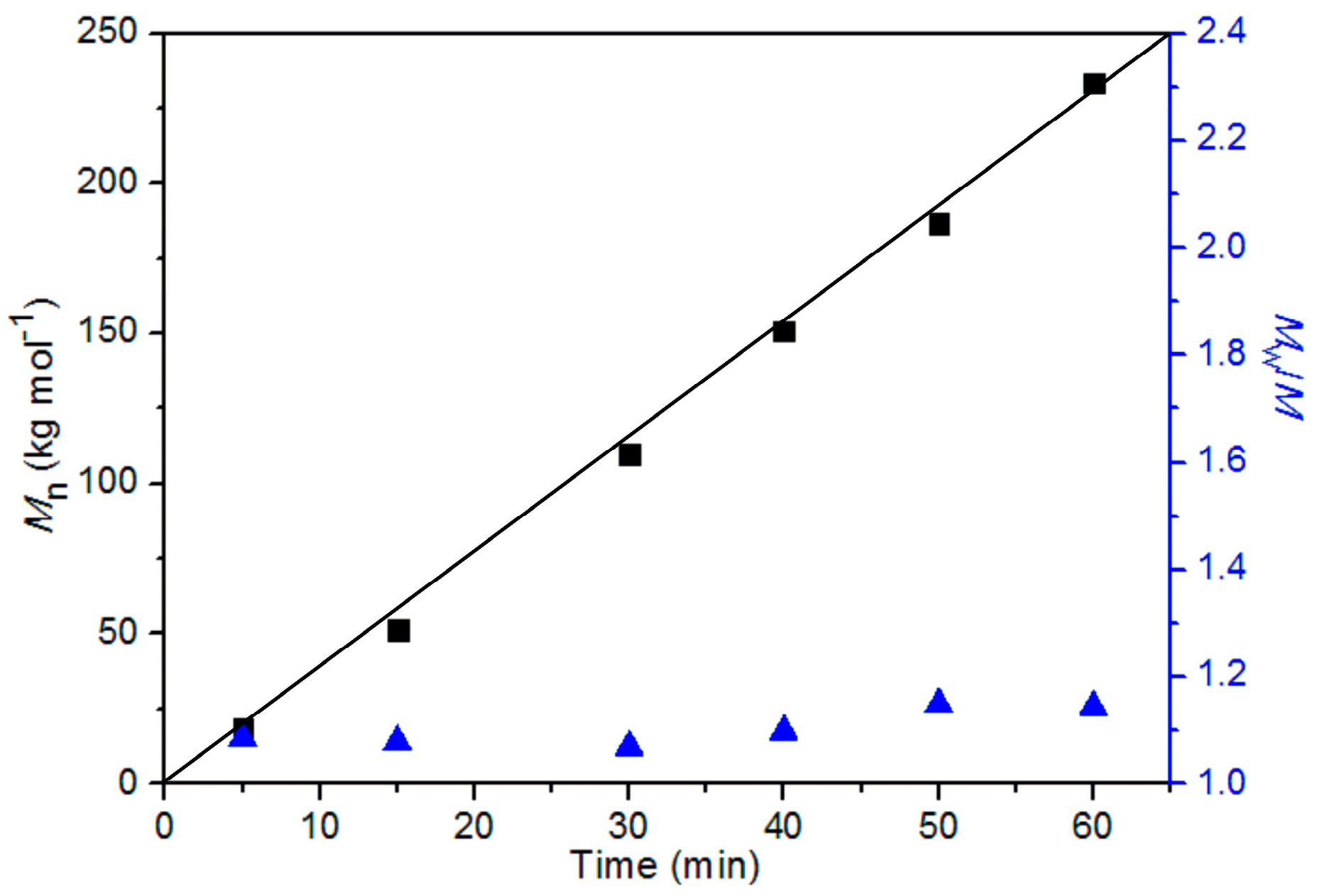
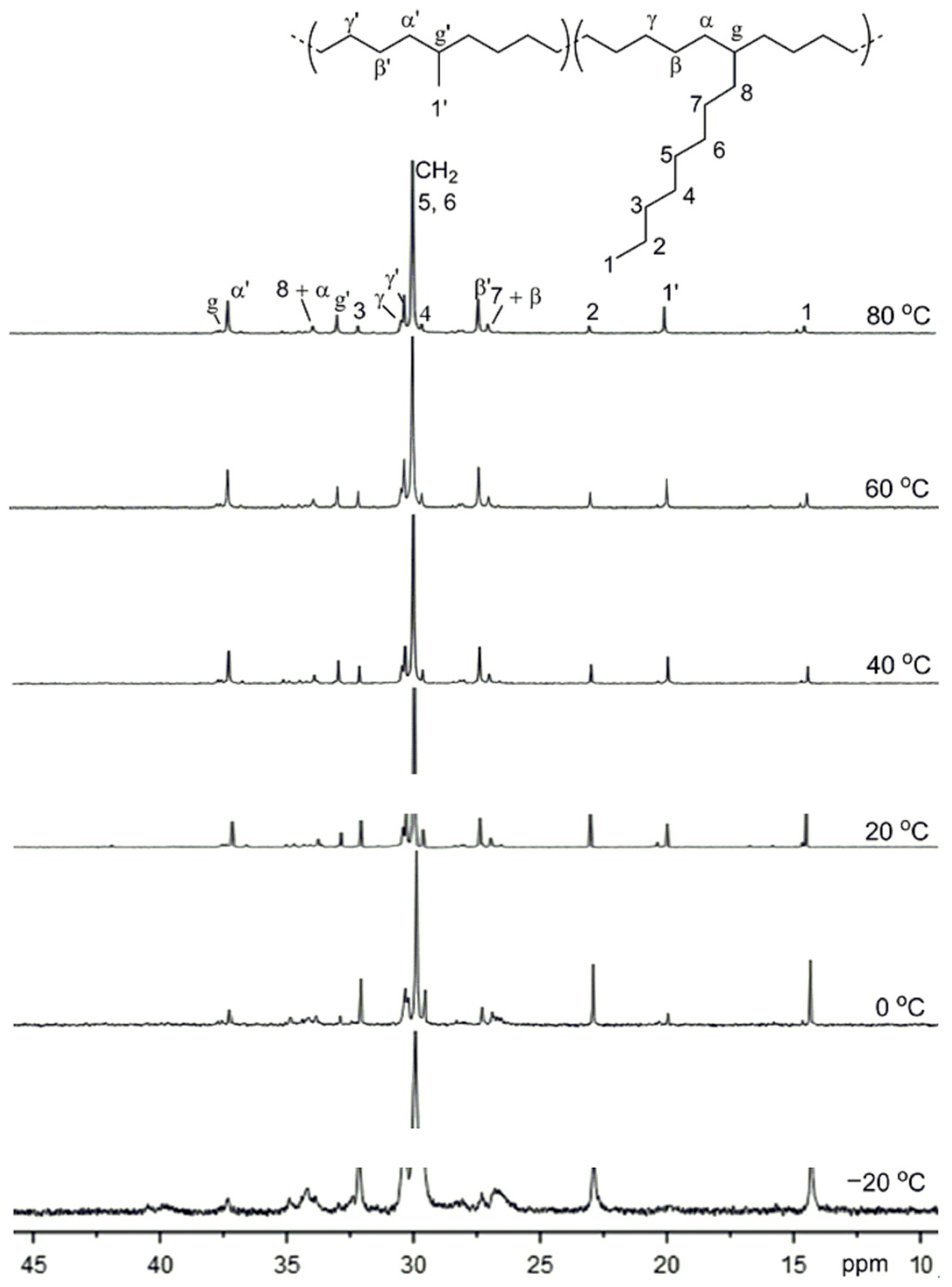
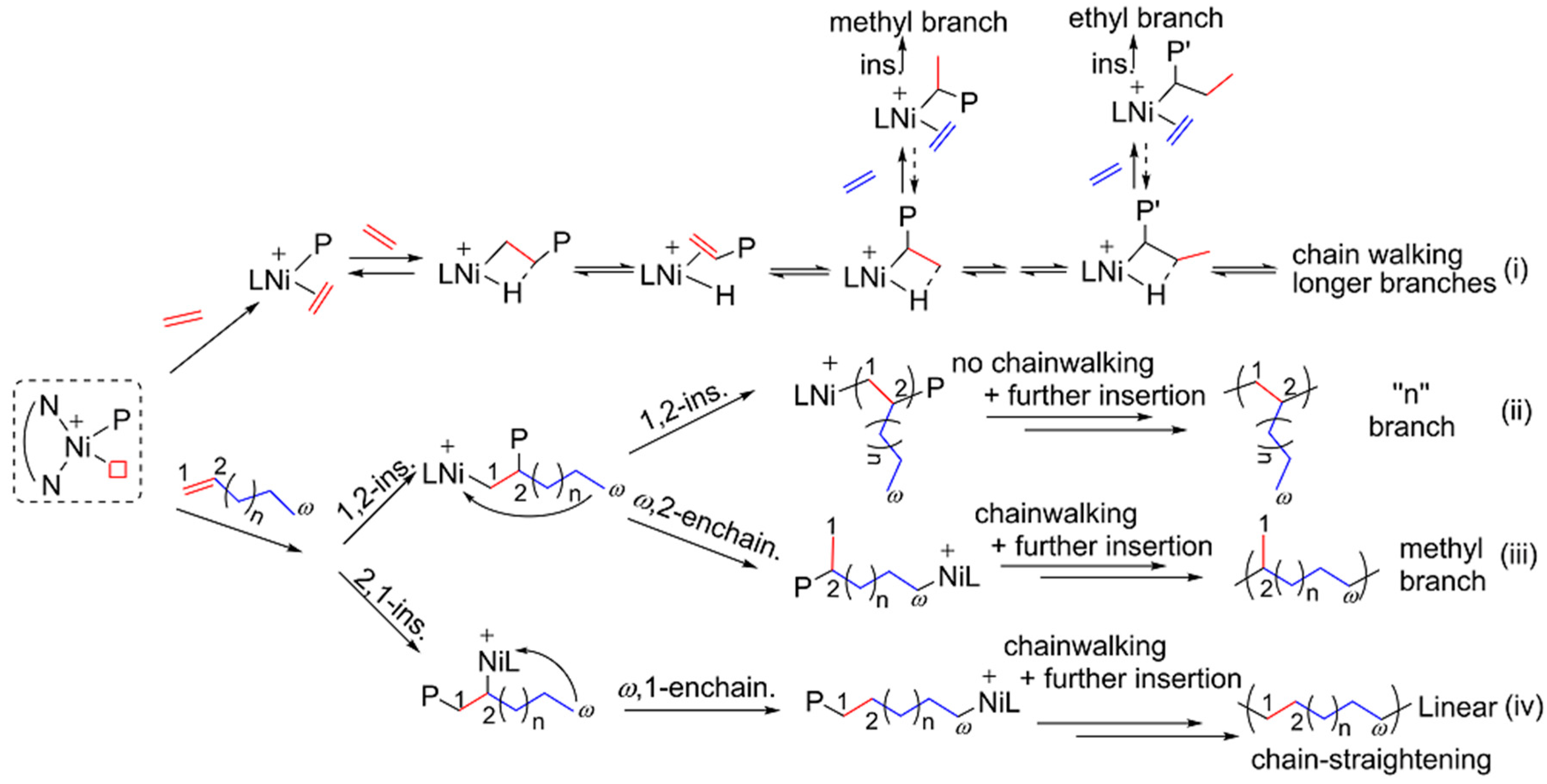
3.3. Ethylene Polymerization
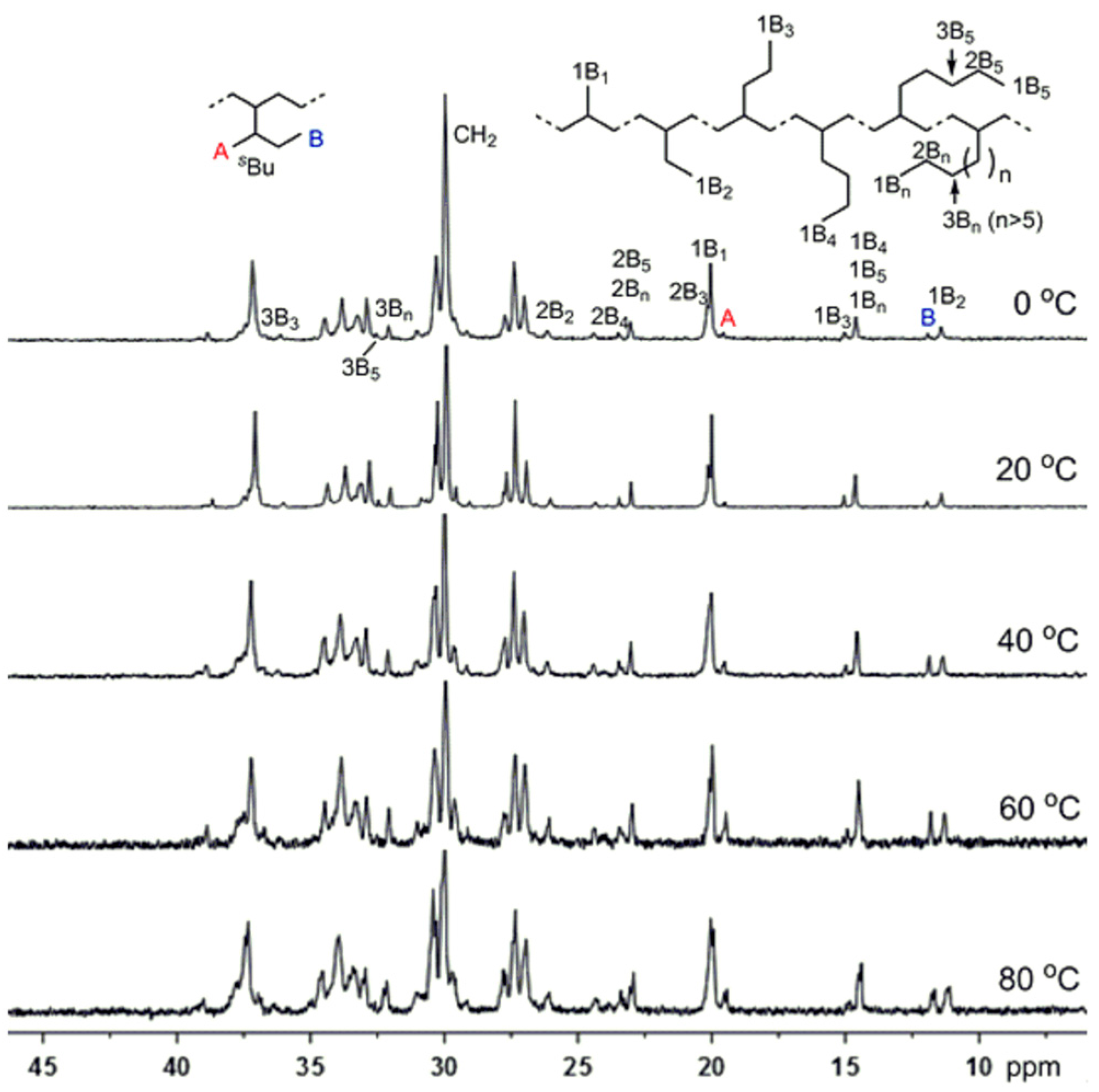
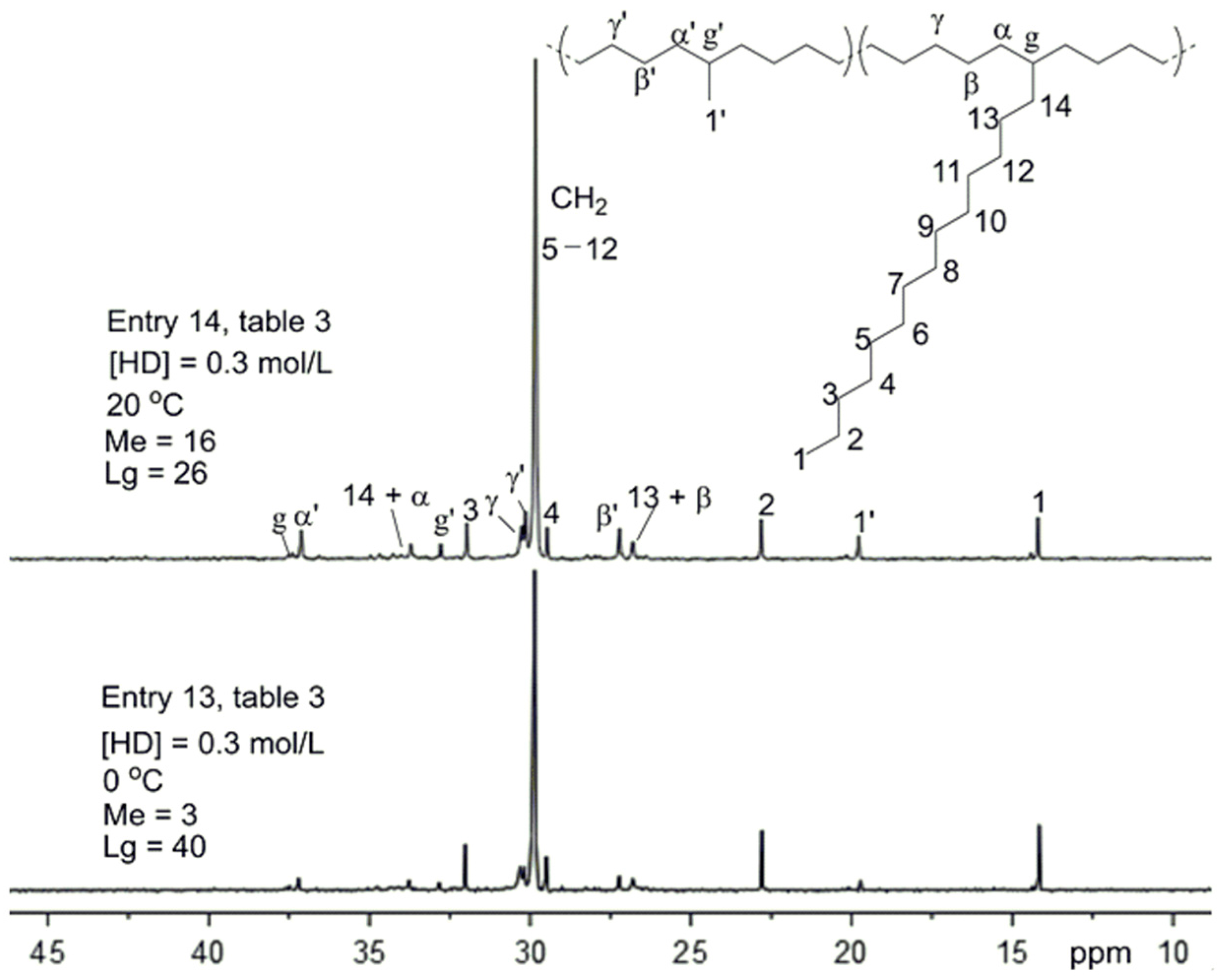
3.4. Higher 1-Alkenes Polymerization
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Malpass, D.B. Introduction to Industrial Polyethylene; Wiley: Hoboken, NJ, USA, 2010. [Google Scholar]
- Ittel, S.D.; Johnson, L.K.; Brookhart, M. Late-metal catalysts for ethylene homo- and copolymerization. Chem. Rev. 2000, 100, 1169–1203. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.K.; Killian, C.M.; Brookhart, M. New Pd(II)- and Ni(II)-based catalysts for polymerization of ethylene and α-olefins. J. Am. Chem. Soc. 1995, 117, 6414–6415. [Google Scholar] [CrossRef]
- Killian, C.M.; Tempel, D.J.; Johnson, L.K.; Brookhart, M. Living polymerization of α-olefins using NiII-α-diimine catalysts. Synthesis of new block polymers based on α-olefins. J. Am. Chem. Soc. 1996, 118, 11664–11665. [Google Scholar] [CrossRef]
- Johnson, L.K.; Mecking, S.; Brookhart, M. Copolymerization of ethylene and propylene with functionalized vinyl monomers by palladium(II) catalysts. J. Am. Chem. Soc. 1996, 118, 267–268. [Google Scholar] [CrossRef]
- Gottfried, A.C.; Brookhart, M. Living and block copolymerization of ethylene and α-olefins using palladium(II)-α-diimine catalysts. Macromolecules 2003, 36, 3085–3100. [Google Scholar] [CrossRef]
- Chen, Y.S.; Wang, Li.; Yu, H.J.; Zhao, Y.L.; Sun, R.L.; Jing, G.H.; Huang, J.; Khalid, H.; Abbasi, N.M.; Akram, M. Synthesis and application of polyethylene-based functionalized hyperbranched polymers. Prog. Polym. Sci. 2015, 45, 23–43. [Google Scholar] [CrossRef]
- Coates, G.W.; Hustad, P.D.; Reinartz, S. Catalysts for the living insertion polymerization of alkenes: Access to new polyolefin architectures using Ziegler–Natta chemistry. Angew. Chem. Int. Ed. 2002, 41, 2236–2257. [Google Scholar] [CrossRef]
- Camacho, D.H.; Guan, Z. Living polymerization of α-olefins at elevated temperatures catalyzed by a highly active and robust cyclophane-based nickel catalyst. Macromolecules 2005, 38, 2544–2546. [Google Scholar] [CrossRef]
- Leone, G.; Mauri, M.; Bertini, F.; Canetti, M.; Piovani, D.; Ricci, G. Ni(II) α-diimine-catalyzed α-olefins polymerization: Thermoplastic elastomers of block copolymers. Macromolecules 2015, 48, 1304–1312. [Google Scholar] [CrossRef]
- Gao, H.Y.; Liu, X.F.; Tang, Y.; Pan, J.; Wu, Q. Living/controlled polymerization of 4-methyl-1-pentene with α-diimine nickel-diethylaluminium chloride: Effect of alkylaluminium cocatalysts. Polym. Chem. 2011, 2, 1398–1403. [Google Scholar] [CrossRef]
- Allen, K.E.; Campos, J.; Daugulis, O.; Brookhart, M. Living polymerization of ethylene and copolymerization of ethylene/methyl acrylate using “sandwich” diimine palladium catalysts. ACS Catal. 2015, 5, 456–464. [Google Scholar] [CrossRef]
- Liu, J.; Chen, D.R.; Wu, H.; Xiao, Z.F.; Gao, H.Y.; Zhu, F.M.; Wu, Q. Polymerization of α-olefins using a camphyl α-diimine nickel catalyst at elevated temperature. Macromolecules 2014, 47, 3325–3331. [Google Scholar] [CrossRef]
- Meinhard, D.; Wegner, M.; Kipiani, G.; Hearley, A.; Reuter, P.; Fischer, S.; Marti, O.; Rieger, B. New nickel(II) diimine complexes and the control of polyethylene microstructure by catalyst design. J. Am. Chem. Soc. 2007, 129, 9182–9191. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, T.; Klimovica, K.; LaPointe, A.M.; Keresztes, I.; Lobkovsky, E.B.; Daugulis, O.; Coates, G.W. Secondary alkene insertion and precision chain-walking: A new route to semicrystalline “polyethylene” from α-olefins by combining two rare catalytic events. J. Am. Chem. Soc. 2014, 136, 7213–7216. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.Y.; Sui, X.L.; Chen, C. Highly robust palladium(II) α-diimine catalysts for slow-chain-walking polymerization of ethylene and copolymerization with methyl acrylate. Angew. Chem. Int. Ed. 2015, 54, 9948–9953. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Nakayama, Y.; Shiono, T. Heterogenization of an anilinonaphthoquinone-chelated nickel complex for ethylene polymerization using silica-supported modified methylaluminoxane. Macromol. Chem. Phys. 2014, 215, 1792–1796. [Google Scholar] [CrossRef]
- Cai, Z.G.; Shen, Z.L.; Zhou, X.Y.; Jordan, R.F. Enhancement of chain growth and chain transfer rates in ethylene polymerization by (phosphine-sulfonate)PdMe catalysts by binding of B(C6F5)3 to the sulfonate group. ACS Catal. 2012, 2, 1187–1195. [Google Scholar] [CrossRef]
- Zhang, D.F.; Nadres, E.T.; Brookhart, M.; Daugulis, O. Synthesis of highly branched polyethylene using “sandwich”(8-p-tolyl naphthyl α-diimine) nickel(II) catalysts. Organometallics 2013, 32, 5136–5143. [Google Scholar] [CrossRef]
- Rhinehart, J.L.; Brown, L.A.; Long, B.K. A robust Ni(II) α-diimine catalyst for high temperature ethylene polymerization. J. Am. Chem. Soc. 2013, 135, 16316–16319. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Zhao, Y.; Gao, H.Y.; Zhang, L.; Zhu, F.; Wu, Q. Synthesis of hyperbranched polyethylene amphiphiles by chain-walking polymerization in tandem with RAFT polymerization and supramolecular self-assembly vesicles. Macromol. Rapid Commun. 2012, 33, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.H.; Song, S.; Li, B.; Redshaw, C.; Hao, X.; Li, Y.S. Ethylene polymerization by 2-iminopyridylnickel halide complexes: Synthesis, characterization and catalytic influence of the benzhydryl group. Dalton Trans. 2012, 41, 11999–12010. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.H.; Dai, S.Y.; Sui, X.L.; Chen, C.L. Palladium and nickel catalyzed chain-walking olefin polymerization and copolymerization. ACS Catal. 2016, 6, 428–441. [Google Scholar] [CrossRef]
- Tao, W.J.; Nakano, R.; Ito, S.; Nozaki, K. Copolymerization of ethylene and polar monomers by using Ni/IzQO catalysts. Angew. Chem. 2016, 128, 2885–2889. [Google Scholar] [CrossRef]
- Wang, F.Z.; Yuan, J.C.; Song, F.Y.; Li, J.; Jia, Z.; Yuan, B.N. New chiral-diimine nickel (II) complexes bearing ortho-sec-phenethyl groups for ethylene polymerization. Appl. Organomet. Chem. 2013, 27, 319–327. [Google Scholar] [CrossRef]
- Wang, F.Z.; Yuan, J.C.; Xu, W.B.; Mei, T.J.; Li, J.; Yuan, B.N.; Song, F.Y.; Jia, Z. Chiral naphthyl-α-diimine nickel (II) catalysts bearing sec-phenethyl groups: Chain-walking polymerization of ethylene at high temperature and stereoselective. Organometallics 2013, 32, 3960–3968. [Google Scholar]
- Yuan, J.C.; Wang, F.Z.; Yuan, B.N.; Jia, Z.; Song, F.Y.; Li, J. Highly active ortho-phenyl substituted α-diimine nickel(II) catalysts for “chain-walking polymerization” of ethylene: Synthesis of the nanosized dendritic polyethylene. J. Mol. Catal. A Chem. 2013, 370, 132–139. [Google Scholar] [CrossRef]
- Wang, F.Z.; Tanaka, R.; Li, Q.S.; Yuan, J.C.; Nakayama, Y.; Shiono, T. Synthesis and application of α-diimine Ni(II) and Pd(II) complexes with bulky steric groups to polymerization of ethylene and methyl methacrylate. J. Mol. Catal. A Chem. 2015, 398, 231–240. [Google Scholar] [CrossRef]
- Wang, F.Z.; Yuan, J.C.; Li, Q.S.; Tanaka, R.; Nakayama, Y.; Shiono, T. New nickel(II) diimine complexes bearing phenyl and sec-phenethyl groups: Synthesis, characterization and ethylene polymerization behaviour. Appl. Organometal. Chem. 2014, 28, 477–483. [Google Scholar] [CrossRef]
- Wang, F.Z.; Cai, Z.G.; Tanaka, R.; Nakayama, Y.; Shiono, T. Room-temperature Suzuki–miyaura cross-coupling reaction with α-diimine Pd(II) catalysts. Appl. Organometal. Chem. 2015, 29, 771–776. [Google Scholar] [CrossRef]
- Moody, L.S.; Mackenzie, P.B.; Killian, C.M.; Christopher, M.; Lavoie, G.G.; Ponasik, J.A., Jr.; Smith, T.W.; Pearson, J.C.; Barrett, A.G.M.; Coates, G.W. Catalysts Containing N-Pyrrolyl Substituted Nitrogen Donors. U.S. Patent US6545108 B1, 8 April 2003. [Google Scholar]
- Liu, J.Y.; Li, Y.G.; Li, Y.S.; Hu, N.H. Ethylene polymerization by (α-diimine)nickel (II) complexes bearing different substituents on para-position of imines activated with MMAO. J. Appl. Polymer. Sci. 2008, 109, 700–707. [Google Scholar] [CrossRef]
- Okada, M.; Nakayama, Y.; Shiono, T. Synthesis of anilinonaphthoquinone-based nickel complexes and their application for olefin polymerization. J. Organomet. Chem. 2007, 692, 5183–5189. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | cat. | co-cat. | [Al]/[Ni] | T (°C) | Yield (g) | act. b | Mn c (×104) | Mw/Mn c | B d |
|---|---|---|---|---|---|---|---|---|---|
| 1 | C1 | Et2AlCl | 200 | 20 | 1.10 | 1.32 | 13.4 | 1.68 | – |
| 2 | C1 | Et2AlCl | 400 | 20 | 1.57 | 1.88 | 14.0 | 1.68 | – |
| 3 | C1 | Et2AlCl | 600 | 20 | 1.73 | 2.08 | 16.3 | 1.73 | 105 |
| 4 | C1 | Et2AlCl | 800 | 20 | 1.66 | 1.99 | 10.2 | 1.78 | – |
| 5 | C1 | Et2AlCl | 1,000 | 20 | 1.45 | 1.74 | 12.9 | 1.82 | – |
| 6 | C1 | Et2AlCl | 600 | 0 | 1.79 | 2.15 | 13.9 | 1.85 | nd e |
| 7 | C1 | Et2AlCl | 600 | 40 | 0.91 | 1.20 | 7.2 | 1.87 | 119 |
| 8 | C1 | Et2AlCl | 600 | 60 | 0.56 | 0.67 | 3.3 | 1.87 | 134 |
| 9 | C1 | Et2AlCl | 600 | 80 | 0.12 | 0.14 | 2.1 | 1.79 | 140 |
| 10 | C1 | MMAO | 600 | 0 | 2.62 | 3.13 | 17.6 | 1.71 | 113 |
| 11 | C1 | MMAO | 600 | 20 | 1.81 | 2.17 | 15.9 | 1.63 | 121 |
| 12 | C1 | MMAO | 600 | 40 | 1.19 | 1.43 | 7.3 | 1.42 | 133 |
| 13 | C1 | MMAO | 600 | 60 | 0.89 | 1.07 | 6.0 | 1.32 | 139 |
| 14 | C1 | MMAO | 600 | 80 | 0.86 | 1.03 | 5.0 | 1.86 | 145 |
| 15 | C2 | MMAO | 600 | 20 | 1.85 | 2.22 | 16.1 | 1.76 | 103 |
| 16 | C3 | MMAO | 600 | 20 | 1.94 | 2.33 | 16.9 | 1.84 | 124 |
| 17 | C4 | MMAO | 600 | 20 | 2.13 | 2.56 | 17.2 | 1.95 | 106 |
| 18 | C5 | MMAO | 600 | 20 | 1.64 | 2.00 | 14.9 | 1.84 | 94 |
| 19 | C6 | MMAO | 600 | 20 | 2.60 | 3.12 | 17.9 | 1.92 | 125 |
| 20 | C7 | MMAO | 600 | 20 | 0.81 | 0.97 | 11.6 | 1.72 | 81 |
| Entry/cocat. | T (°C) | Tg (°C) | Total CH3 b | CH3/1000 C b | |||||
|---|---|---|---|---|---|---|---|---|---|
| Me | Et | Pr | Bu | sec-Bu | Lg (n ≥ 5) | ||||
| 3/Et2AlCl | 20 | –57.0 | 105 | 74 | 7 | 5 | 5 | 0 | 14 |
| 7/Et2AlCl | 40 | –58.8 | 120 | 84 | 8 | 3 | 6 | 1 | 18 |
| 10/MMAO | 0 | –56.7 | 114 | 88 | 7 | 1 | 3 | 1 | 14 |
| 11/MMAO | 20 | –58.3 | 122 | 88 | 9 | 3 | 3 | 2 | 17 |
| 12/MMAO | 40 | –60.6 | 131 | 86 | 11 | 2 | 6 | 7 | 19 |
| 13/MMAO | 60 | –65.5 | 143 | 82 | 15 | 3 | 8 | 12 | 23 |
| 14/MMAO | 80 | –68.2 | 146 | 88 | 16 | 2 | 8 | 10 | 22 |
| Entry | Monomer | T (°C) | Yield (g) | act. b | Mn c (×104) | Mw/Mn c | B d | ω,1 e (%) | CH3/1000C f | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Type | [M] | Me | Pr | Bu | Lg | CH3 | ||||||||
| 1 | H | 1.5 | 20 | 0.63 | 126 | 31.2 | 1.32 | 142 | 34 | 13 | 5 | 123 | 2 | 143 |
| 2 | H | 0.3 | 20 | 0.41 | 82 | 12.9 | 1.09 | 121 | 42 | 62 | 1 | 54 | 3 | 120 |
| 3 | O | 1.5 | 20 | 0.89 | 178 | 19.0 | 1.14 | 107 | 29 | 23 | 1 | 84 | 108 | |
| 4 | O | 0.3 | 20 | 0.61 | 122 | 12.0 | 1.06 | 97 | 35 | 53 | 1 | 42 | 96 | |
| 5 | D | 1.5 | 20 | 1.13 | 226 | 15.2 | 1.10 | 93 | 22 | 12 | 79 | 91 | ||
| 6 | D | 0.3 | −20 | 0.12 | 24 | 4.2 | 1.18 | 91 | 23 | 2 | 0 | 89 | 86 | |
| 7 | D | 0.3 | 0 | 0.23 | 46 | 5.2 | 1.08 | 87 | 26 | 18 | 1 | 67 | 85 | |
| 8 | D | 0.3 | 20 | 0.68 | 136 | 10.8 | 1.09 | 85 | 27 | 40 | 2 | 39 | 81 | |
| 9 | D | 0.3 | 40 | 0.56 | 112 | 12.3 | 1.22 | 81 | 30 | 44 | 2 | 33 | 79 | |
| 10 | D | 0.3 | 60 | 0.41 | 82 | 6.7 | 1.33 | 79 | 32 | 46 | 4 | 26 | 76 | |
| 11 | D | 0.3 | 80 | 0.23 | 43 | 3.7 | 1.41 | 76 | 34 | 51 | 4 | 18 | 73 | |
| 12 | HD | 1.5 | 20 | 1.19 | 238 | 30.3 | 1.08 | 50 | 27 | 8 | 38 | 46 | ||
| 13 | HD | 0.3 | 0 | 0.37 | 74 | 6.4 | 1.06 | 48 | 30 | 3 | 40 | 43 | ||
| 14 | HD | 0.3 | 20 | 0.55 | 110 | 11.0 | 1.04 | 46 | 33 | 16 | 26 | 42 | ||
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, F.; Tanaka, R.; Cai, Z.; Nakayama, Y.; Shiono, T. Synthesis of Highly Branched Polyolefins Using Phenyl Substituted α-Diimine Ni(II) Catalysts. Polymers 2016, 8, 160. https://doi.org/10.3390/polym8040160
Wang F, Tanaka R, Cai Z, Nakayama Y, Shiono T. Synthesis of Highly Branched Polyolefins Using Phenyl Substituted α-Diimine Ni(II) Catalysts. Polymers. 2016; 8(4):160. https://doi.org/10.3390/polym8040160
Chicago/Turabian StyleWang, Fuzhou, Ryo Tanaka, Zhengguo Cai, Yuushou Nakayama, and Takeshi Shiono. 2016. "Synthesis of Highly Branched Polyolefins Using Phenyl Substituted α-Diimine Ni(II) Catalysts" Polymers 8, no. 4: 160. https://doi.org/10.3390/polym8040160
APA StyleWang, F., Tanaka, R., Cai, Z., Nakayama, Y., & Shiono, T. (2016). Synthesis of Highly Branched Polyolefins Using Phenyl Substituted α-Diimine Ni(II) Catalysts. Polymers, 8(4), 160. https://doi.org/10.3390/polym8040160