
Papain-Catalyzed Synthesis of Polyglutamate Containing a Nylon Monomer Unit

Abstract
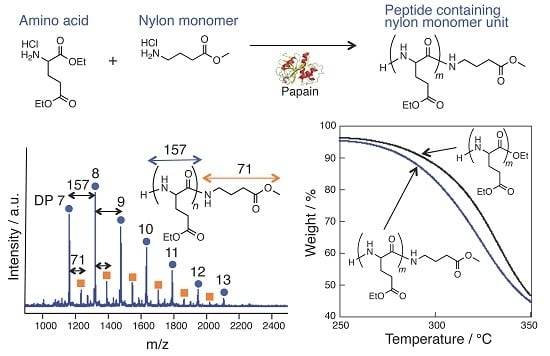
:
1. Introduction
2. Materials and Methods
2.1. Materials
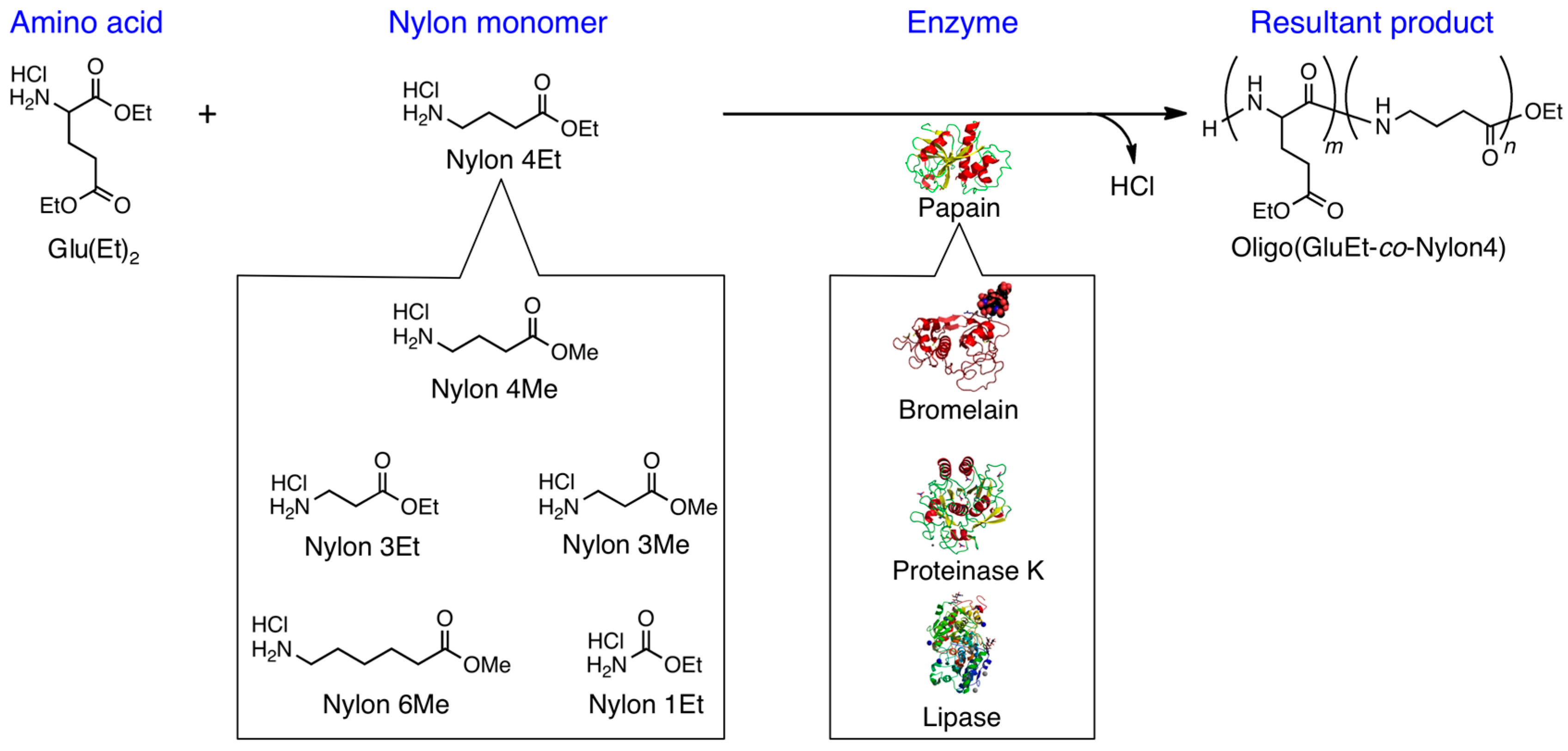
2.2. Chemoenzymatic Peptide Synthesis
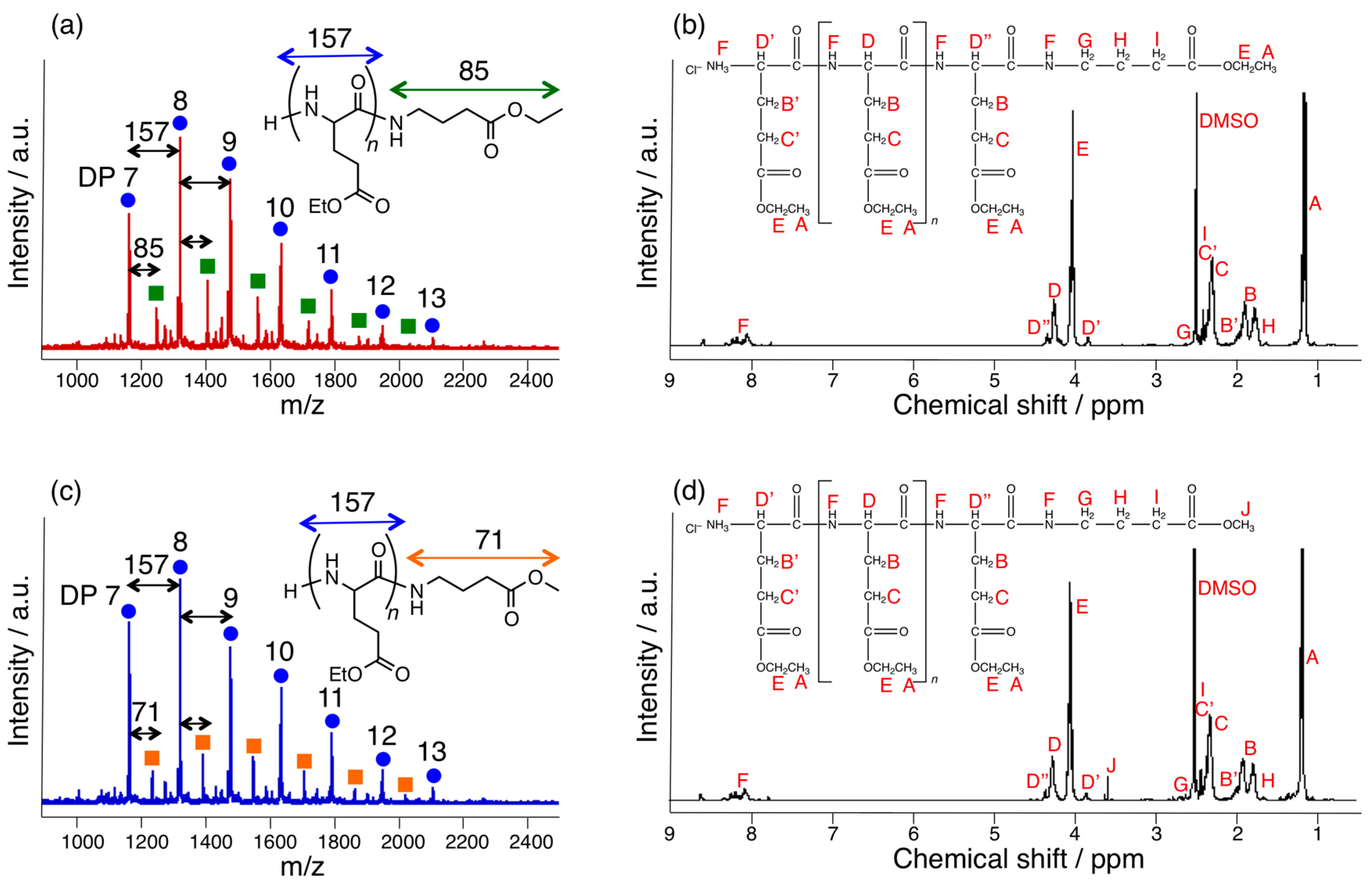
2.3. MALDI-TOF MS
2.4. 1H-NMR
2.5. Thermal Analysis
3. Results and Discussion
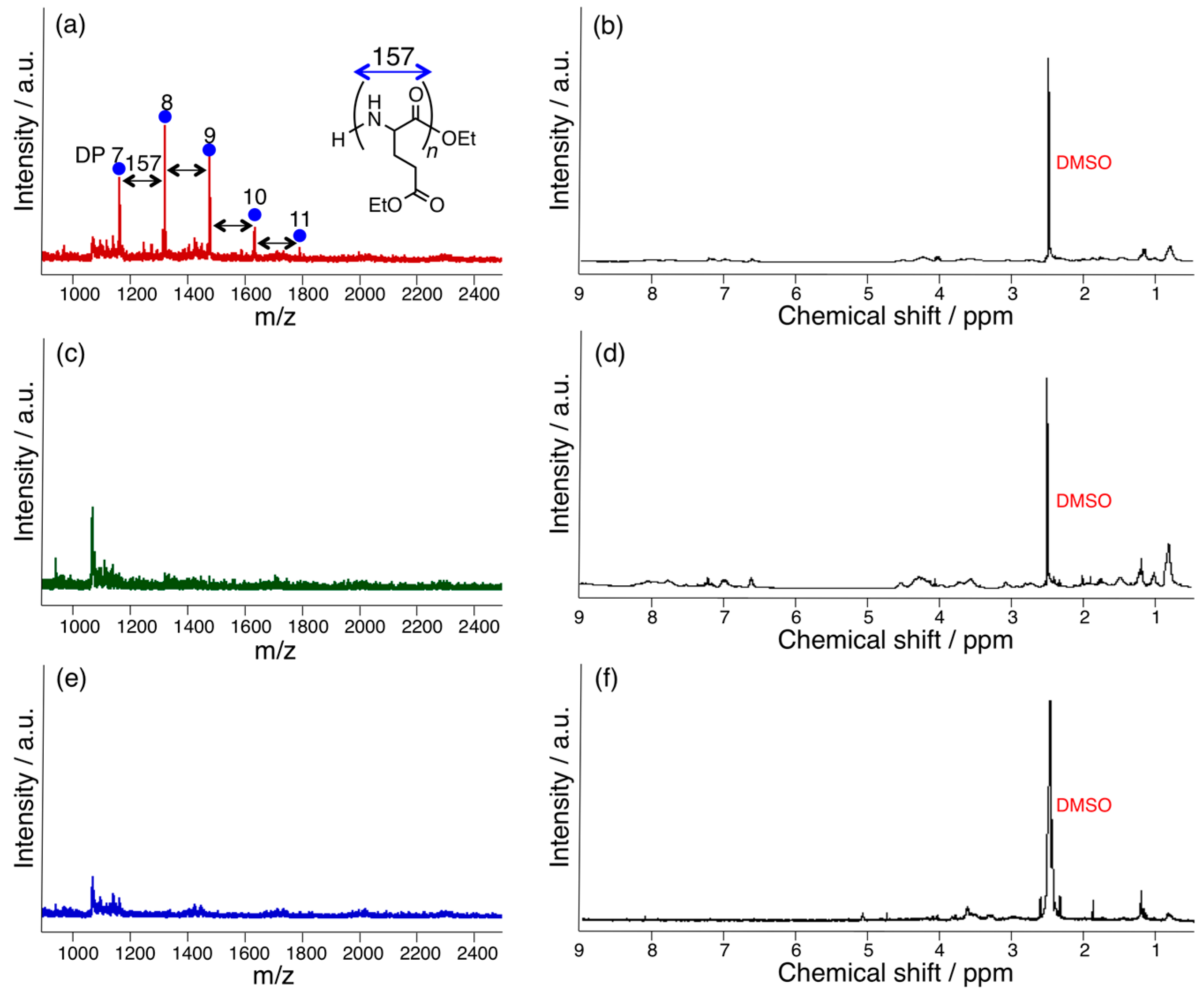
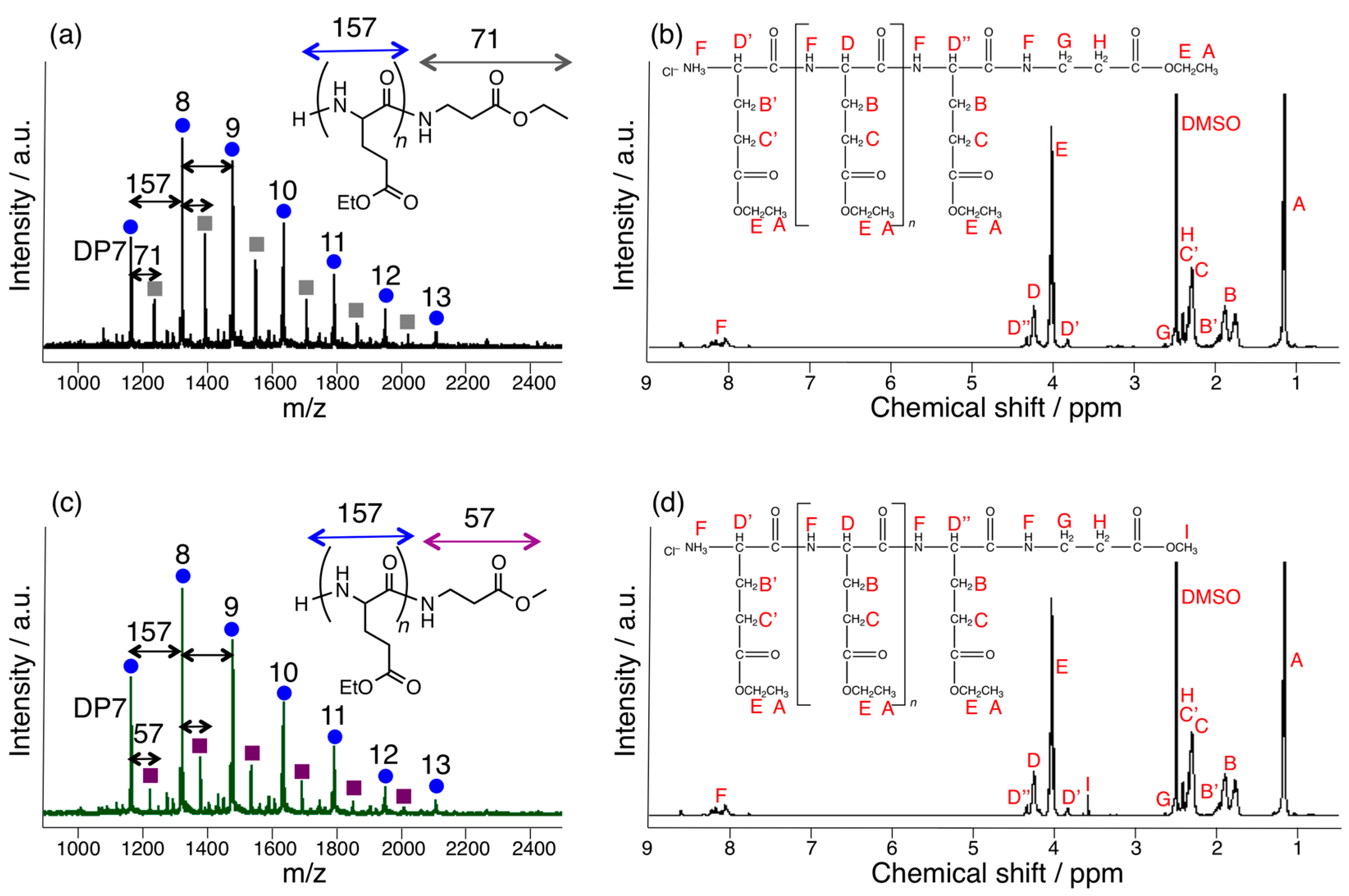
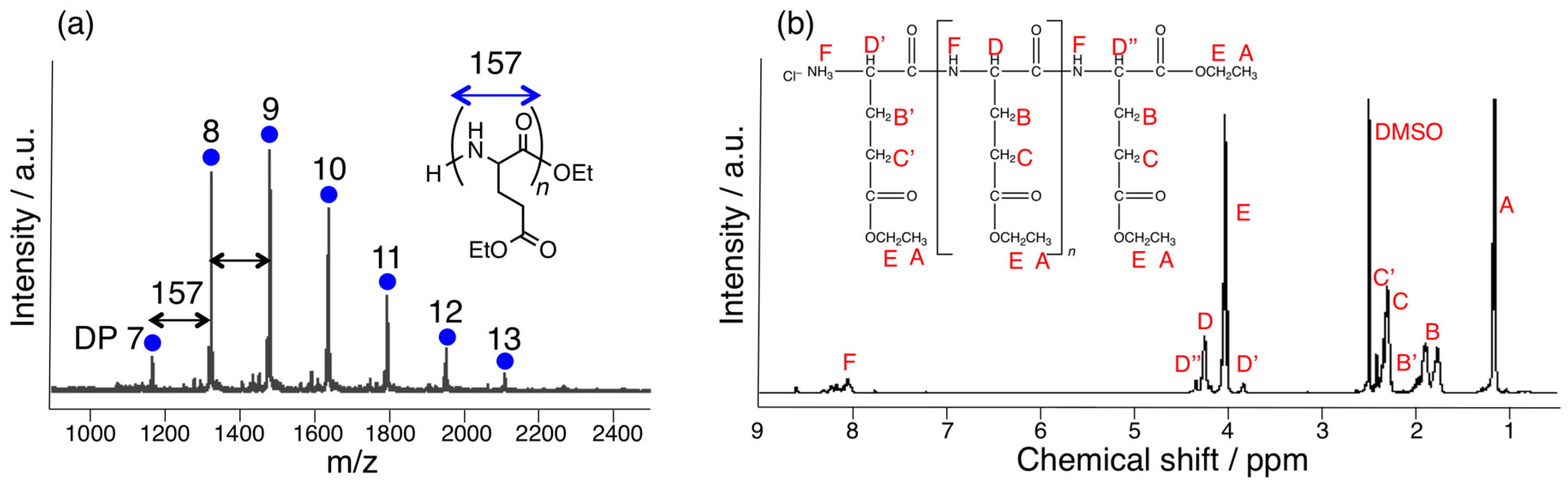
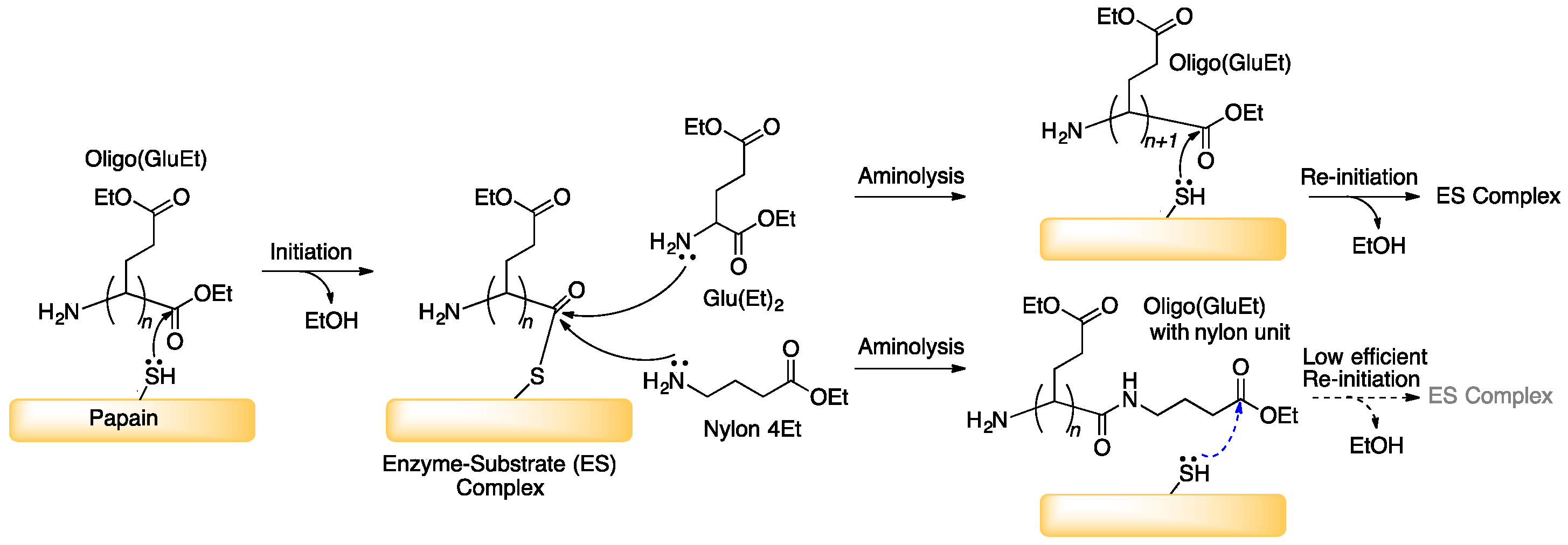
3.1. Papain-Catalyzed Copolymerization of Peptides with a Nylon 4 Unit
3.2. Bromelain, Proteinase K, and Lipase-Catalyzed Chemoenzymatic Synthesis Using Glu(Et)2 and Nylon 4Et
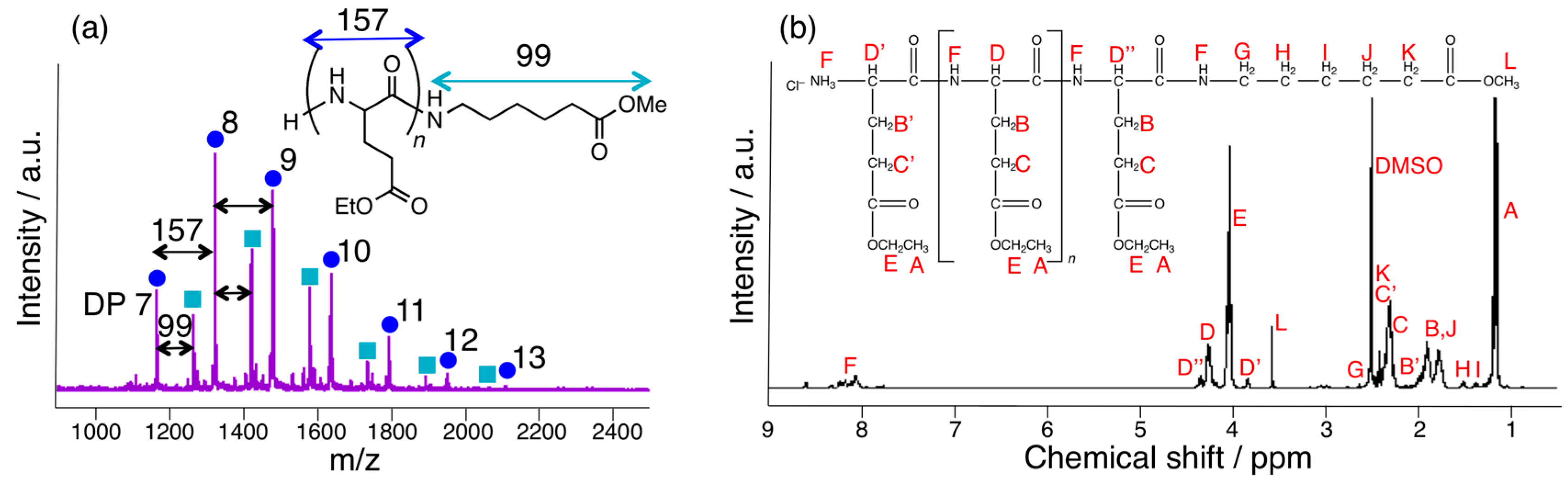
3.3. Papain-Catalyzed Synthesis of Peptides with Nylon 1, 3, or 6 Monomers
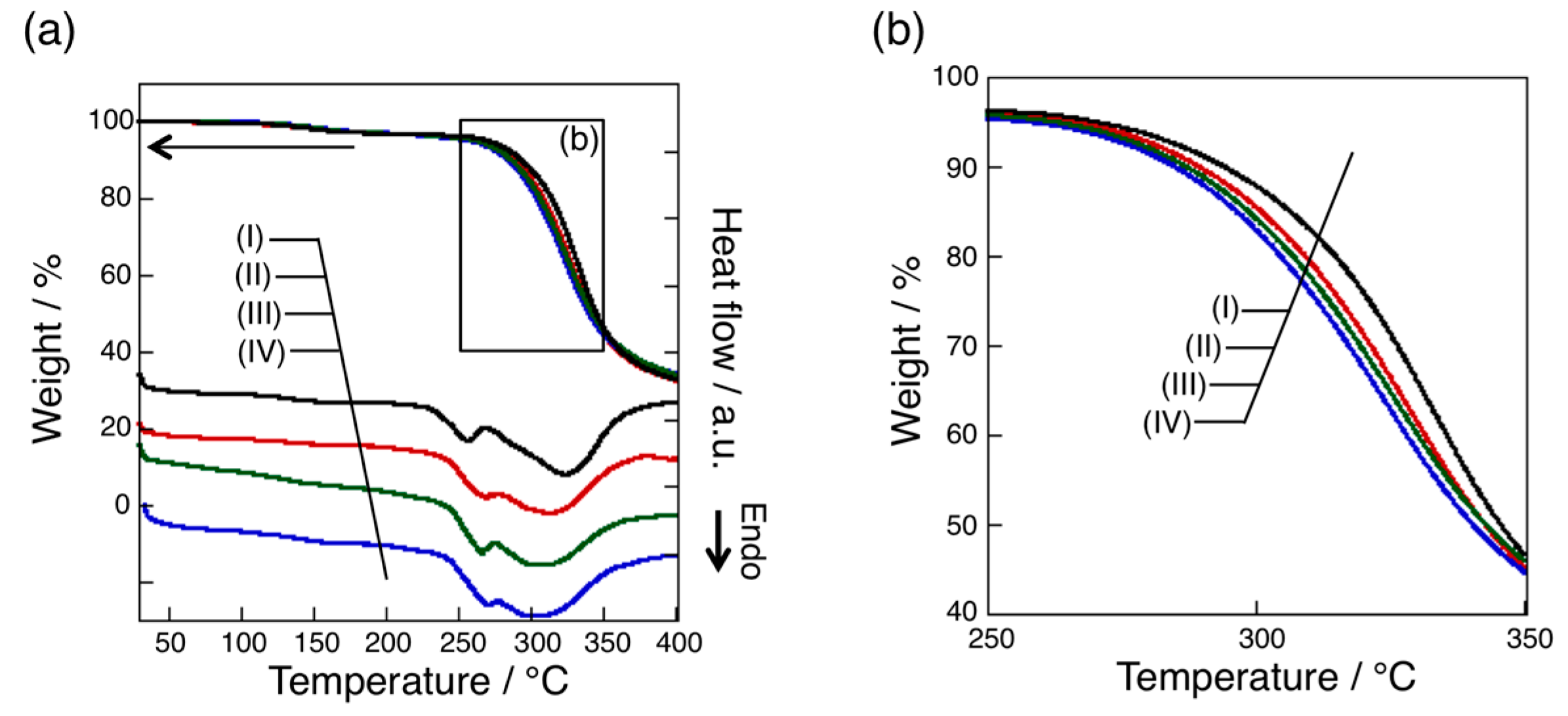
3.4. Thermal Analysis of oligo(GluEt) with or without Nylon Units
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| NMR | nuclear magnetic resonance |
| MALDI-TOF MS | matrix assisted laser desorption ionization time of flight mass spectrometry |
| nylon 4Me | methyl 4-aminobutyrate hydrochloride |
| nylon 4Et | ethyl 4-aminobutyrate hydrochloride |
| nylon 3Me | β-alanine methyl ester hydrochloride |
| nylon 3Et | β-alanine ethyl ester hydrochloride |
| nylon 6Me | methyl 6-aminohexanoate hydrochloride |
| nylon 1Et | ethyl carbamate |
| glu(Et)2 | l-glutamic acid diethyl ester hydrochloride |
| oligo(GluEt) | oligo(glutamic acid-γ-ethyl ester) |
| DP | degree of polymerization |
| HABA | 2-(4-hydroxyphenylazo)benzoic acid |
| Na-TFA | sodium trifluoroacetate |
| DMSO-d6 | dimethyl sulfoxide-d6 |
References
- Numata, K. Poly(amino acid)s/polypeptides as potential functional and structural materials. Polym. J. 2015, 47, 537–545. [Google Scholar] [CrossRef]
- Cebe, P.; Hu, X.; Kaplan, D.L.; Zhuravlev, E.; Wurm, A.; Arbeiter, D.; Schick, C. Beating the heat-fast scanning melts silk β sheet crystals. Sci. Rep. 2013, 3, 1130. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, K.; Urano, Y.; Numata, K. Biodegradability of nylon 4 film in a marine environment. Polym. Degrad. Stab. 2013, 98, 1847–1851. [Google Scholar] [CrossRef]
- Merrifield, R.B. Solid-phase peptide synthesis. 3. An improved synthesis of bradykinin. Biochemistry 1964, 3, 1385–1390. [Google Scholar] [CrossRef] [PubMed]
- Arlinghaus, R.; Shaefer, J.; Schweet, R. Mechanism of peptide bond formation in polypeptide synthesis. Proc. Natl. Acad. Sci. USA 1964, 51, 1291–1299. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-D.; Meienhofer, J. Solid-phase peptide synthesis using mild base cleavage of Nαfluorenylmethyloxycarbonylamino acids, exemplified by a synthesis of dihydrosomatostatin. Int. J. Pept. Protein Res. 1978, 11, 246–249. [Google Scholar] [CrossRef] [PubMed]
- Arcidiacono, S.; Mello, C.; Kaplan, D.; Cheley, S.; Bayley, H. Purification and characterization of recombinant spider silk expressed in Escherichia coli. Appl. Microbiol. Biotechnol. 1998, 49, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Prince, J.T.; McGrath, K.P.; DiGirolamo, C.M.; Kaplan, D.L. Construction, cloning, and expression of synthetic genes encoding spider dragline silk. Biochemistry 1995, 34, 10879–10885. [Google Scholar] [CrossRef] [PubMed]
- Winkler, S.; Szela, S.; Avtges, P.; Valluzzi, R.; Kirschner, D.A.; Kaplan, D. Designing recombinant spider silk proteins to control assembly. Int. J. Biol. Macromol. 1999, 24, 265–270. [Google Scholar] [CrossRef]
- Rising, A.; Widhe, M.; Johansson, J.; Hedhammar, M. Spider silk proteins: Recent advances in recombinant production, structure-function relationships and biomedical applications. Cell. Mol. Life Sci. 2011, 68, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Ageitos, J.M.; Chuah, J.-A.; Numata, K. Chemo-enzymatic synthesis of linear and branched cationic peptides: Evaluation as gene carriers. Macromol. Biosci. 2015, 15, 990–1003. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, K.; Numata, K. Papain-catalyzed chemoenzymatic synthesis of telechelic polypeptides using bis(leucine ethyl ester) initiator. Macromol. Biosci. 2016. [CrossRef] [PubMed]
- Van Rantwijk, F.; Hacking, J.M.A.P.; Sheldon, A.R. Lipase-catalyzed synthesis of carboxylic amides: Nitrogen nucleophiles as acyl acceptor. Monatsh. Chem. 2000, 131, 549–569. [Google Scholar] [CrossRef]
- Yazawa, K.; Numata, K. Recent advances in chemoenzymatic peptide syntheses. Molecules 2014, 19, 13755–13774. [Google Scholar] [CrossRef] [PubMed]
- Baker, P.J.; Numata, K. Chemoenzymatic synthesis of poly(l-alanine) in aqueous environment. Biomacromolecules 2012, 13, 947–951. [Google Scholar] [CrossRef] [PubMed]
- De Beer, R.J.; Zarzycka, B.; Amatdjais-Groenen, H.I.; Jans, S.C.; Nuijens, T.; Quaedflieg, P.J.; van Delft, F.L.; Nabuurs, S.B.; Rutjes, F.P. Papain-catalyzed peptide bond formation: Enzyme-specific activation with guanidinophenyl esters. ChemBioChem 2011, 12, 2201–2207. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Vaidya, A.; Viswanathan, K.; Cui, J.; Xie, W.; Gao, W.; Gross, R.A. Rapid regioselective oligomerization of l-glutamic acid diethyl ester catalyzed by papain. Macromolecules 2006, 39, 7915–7921. [Google Scholar] [CrossRef]
- Schwab, L.W.; Kloosterman, W.M.J.; Konieczny, J.; Loos, K. Papain catalyzed (co)oligomerization of α-amino acids. Polymers 2012, 4, 710–740. [Google Scholar] [CrossRef]
- Fagerland, J.; Finne-Wistrand, A.; Numata, K. Short one-pot chemo-enzymatic synthesis of l-lysine and l-alanine diblock co-oligopeptides. Biomacromolecules 2014, 15, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Numata, K.; Baker, P.J. Synthesis of adhesive peptides similar to those found in blue mussel (mytilus edulis) using papain and tyrosinase. Biomacromolecules 2014, 15, 3206–3212. [Google Scholar] [CrossRef] [PubMed]
- Ageitos, J.M.; Yazawa, K.; Tateishi, A.; Tsuchiya, K.; Numata, K. The benzyl ester group of amino acid monomers enhances substrate affinity and broadens the substrate specificity of the enzyme catalyst in chemoenzymatic copolymerization. Biomacromolecules 2016, 17, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Xie, W.; Su, Q.; Du, W.; Gross, R.A. Protease-catalyzed oligomerization of l-lysine ethyl ester in aqueous solution. ACS Catal. 2011, 1, 1022–1034. [Google Scholar] [CrossRef]
- Qin, X.; Xie, W.; Tian, S.; Ali, M.A.; Shirke, A.; Gross, R.A. Influence of Nε-protecting groups on the protease-catalyzed oligomerization of l-lysine methyl ester. ACS Catal. 2014, 4, 1783–1792. [Google Scholar] [CrossRef]
- Viswanathan, K.; Li, G.; Gross, R.A. Protease catalyzed in situ C-terminal modification of oligoglutamate. Macromolecules 2010, 43, 5245–5255. [Google Scholar] [CrossRef]
- Viswanathan, K.; Schofield, M.H.; Teraoka, I.; Gross, R.A. Surprising metal binding properties of phytochelatin-like peptides prepared by protease-catalysis. Green Chem. 2012, 14, 1020. [Google Scholar] [CrossRef]
- Baker, P.J.; Patwardhan, S.V.; Numata, K. Synthesis of homopolypeptides by aminolysis mediated by proteases encapsulated in silica nanospheres. Macromol. Biosci. 2014, 14, 1619–1626. [Google Scholar] [CrossRef] [PubMed]
- Ageitos, J.M.; Baker, P.J.; Sugahara, M.; Numata, K. Proteinase K-catalyzed synthesis of linear and star oligo(l-phenylalanine) conjugates. Biomacromolecules 2013, 14, 3635–3642. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Sato, R.; Li, Z.; Numata, K. Chemoenzymatic synthesis of oligo(l-cysteine) for use as a thermostable bio-based material. Macromol. Biosci. 2016, 16, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Yazawa, K.; Sugahara, M.; Yutani, K.; Takehira, M.; Numata, K. Derivatization of proteinase K with heavy atoms enhances its thermal stability. ACS Catal. 2016, 6, 3036–3046. [Google Scholar] [CrossRef]
- Gotor-Fernández, V.; Busto, E.; Gotor, V. Candida antarctica lipase B: An ideal biocatalyst for the preparation of nitrogenated organic compounds. Adv. Synth. Catal. 2006, 348, 797–812. [Google Scholar] [CrossRef]
- Kirk, O.; Christensen, M.W. Lipases from Candida antarctica: Unique biocatalysts from a unique origin. Org. Process Res. Dev. 2002, 6, 446–451. [Google Scholar] [CrossRef]
- Kobayashi, S. Lipase-catalyzed polyester synthesis—A green polymer chemistry. Proc. Jpn. Acad. Ser. B 2010, 86, 338–365. [Google Scholar] [CrossRef]
- Numata, K.; Srivastava, R.K.; Finne-Wistrand, A.; Albertsson, A.-C.; Doi, Y.; Abe, H. Branched poly(lactide) synthesized by enzymatic polymerization: Effects of molecular branches and stereochemistry on enzymatic degradation and alkaline hydrolysis. Biomacromolecules 2007, 8, 3115–3125. [Google Scholar] [CrossRef] [PubMed]
- Stavila, E.; Alberda van Ekenstein, G.O.; Woortman, A.J.; Loos, K. Lipase-catalyzed ring-opening copolymerization of epsilon-caprolactone and β-lactam. Biomacromolecules 2014, 15, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Stavila, E.; Loos, K. Synthesis of lactams using enzyme-catalyzed aminolysis. Tetrahedron Lett. 2013, 54, 370–372. [Google Scholar] [CrossRef]
- Cheng, H.N.; Gu, Q.M.; Maslanka, W.W. Enzyme-catalyzed polyamides and compositions and processes of preparing and using the same. US Patents US 6677427 B1, 13 January 2004. [Google Scholar]
- Numata, K.; Sato, R.; Yazawa, K.; Hikima, T.; Masunaga, H. Crystal structure and physical properties of Antheraea yamamai silk fibers: Long poly(alanine) sequences are partially in the crystalline region. Polymer 2015, 77, 87–94. [Google Scholar] [CrossRef]
- Viswanathan, K.; Omorebokhae, R.; Li, G.; Gross, R.A. Protease-catalyzed oligomerization of hydrophobic amino acid ethyl esters in homogeneous reaction media using l-phenylalanine as a model system. Biomacromolecules 2010, 11, 2152–2160. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-B.; Cai, Y.; Yang, S.; Wang, H.; Hou, R.-Z.; Xu, L.; Xiao-Xia, W.; Zhang, X.-Z. Synthesis of tetrapeptide Bz-RGDS-NH2 by a combination of chemical and enzymatic methods. J. Biotechnol. 2006, 125, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Sanda, F.; Fujiyama, T.; Endo, T. Chemical synthesis of poly-γ-glutamic acid by polycondensation of γ-glutamic acid dimer: Synthesis and reaction of poly-γ-glutamic acid methyl ester. J. Polym. Sci. A Polym. Chem. 2001, 39, 732–741. [Google Scholar] [CrossRef]
- Uyama, H.; Fukuoka, T.; Komatsu, I.; Watanabe, T.; Kobayashi, S. Protease-catalyzed regioselective polymerization and copolymerization of glutamic acid diethyl ester. Biomacromolecules 2002, 3, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Bajorath, J.; Hinrichs, W.; Saenger, W. The enzymatic activity of proteinase K is controlled by calcium. Eur. J. Biochem. 1988, 176, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Berti, P.J.; Faerman, C.H.; Storer, A.C. Cooperativity of papain-substrate interaction energies in the S2 to S2′ subsites. Biochemistry 1991, 30, 1394–1402. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, V.C.; Moodie, R.B. Protonation equilibria of amides and related compounds. Part II. Butyramide, n-butyrylglycine, and some carbamic acid esters. J. Chem. Soc. B 1968, 275–277. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nylon unit | Yield/mol % | Monomeric composition of nylon unit/mol % |
|---|---|---|
| Nylon 4Et | 53 ± 2 | 13 ± 6 |
| Nylon 4Me | 45 ± 4 | 12 ± 3 |
| Nylon 3Et | 49 ± 5 | 18 ± 4 |
| Nylon 3Me | 45 ± 2 | 15 ± 6 |
| Nylon 6Me | 35 ± 3 | 12 ± 2 |
| Nylon 1Et | 40 ± 4 | Not detected |
| Product | 1% Weight loss/°C | 5% Weight loss/°C | 10% Weight loss/°C |
|---|---|---|---|
| Oligo(GluEt-co-15 mol % nylon 3Me) | 130 ± 6 | 265 ± 3 | 289 ± 1 |
| Oligo(GluEt-co-12 mol % nylon 4Me) | 145 ± 7 | 267 ± 9 | 289 ± 4 |
| Oligo(GluEt-co-12 mol % nylon 6Me) | 135 ± 7 | 267 ± 6 | 289 ± 4 |
| Oligo(GluEt) | 134 ± 6 | 271 ± 2 | 295 ± 1 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yazawa, K.; Numata, K. Papain-Catalyzed Synthesis of Polyglutamate Containing a Nylon Monomer Unit. Polymers 2016, 8, 194. https://doi.org/10.3390/polym8050194
Yazawa K, Numata K. Papain-Catalyzed Synthesis of Polyglutamate Containing a Nylon Monomer Unit. Polymers. 2016; 8(5):194. https://doi.org/10.3390/polym8050194
Chicago/Turabian StyleYazawa, Kenjiro, and Keiji Numata. 2016. "Papain-Catalyzed Synthesis of Polyglutamate Containing a Nylon Monomer Unit" Polymers 8, no. 5: 194. https://doi.org/10.3390/polym8050194
APA StyleYazawa, K., & Numata, K. (2016). Papain-Catalyzed Synthesis of Polyglutamate Containing a Nylon Monomer Unit. Polymers, 8(5), 194. https://doi.org/10.3390/polym8050194