Reorganizing Polymer Chains with Cyclodextrins

 , , and
, , and 
Abstract
:1. Introduction
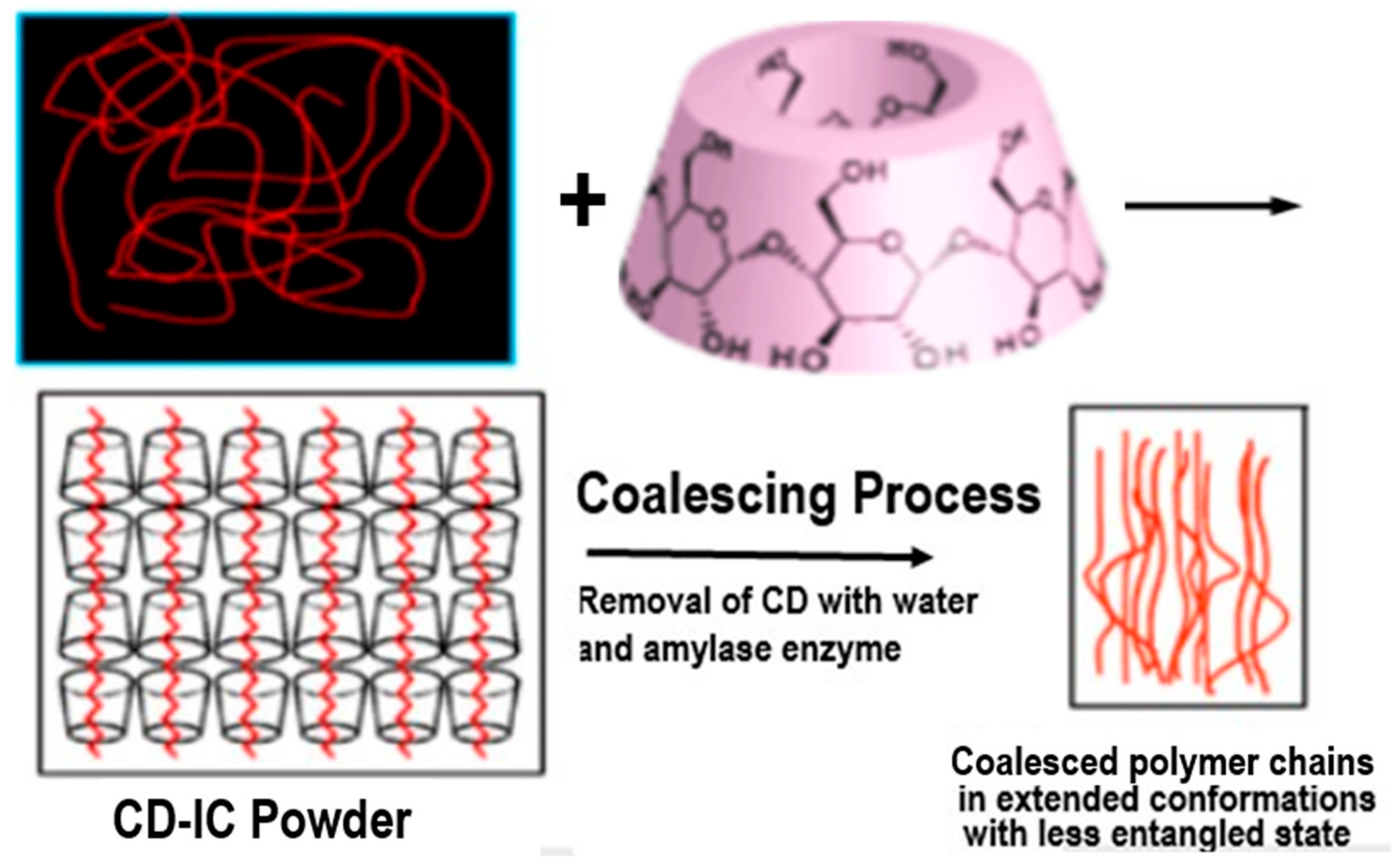
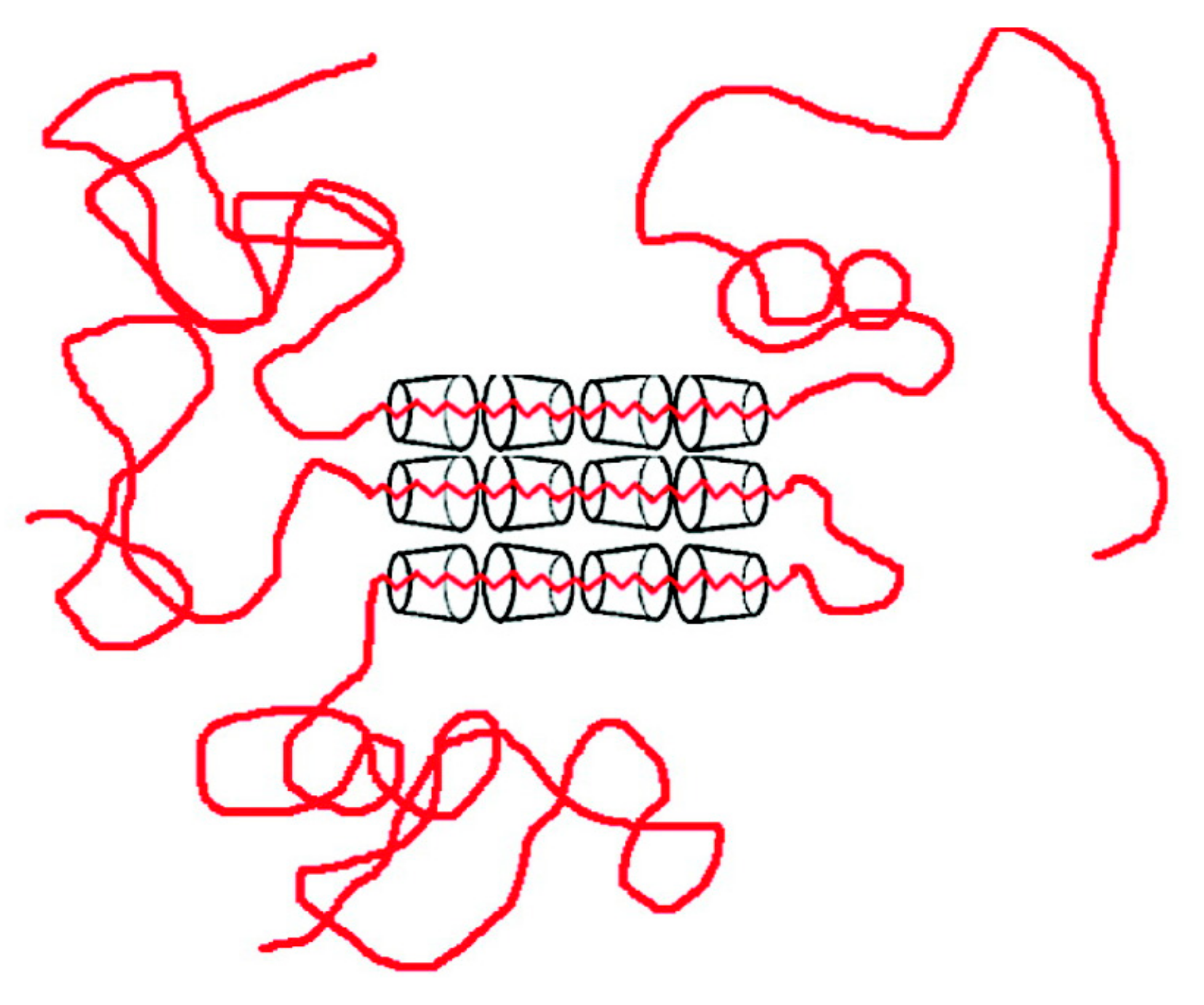
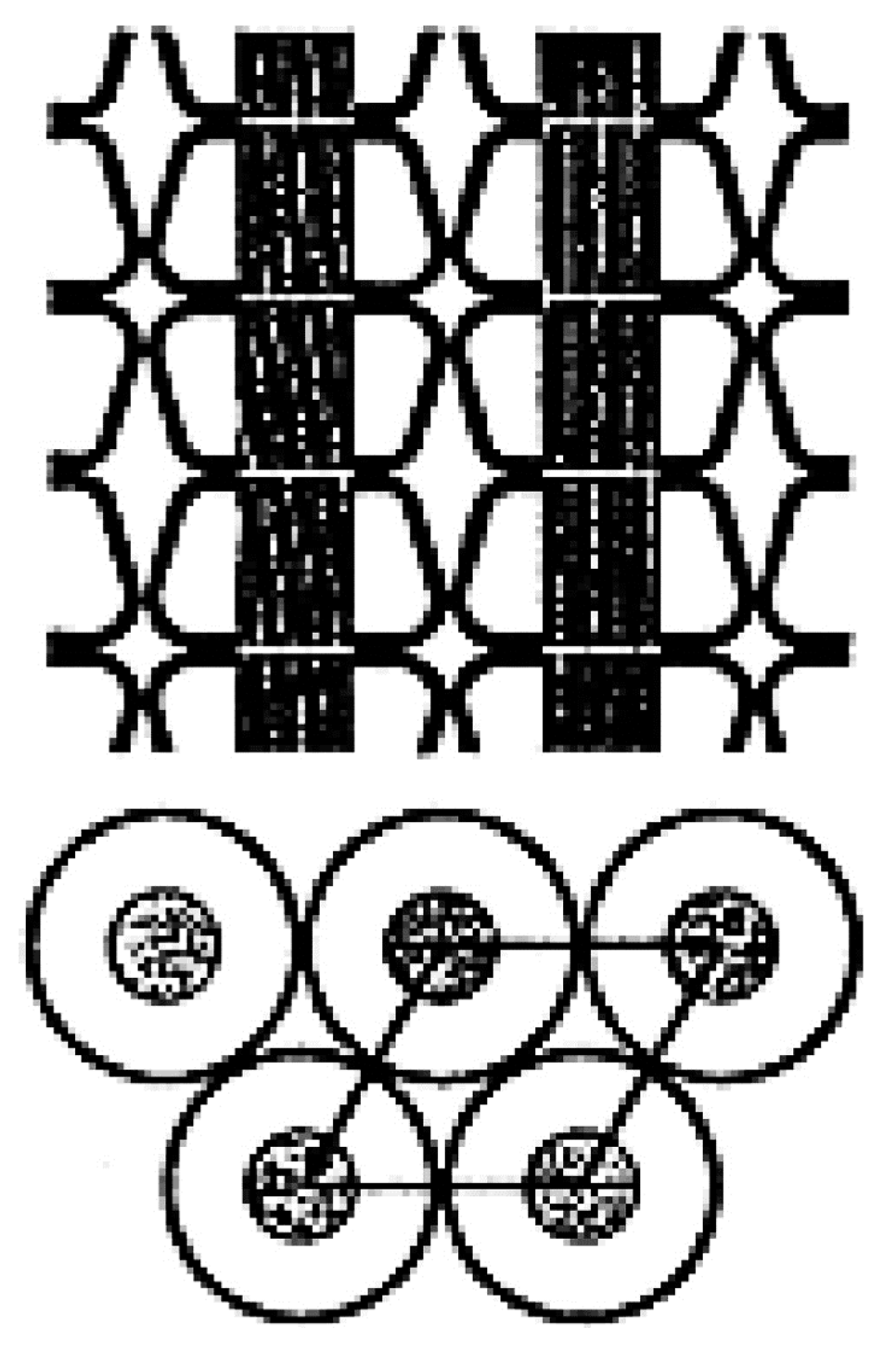
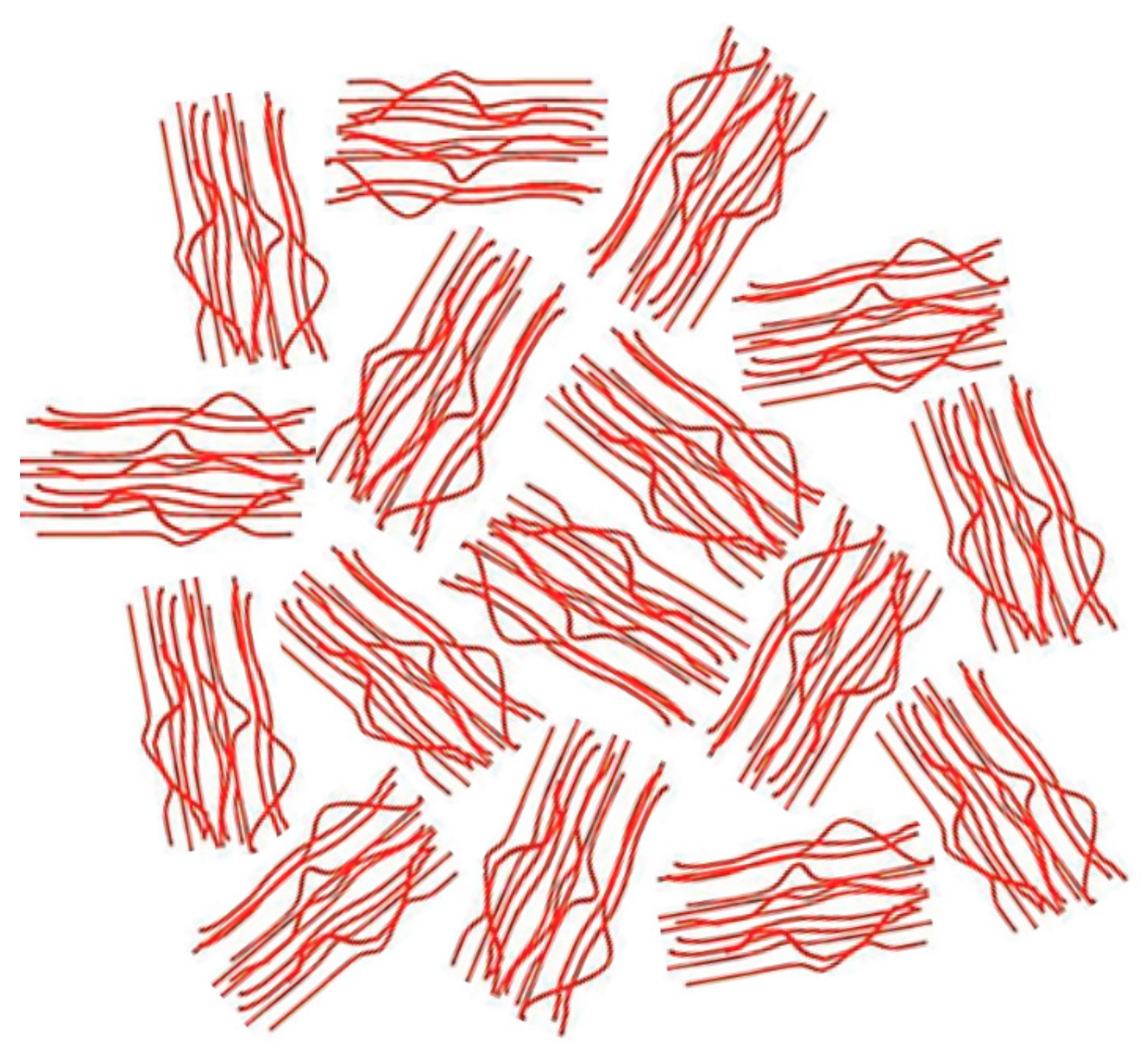
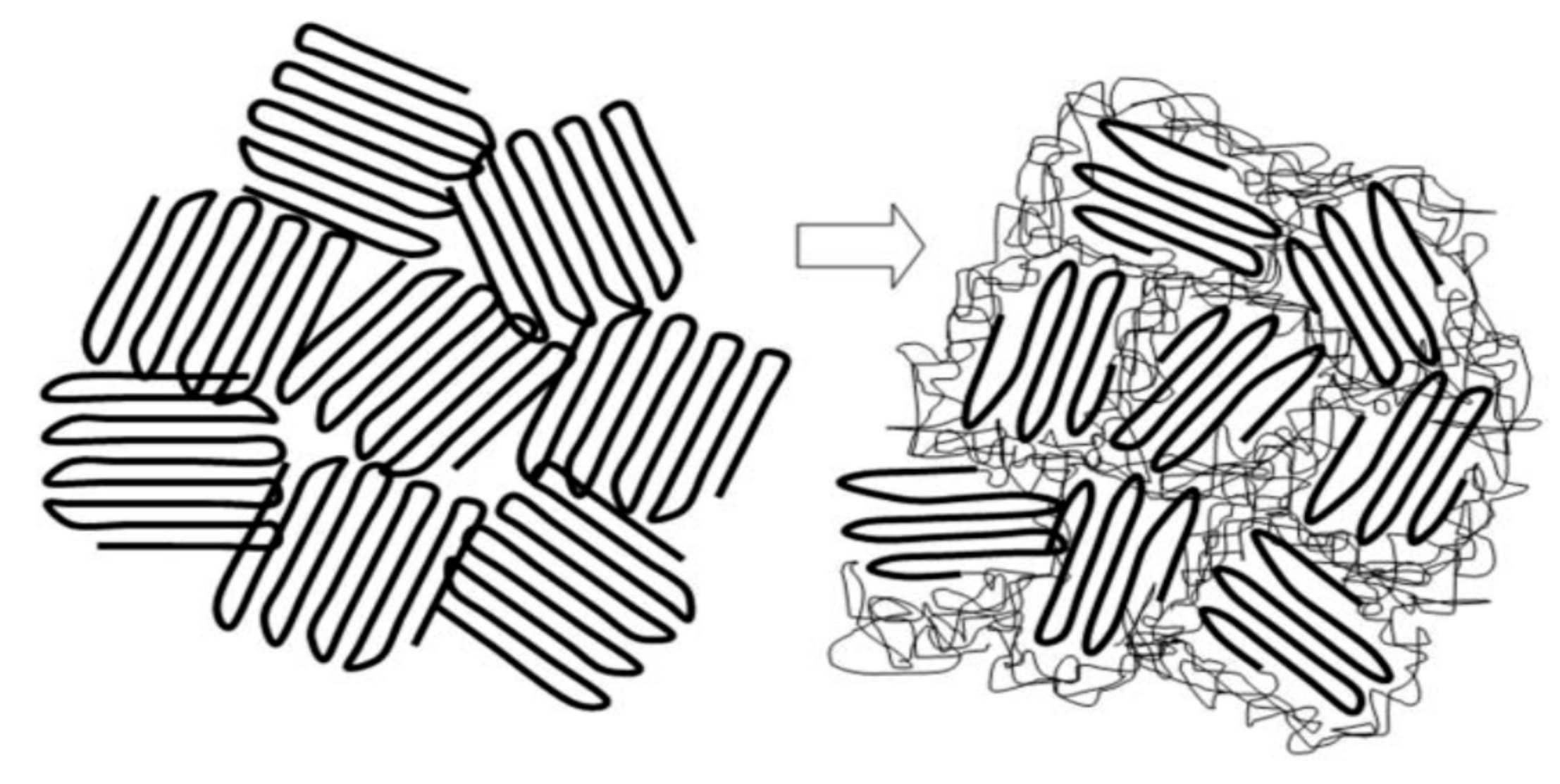
2. Polymers Reorganized through Complexation with and Coalescence from Their CD-ICs
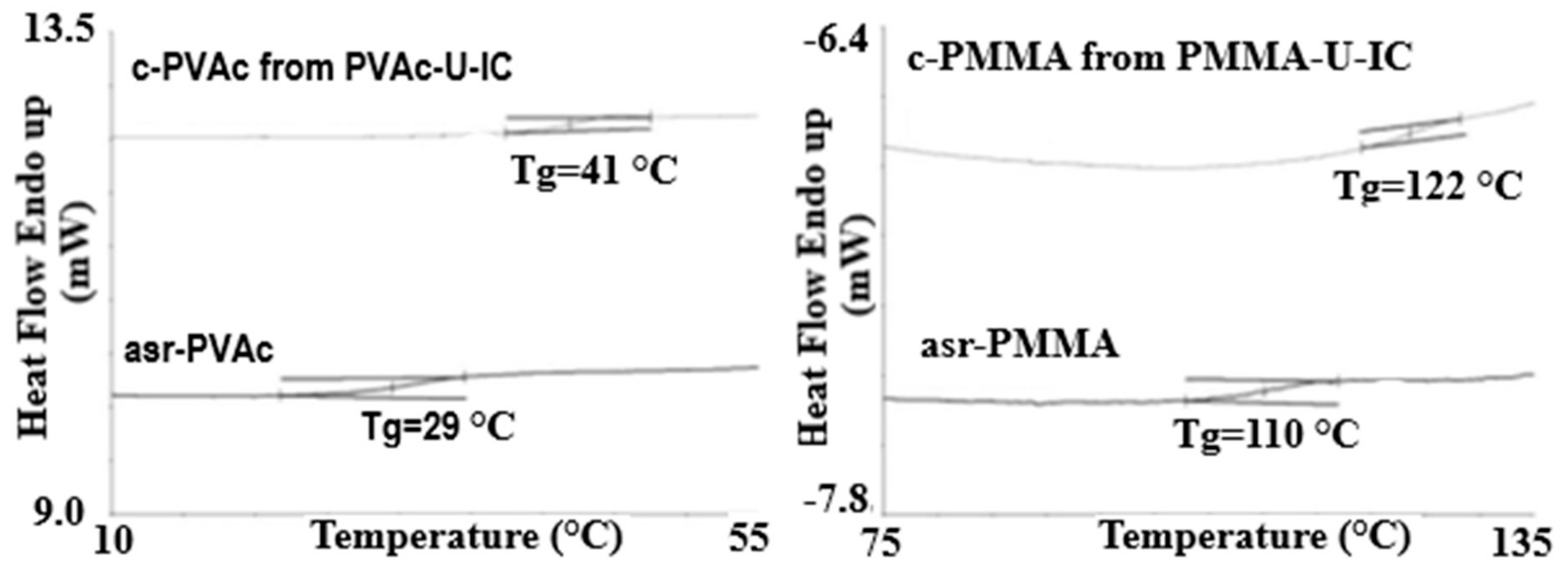
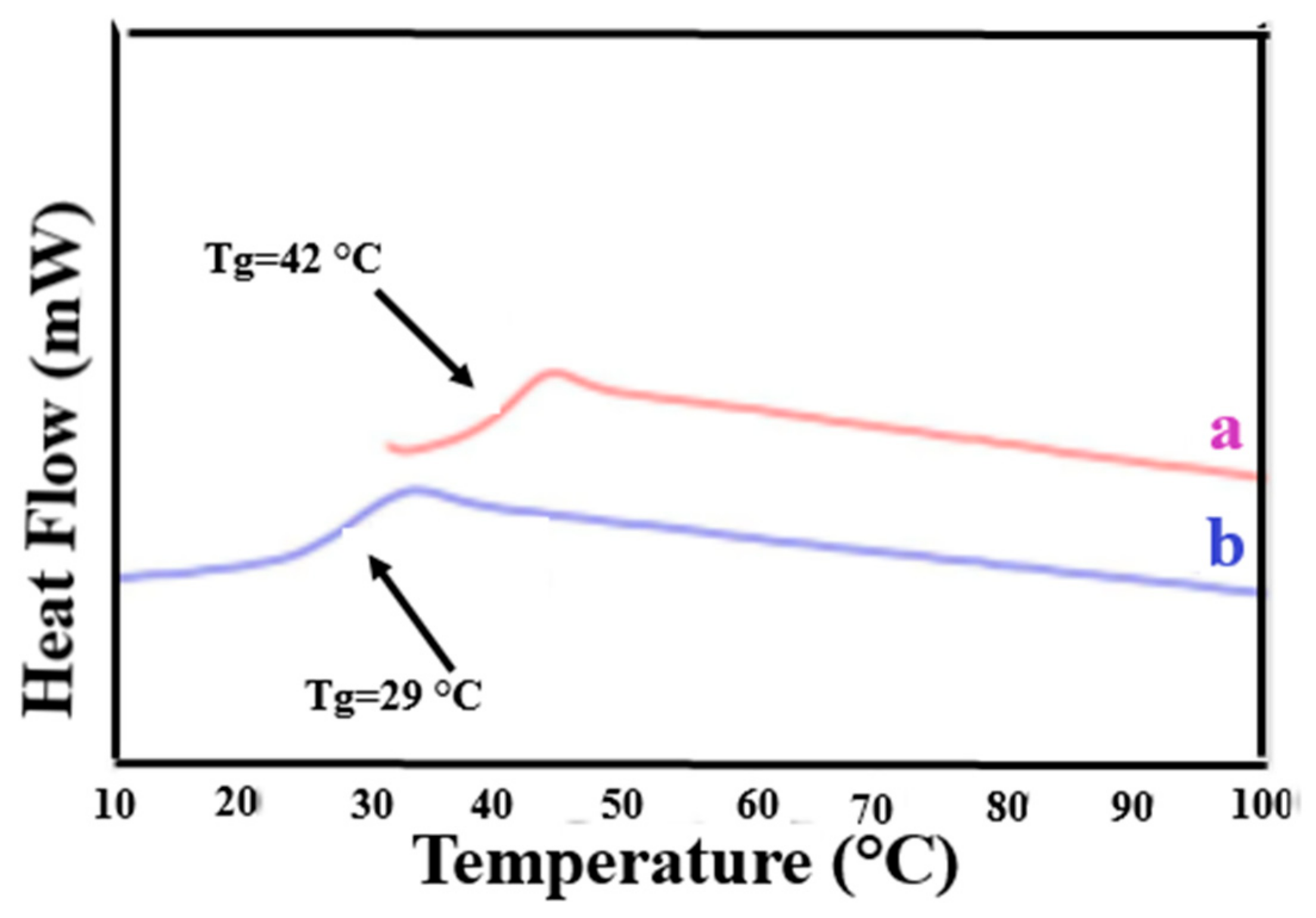
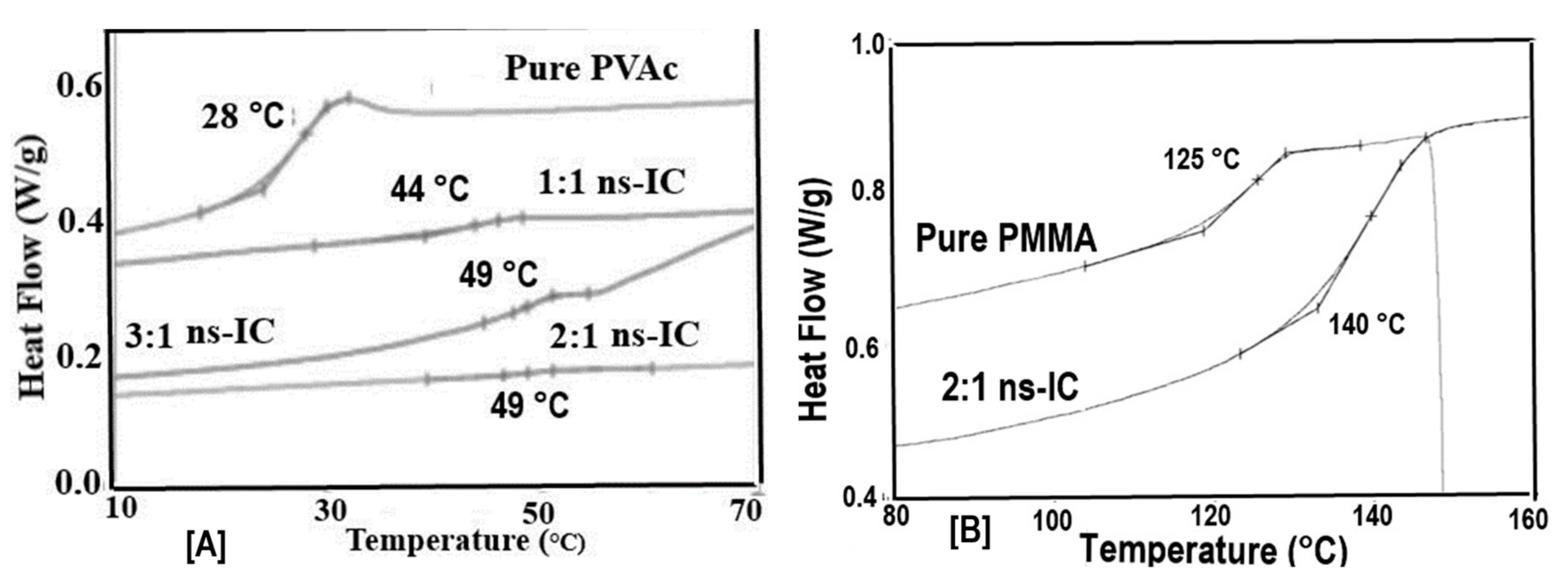
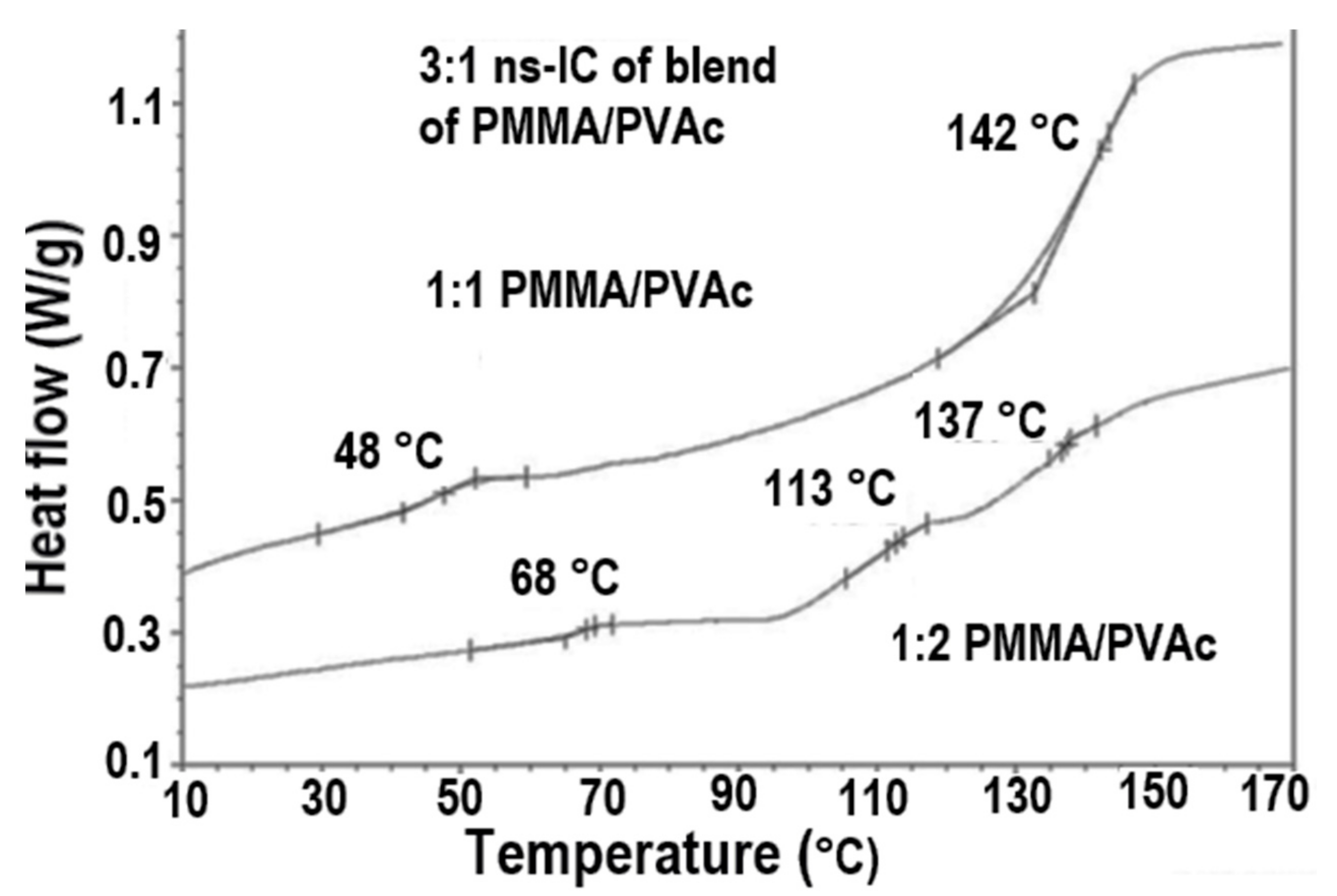
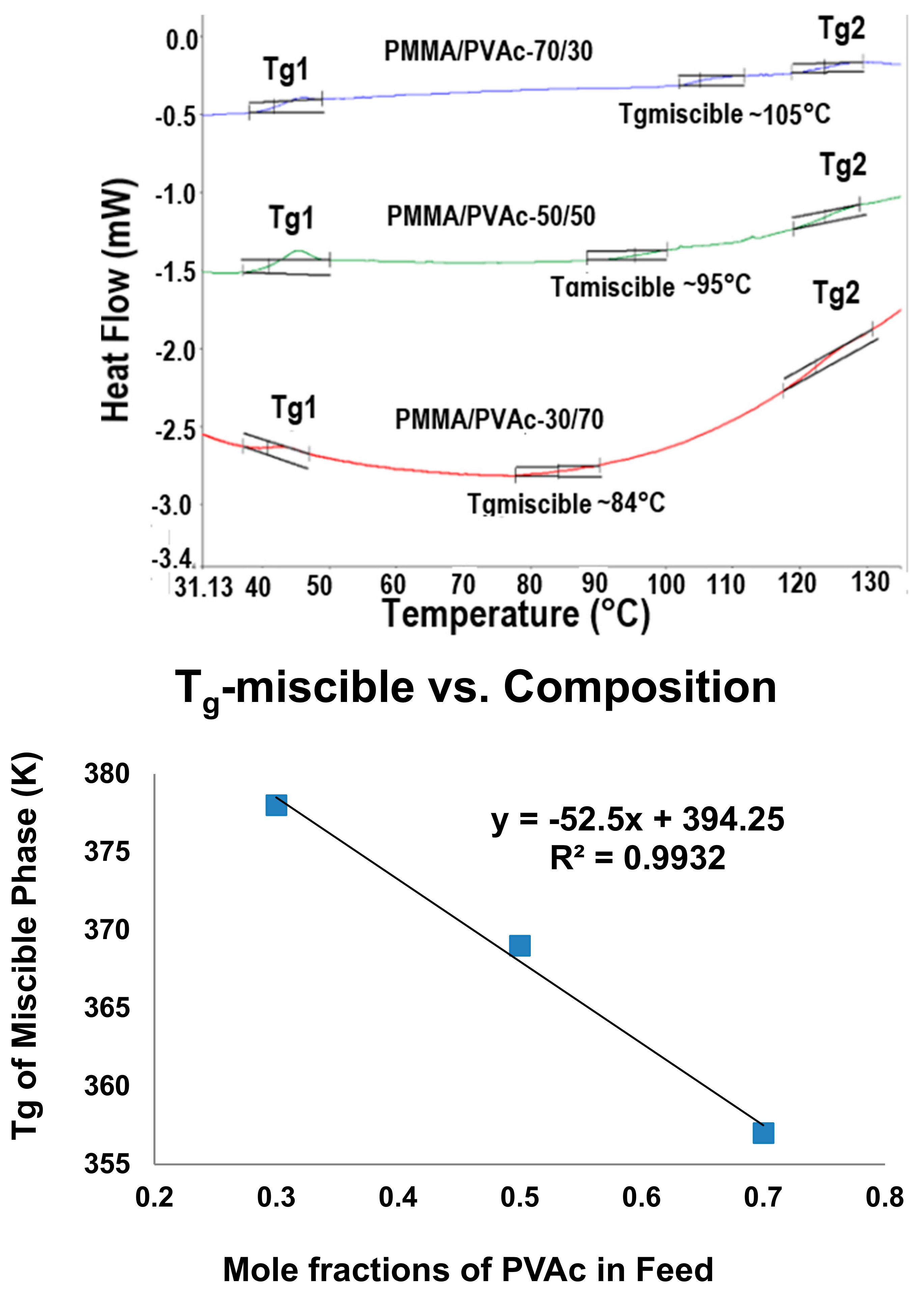
2.1. Amorphous Polymers
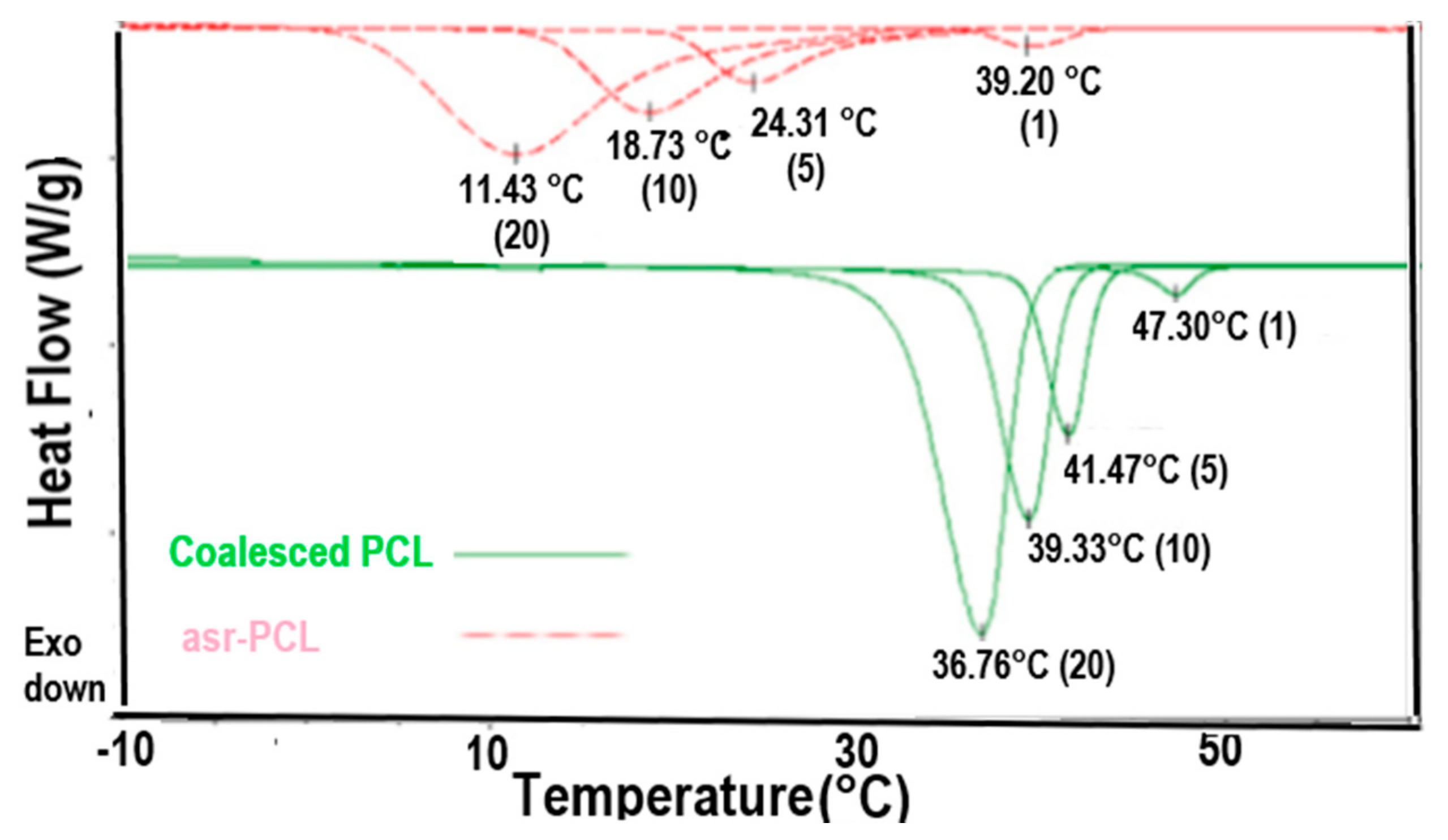
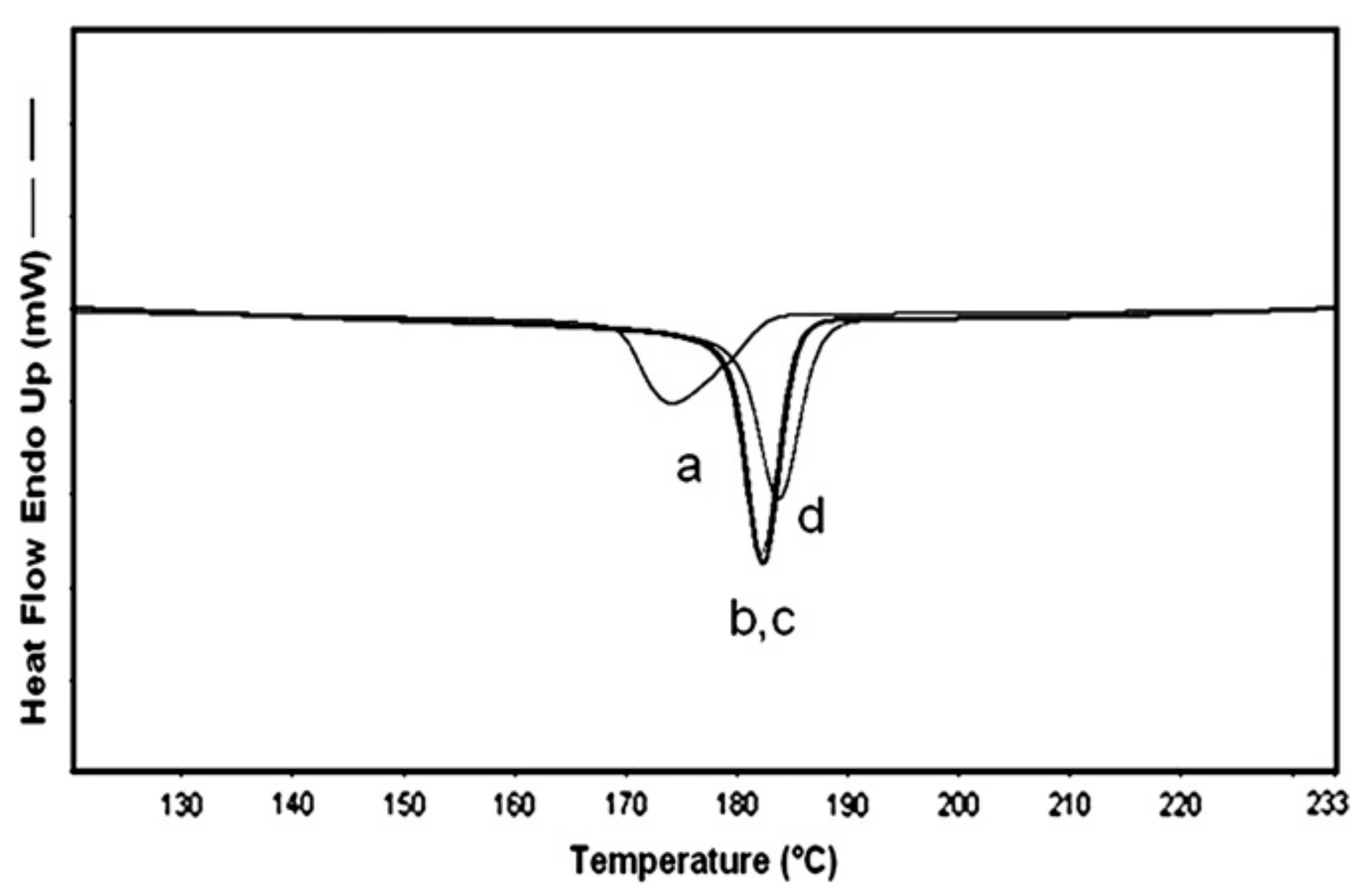
2.2. Semi-Crystalline Polymers
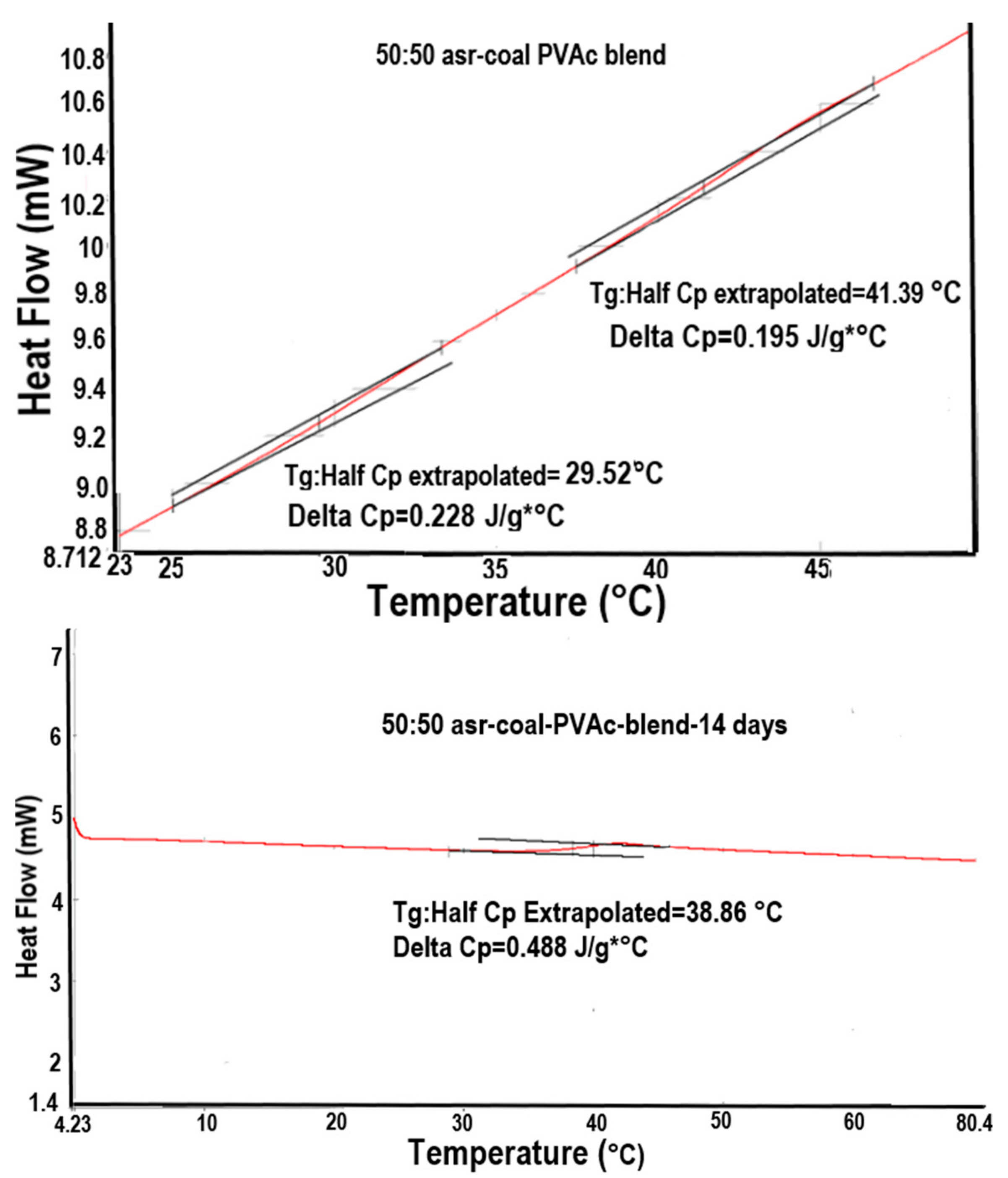
3. Thermal Stability of the Chain Organization in Coalesced Polymer Samples
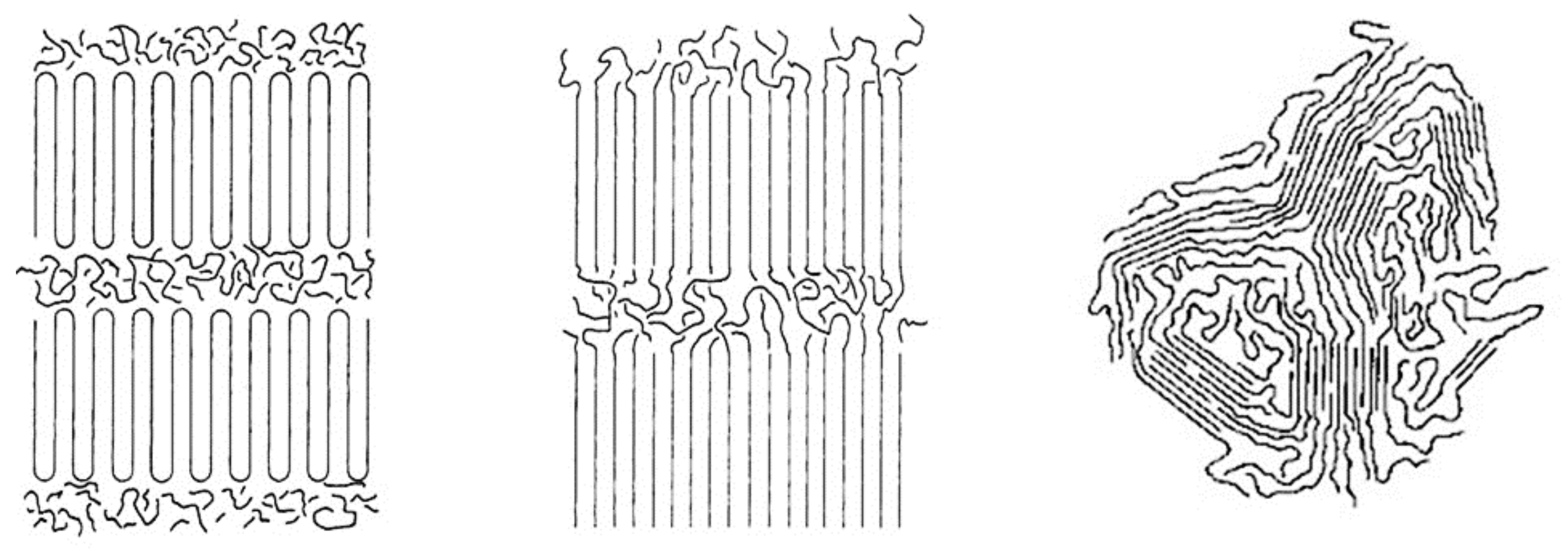
4. Polarized Optical Examination of Solid and Molten asr- and c-PCL Films
5. Summary and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Harada, A.; Kamachi, M. Complex formation between poly (ethylene glycol) and α-cyclodextrin. Macromolecules 1990, 23, 2821. [Google Scholar] [CrossRef]
- Tonelli, A.E. Molecular processing of polymers with cyclodextrins. Adv. Polym. Sci. 2009, 222, 55–77. [Google Scholar]
- Huang, L.; Tonelli, A. On the dynamics of dense polymer systems. J. Macromol. Sci. Rev. Macromol. Chem. Phys. 1998, 38, 781. [Google Scholar] [CrossRef]
- Huang, L.; Vasanthan, N.; Tonelli, A.E. Polymer-polymer composites fabricated by the in situ release and coalescence of polymer chains from their inclusion compounds with urea into a carrier polymer phase. J. Appl. Polym. Sci. 1997, 64, 281–287. [Google Scholar] [CrossRef]
- Huang, L.; Allen, E.G.; Tonelli, A.E. Recent Research Developments in Macromolecular Research; Pandalai, S.G., Ed.; Research Sign-Post: Trivandrum, India, 1997; Volume 2, p. 175. [Google Scholar]
- Huang, L.; Allen, E.; Tonelli, A.E. Study of the Inclusion Compounds Formed between α-Cyclodextrin and High Molecular Weight Poly (ethylene oxide) and Poly (∈-caprolactone). Polymer 1998, 39, 4857–4865. [Google Scholar] [CrossRef]
- Huang, L.; Tonelli, A.E. Inclusion Compounds as a Means to Fabricate Controlled Release Materials. In Intelligent Materials for Controlled Release; Dinh, S.M., DeNuzzio, J.D., Comfort, A.R., Eds.; ACS Symposium Series No. 728; American Chemical Society: Washington, DC, USA, 1999; Chapter 10. [Google Scholar]
- Rusa, C.C.; Tonelli, A.E. Polymer/polymer inclusion compounds as a novel approach to obtaining a PLLA/PCL intimately compatible blend. Macromolecules 2000, 33, 5321–5324. [Google Scholar] [CrossRef]
- Huang, L.; Gerber, M.; Taylor, H.; Lu, J.; Tapaszi, E.; Wutkowski, M.; Hill, M.; Nunalee, F.N.; Harvey, A.; Rusa, C.C.; et al. Creation of Polymer Films with Novel Structures by Processing with Inclusion Compounds. In Film Formation in Coatings: Mechanisms, Properties, and Morphology; Povder, T., Urban, M.W., Eds.; ACS Symposium Series No. 790; American Chemical Society: Washington, DC, USA, 2001; Chapter 14. [Google Scholar]
- Lu, J.; Mirau, P.A.; Rusa, C.C.; Tonelli, A.E. Cyclodextrin: From Basic Research to Market. In Proceedings of the 10th International Cyclodextrin Symposium (CD-2000), Ann Arbor, MI, USA, 21–24 May 2000; Szejtli, J., Ed.; Mira Digital Publishing: Saint Louis, MO, USA, 2001. [Google Scholar]
- Rusa, C.C.; Lu, J.; Huang, L.; Tonelli, A.E. Cyclodextrin: From Basic Research to Market. In Proceedings of the 10th International Cyclodextrin Symposium (CD-2000), Ann Arbor, MI, USA, 21–24 May 2000; Szejtli, J., Ed.; Mira Digital Publishing: Saint Louis, MO, USA, 2001. [Google Scholar]
- Rusa, C.C.; Luca, C.; Tonelli, A.E. Polymer−Cyclodextrin Inclusion Compounds: Toward New Aspects of Their Inclusion Mechanism. Macromolecules 2001, 34, 1318–1322. [Google Scholar] [CrossRef]
- Wei, M.; Tonelli, A.E. Compatiblization of polymers via coalescence from their common cyclodextrin inclusion compounds. Macromolecules 2001, 34, 4061–4065. [Google Scholar] [CrossRef]
- Shuai, X.; Porbeni, F.E.; Wei, M.; Shin, I.D.; Tonelli, A.E. Formation of and coalescence from the inclusion complex of a biodegradable block copolymer and α-cyclodextrin: A novel means to modify the phase structure of biodegradable block copolymers. Macromolecules 2001, 34, 7355–7361. [Google Scholar] [CrossRef]
- Huang, L.; Gerber, M.; Taylor, H.; Lu, J.; Tapazsi, E.; Wutkowski, M.; Hill, M.; Lewis, C.; Harvey, A.; Wei, M.; et al. Creation of novel polymer materials by processing with inclusion compounds. Macromol. Symp. 2001, 176, 129–144. [Google Scholar] [CrossRef]
- Shuai, X.; Wei, M.; Porbeni, F.E.; Bullions, T.A.; Tonelli, A.E. Formation of and Coalescence from the Inclusion Complex of a Biodegradable Block Copolymer and α-Cyclodextrin. 2: A Novel Way To Regulate the Biodegradation Behavior of Biodegradable Block Copolymers. Biomacromolecules 2002, 3, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Bullions, T.A.; Wei, M.; Porbeni, F.E.; Gerber, M.J.; Peet, J.; Balik, M.; White, J.L.; Tonelli, A.E. Reorganization of the structures, morphologies, and conformations of bulk polymers via coalescence from polymer–cyclodextrin inclusion compounds. J. Polym. Sci. Part B Polym. Phys. 2002, 40, 992–1012. [Google Scholar] [CrossRef]
- Shuai, X.; Porbeni, F.E.; Wei, M.; Bullions, T.; Tonelli, A.E. Formation of Inclusion Complexes of Poly (3-hydroxybutyrate) s with Cyclodextrins. 1. Immobilization of Atactic Poly (R,S-3-hydroxybutyrate) and Miscibility Enhancement between Poly (R,S-3-hydroxybutyrate) and Poly (∈-caprolactone). Macromolecules 2002, 35, 3126–3132. [Google Scholar] [CrossRef]
- Wei, M.; Davis, W.; Urban, B.; Song, Y.; Porbeni, F.E.; Wang, X.; White, J.L.; Balik, C.M.; Rusa, C.C.; Fox, J.; et al. Manipulation of Nylon-6 crystal structures with its α-Cyclodextrin inclusion complex. Macromolecules 2002, 35, 8039–8044. [Google Scholar] [CrossRef]
- Rusa, C.C.; Bullions, T.A.; Fox, J.; Porbeni, F.E.; Wang, X.; Tonelli, A.E. Inclusion compound formation with a new columnar cyclodextrin host. Langmuir 2002, 18, 10016–10032. [Google Scholar] [CrossRef]
- Shuai, X.; Porbeni, F.E.; Wei, M.; Bullions, T.; Tonelli, A.E. Inclusion complex formation between α,γ-cyclodextrins and a triblock copolymer and the cyclodextrin-type-dependent microphase structures of their coalesced samples. Macromolecules 2002, 35, 2401–2405. [Google Scholar] [CrossRef]
- Wei, M.; Shuai, X.; Tonelli, A.E. Melting and crystallization behaviors of biodegradable polymers enzymatically coalesced from their cyclodextrin inclusion complexes. Biomacromolecules 2003, 4, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Bullions, T.A.; Edeki, E.M.; Porbeni, F.E.; Wei, M.; Shuai, X.; Rusa, C.C.; Tonelli, A.E. Intimate blend of poly (ethylene terephthalate) and poly (ethylene 2,6-naphthalate) via formation with and coalescence from their common inclusion compound with γ-cyclodextrin. J. Polym. Sci. Part B Polym. Phys. 2003, 41, 139–148. [Google Scholar] [CrossRef]
- Abdala, A.A.; Tonelli, A.E.; Khan, S.A. Modulation of hydrophobic interactions in associative polymers using inclusion compounds and surfactants. Macromolecules 2003, 36, 7833–7841. [Google Scholar] [CrossRef]
- Tonelli, A.E. The potential for improving medical textiles with cyclodextrin inclusion compounds. J. Text. Appar. Technol. Manag. 2003, 3, 12. [Google Scholar]
- Tonelli, A.E. Reorganization of the structures, morphologies, and conformations of polymers by coalescence from their crystalline inclusion compounds formed with cyclodextrins. Macromol. Symp. 2003, 203, 71–88. [Google Scholar] [CrossRef]
- Wei, M.; Bullions, T.A.; Rusa, C.C.; Wang, X.; Tonelli, A.E. Unique morphological and thermal behaviors of reorganized poly (ethylene terephthalates). J. Polym. Sci. Part B Polym. Phys. 2004, 42, 386–394. [Google Scholar] [CrossRef]
- Rusa, C.C.; Uyar, T.; Rusa, M.; Hunt, M.A.; Wang, X.; Tonelli, A.E. An intimate polycarbonate/poly (methyl methacrylate)/poly (vinyl acetate) ternary blend via coalescence from their common inclusion compound with γ-cyclodextrin. J. Polym. Sci. Part B Polym. Phys. 2004, 42, 4182–4194. [Google Scholar] [CrossRef]
- Abdala, A.A.; Wu, W.; Olesen, K.R.; Jenkins, R.D.; Tonelli, A.E.; Khan, S. Solution rheology of hydrophobically modified associative polymers: Effects of backbone composition and hydrophobe concentration. J. Rheol. 2004, 48, 979–994. [Google Scholar] [CrossRef]
- Wei, M.; Shin, I.D.; Urban, B.; Tonelli, A.E. Partial miscibility in a nylon-6/nylon-66 blend coalesced from their common α-cyclodextrin inclusion complex. J. Polym. Sci. Part B Polym. Phys. 2004, 42, 1369–1378. [Google Scholar] [CrossRef]
- Rusa, C.C.; Shuai, X.; Bullions, T.A.; Wei, M.; Porbeni, F.E.; Lu, J.; Huang, L.; Fox, J.; Tonelli, A.E. Controlling the behaviors of biodegradable/bioabsorbable polymers with cyclodextrins. J. Polym. Environ. 2004, 12, 157–163. [Google Scholar] [CrossRef]
- Uyar, T.; Rusa, M.; Tonelli, A.E. Polymerization of Styrene in Cyclodextrin Channels: Can Confined Free-Radical Polymerization Yield Stereoregular Polystyrene? Makromol. Rapid Commun. 2004, 25, 1382–1386. [Google Scholar] [CrossRef]
- Rusa, C.C.; Wei, M.; Bullions, T.A.; Rusa, M.; Gomez, M.A.; Porbeni, F.E.; Wang, X.; Shin, I.D.; Balik, C.M.; White, J.L.; et al. Controlling the polymorphic behaviors of semicrystalline polymers with cyclodextrins. Cryst. Growth Des. 2004, 4, 1431–1441. [Google Scholar] [CrossRef]
- Rusa, C.C.; Wei, M.; Shuai, X.; Bullions, T.A.; Wang, X.; Rusa, M.; Uyar, T.; Tonelli, A.E. Molecular mixing of incompatible polymers through formation of and coalescence from their common crystalline cyclodextrin inclusion compounds. J. Polym. Sci. Part B Polym. Phys. 2004, 42, 4207–4224. [Google Scholar] [CrossRef]
- Rusa, M.; Aboelfotoh, O.; Kolbas, R.M.; Tonelli, A.E. Conformable nanoscale polymers through formation of cyclodextrin inclusion compounds. PMSE Prepr. 2004, 90, 620. [Google Scholar]
- Rusa, M.; Wang, X.; Tonelli, A.E. Fabrication of Inclusion Compounds with Solid Host γ-Cyclodextrins and Water-Soluble Guest Polymers: Inclusion of Poly (N-acylethylenimine) s in γ-Cyclodextrin Channels As Monitored by Solution 1H NMR. Macromolecules 2004, 37, 6898–6903. [Google Scholar] [CrossRef]
- Rusa, C.C.; Wei, M.; Bullions, T.A.; Shuai, X.; Uyar, T.; Tonelli, A.E. Nanostructuring polymers with cyclodextrins. Polym. Adv. Technol. 2005, 16, 269–275. [Google Scholar] [CrossRef]
- Rusa, C.C.; Rusa, M.; Gomez, M.; Shin, I.D.; Fox, J.D.; Tonelli, A.E. Nanostructuring High Molecular Weight Isotactic Polyolefins via Processing with γ-Cyclodextrin Inclusion Compounds. Formation and Characterization of Polyolefin-γ-Cyclodextrin Inclusion Compounds. Macromolecules 2004, 37, 7992–7999. [Google Scholar] [CrossRef]
- Hernández, R.; Rusa, M.; Rusa, C.C.; López, D.; Mijangos, C.; Tonelli, A.E. Controlling PVA hydrogels with γ-cyclodextrin. Macromolecules 2004, 37, 9620–9625. [Google Scholar] [CrossRef]
- Jia, X.; Wang, X.; Tonelli, A.E.; White, J.L. Two-dimensional spin-diffusion NMR reveals differential mixing in biodegradable polymer blends. Macromolecules 2005, 38, 2775–2780. [Google Scholar] [CrossRef]
- Uyar, T.; Rusa, C.C.; Wang, X.; Rusa, M.; Hacaloglu, J.; Tonelli, A.E. Intimate blending of binary polymer systems from their common cyclodextrin inclusion compounds. J. Polym. Sci. Part B Polym. Phys. 2005, 43, 2578–2593. [Google Scholar] [CrossRef]
- Rusa, C.C.; Bridges, C.; Ha, S.-W.; Tonelli, A.E. Conformational Changes Induced in Bombyx m ori Silk Fibroin by Cyclodextrin Inclusion Complexation. Macromolecules 2005, 38, 5640–5646. [Google Scholar] [CrossRef]
- Uyar, T.; Rusa, C.C.; Hunt, M.A.; Aslan, E.; Hacaloglu, J.; Tonelli, A.E. Reorganization and improvement of bulk polymers by processing with their cyclodextrin inclusion compounds. Polymer 2005, 46, 4762–4775. [Google Scholar] [CrossRef]
- Uyar, T.; Aslan, E.; Tonelli, A.E.; Hacaloglu, J. Thermal degradation of polycarbonate, poly (vinyl acetate) and their blends. Polym. Degrad. Stab. 2006, 91, 2960–2967. [Google Scholar] [CrossRef]
- Uyar, T.; Hunt, M.A.; Gracz, H.S.; Tonelli, A.E. Crystalline cyclodextrin inclusion compounds formed with aromatic guests: Guest-dependent stoichiometries and hydration-sensitive crystal structures. Cryst. Growth Des. 2006, 6, 1113–1119. [Google Scholar] [CrossRef]
- Rusa, C.C.; Rusa, M.; Peet, J.; Uyar, T.; Fox, J.; Hunt, M.A.; Wang, X.; Balik, C.M.; Tonelli, A.E. The nano-threading of polymers. J. Incl. Phenom. Macrocycl. Chem. 2006, 55, 185–192. [Google Scholar] [CrossRef]
- Pang, K.; Schmidt, B.; Kotek, R.; Tonelli, A.E. Reorganization of the chain packing between poly (ethylene isophthalate) chains via coalescence from their inclusion compound formed with γ-cyclodextrin. J. Appl. Polym. Sci. 2006, 102, 6049–6053. [Google Scholar] [CrossRef]
- Uyar, T.; Gracz, H.S.; Rusa, M.; Shin, I.D.; El-Shafei, A.; Tonelli, A.E. Polymerization of styrene in γ-cyclodextrin channels: Lightly rotaxanated polystyrenes with altered stereosequences. Polymer 2006, 47, 6948–6955. [Google Scholar] [CrossRef]
- Uyar, T.; Oguz, G.; Tonelli, A.E.; Hacaloglu, J. Thermal degradation processes of poly(carbonate) and poly(methyl methacrylate) in blends coalesced either from their common inclusion compound formed with γ-cyclodextrin or precipitated from their common solution. Polym. Degrad. Stab. 2006, 91, 2471–2481. [Google Scholar] [CrossRef]
- Tonelli, A.E. Nanofibers and Nanotechnology in Textiles; Brown, P., Stevens, K., Eds.; Woodhead Publ. Ltd.: Cambridge, UK, 2007. [Google Scholar]
- Vedula, J.; Tonelli, A.E. Reorganization of poly (ethylene terephthalate) structures and conformations to alter properties. J. Polym. Sci. Part B Polym. Phys. 2007, 45, 735–746. [Google Scholar] [CrossRef]
- Uyar, T.; Rusa, C.C.; Tonelli, A.E.; Hacaloğlu, J. Pyrolysis mass spectrometry analysis of polycarbonate/poly (methyl methacrylate)/poly (vinyl acetate) ternary blends. Polym. Degrad. Stab. 2007, 92, 32–43. [Google Scholar] [CrossRef]
- Martínez, G.; Gómez, M.A.; Villar-Rodil, S.; Garrido, L.; Tonelli, A.E.; Balik, C.M. Formation of crystalline inclusion compounds of poly (vinyl chloride) of different stereoregularity with γ-cyclodextrin. J. Polym. Sci. Part A Polym. Chem. 2007, 45, 2503–2513. [Google Scholar] [CrossRef]
- Tonelli, A.E. Cyclodextrins as a means to nanostructure and functionalize polymers. J. Incl. Phenom. Macrocycl. Chem. 2008, 60, 197–202. [Google Scholar] [CrossRef]
- Tonelli, A.E. Nanostructuring and functionalizing polymers with cyclodextrins. Polymer 2008, 49, 1725–1736. [Google Scholar] [CrossRef]
- Tonelli, A.E. Organizational stabilities of bulk neat and well-mixed, blended polymer samples coalesced from their crystalline inclusion compounds formed with cyclodextrins. J. Polym. Sci. Part B Polym. Phys. 2009, 47, 1543–1553. [Google Scholar] [CrossRef]
- Mohan, A.; Joyner, X.; Kotek, R.; Tonelli, A.E. Constrained/directed crystallization of nylon-6. I. nonstoichiometric inclusion compounds formed with cyclodextrins. Macromolecules 2009, 42, 8983–8991. [Google Scholar] [CrossRef]
- Busche, B.J.; Tonelli, A.E.; Balik, C.M. Compatibilization of polystyrene/poly (dimethyl siloxane) solutions with star polymers containing a γ-cyclodextrin core and polystyrene arms. Polymer 2010, 51, 454–462. [Google Scholar] [CrossRef]
- Busche, B.J.; Tonelli, A.E.; Balik, C.M. Morphology of polystyrene/poly (dimethyl siloxane) blends compatibilized with star polymers containing a γ-cyclodextrin core and polystyrene arms. Polymer 2010, 51, 1465–1471. [Google Scholar] [CrossRef]
- Busche, B.J.; Tonelli, A.E.; Balik, C.M. Properties of polystyrene/poly (dimethyl siloxane) blends partially compatibilized with star polymers containing a γ-cyclodextrin core and polystyrene arms. Polymer 2010, 51, 6013–6020. [Google Scholar] [CrossRef]
- Mohan, A.; Gurarslan, A.; Joyner, X.; Child, R.; Tonelli, A.E. Melt-crystallized nylon-6 nucleated by the constrained chains of its non-stoichiometric cyclodextrin inclusion compounds and the nylon-6 coalesced from them. Polymer 2011, 52, 1055–1062. [Google Scholar] [CrossRef]
- Williamson, B.R.; Tonelli, A.E. Constrained polymer chain behavior observed in their non-stoichiometric cyclodextrin inclusion complexes. J. Incl. Phenom. Macrocycl. Chem. 2012, 72, 71–78. [Google Scholar] [CrossRef]
- Gurarslan, A.; Tonelli, A.E. Single-component polymer composites. Macromolecules 2011, 44, 3856–3861. [Google Scholar] [CrossRef]
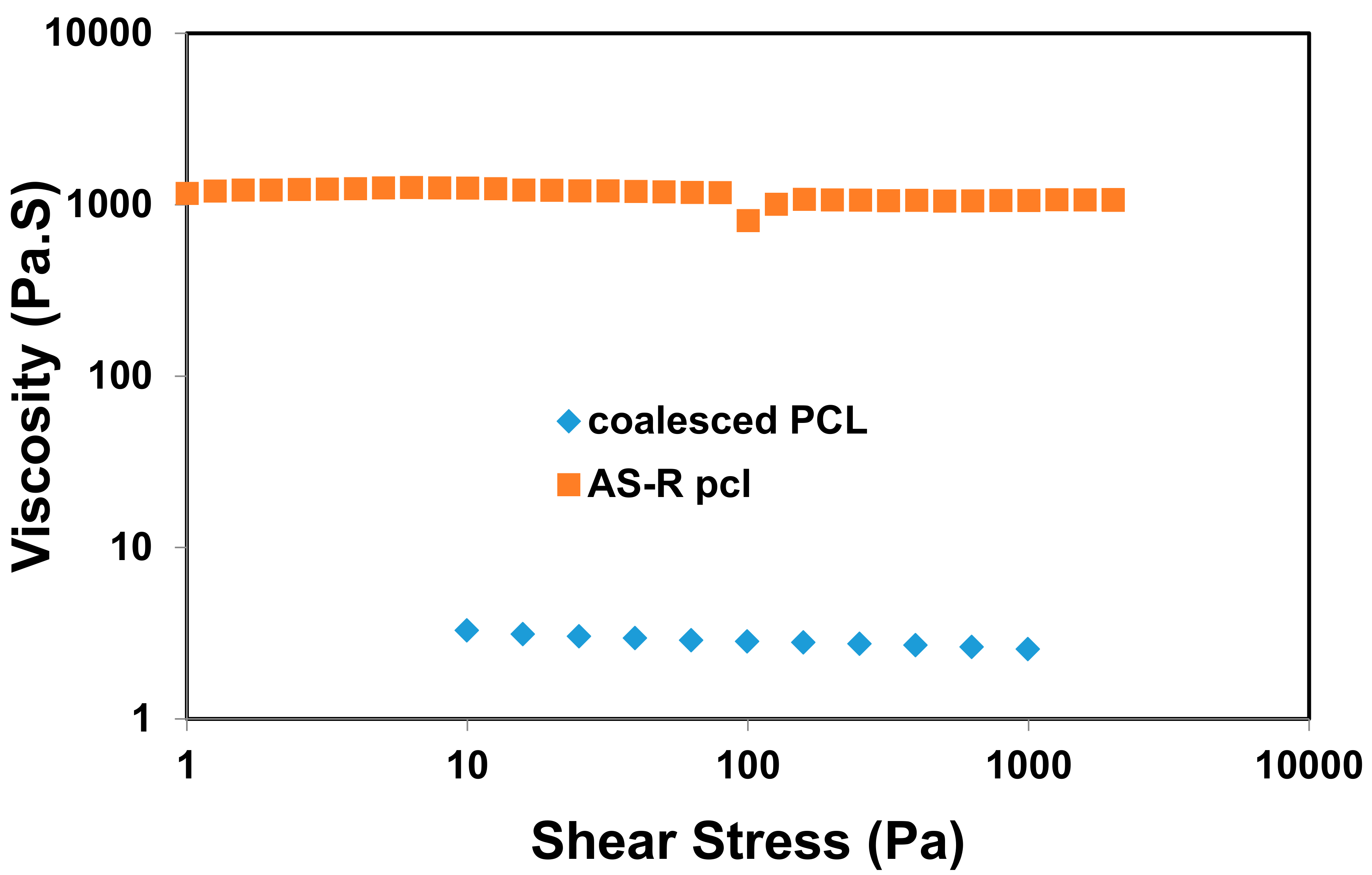
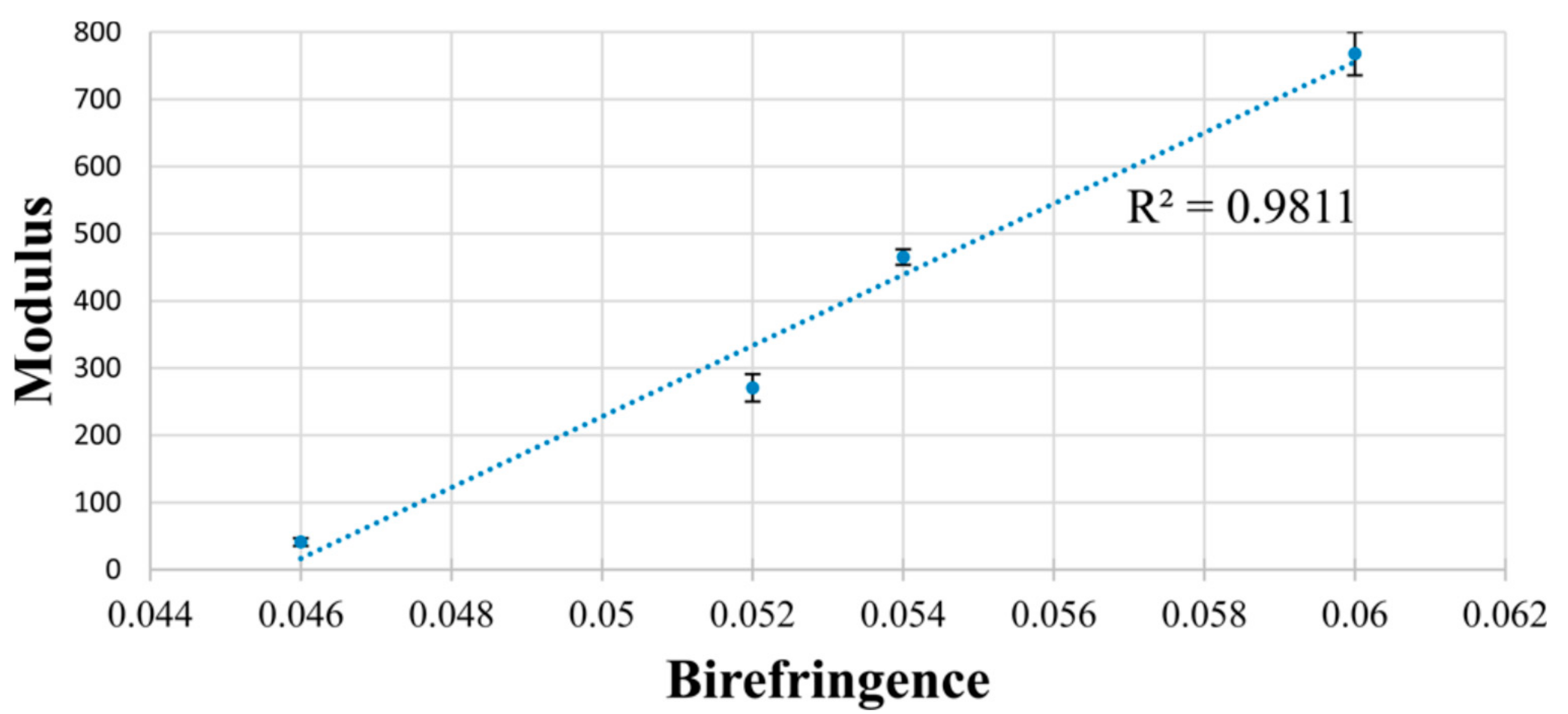
- Williamson, B.R.; Krishnaswany, R.; Tonelli, A.E. Physical properties of poly (ε-caprolactone) coalesced from its α-cyclodextrin inclusion compound. Polymer 2011, 52, 4517–4527. [Google Scholar] [CrossRef]
- Peet, J.; Rusa, C.C.; Hunt, M.A.; Tonelli, A.E.; Balik, C.M. Solid-State Complexation of Poly (Ethylene Glycol) with α-Cyclodextrin. Macromolecules 2005, 38, 537–541. [Google Scholar] [CrossRef]
- Rusa, C.C.; Fox, J.; Tonelli, A.E. Competitive Formation of Polymer−Cyclodextrin Inclusion Compounds. Macromolecules 2003, 36, 2742–2747. [Google Scholar] [CrossRef]
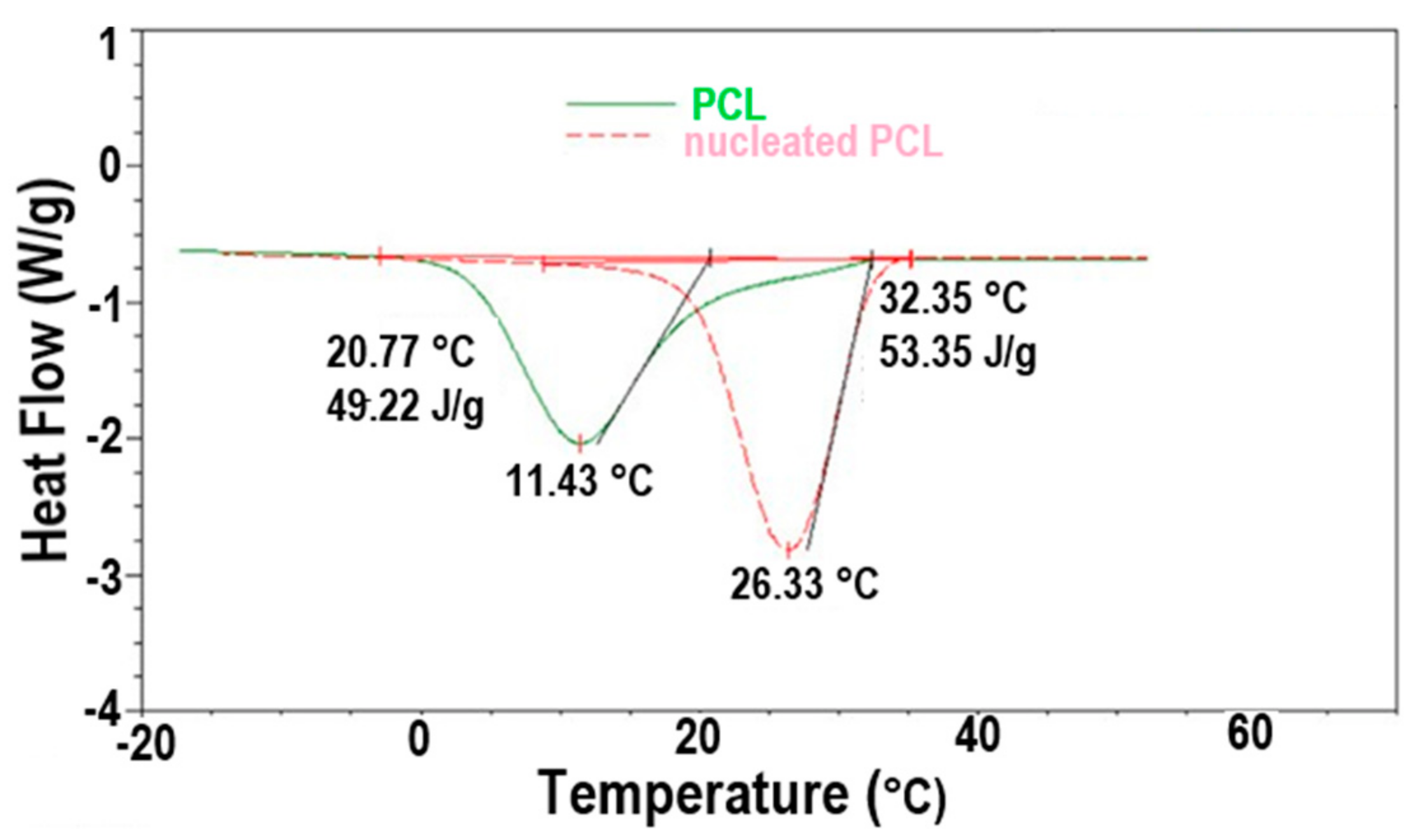
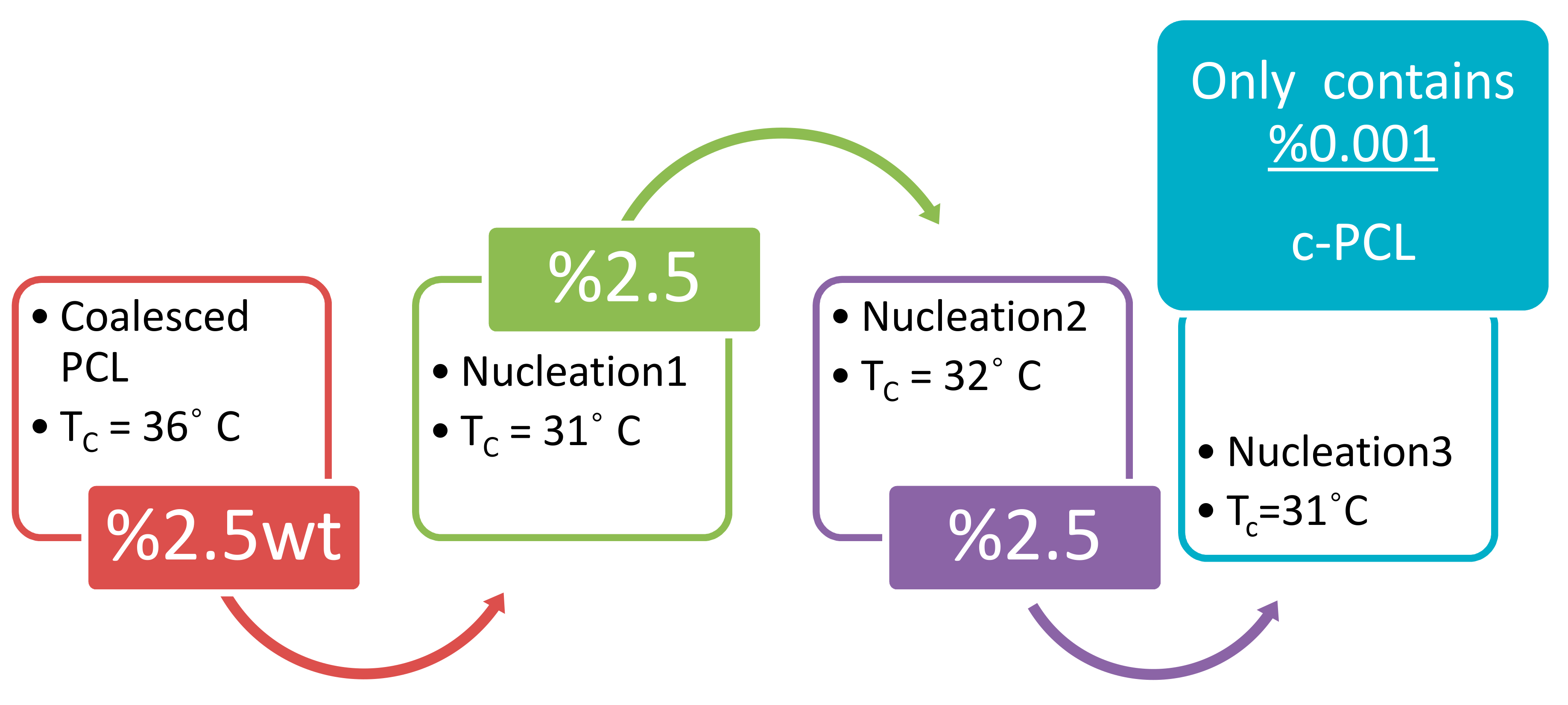
- Gurarslan, A.; Shen, J.; Tonelli, A.E. Behavior of poly (ε-caprolactone) s (PCLs) coalesced from their stoichiometric urea inclusion compounds and their use as nucleants for crystallizing PCL melts: Dependence on PCL molecular weights. Macromolecules 2012, 45, 2835–2840. [Google Scholar] [CrossRef]
- Gurarslan, A.; Tonelli, A.E. Self-reinforced PCL/PCL composites. In Proceedings of the ACS 2012 National Meeting, Anaheim, CA, USA, 27–31 March 2011. [Google Scholar]
- Joijode, A.S.; Hawkins, K.; Tonelli, A.E. Improving Poly (ethylene terephthalate) Through Self-nucleation. Macromol. Mater. Eng. 2013, 298, 1190–1200. [Google Scholar] [CrossRef]
- Joijode, A.S.; Antony, G.J.; Tonelli, A.E.J. Glass-transition temperatures of nanostructured amorphous bulk polymers and their blends. Polym. Sci. Part B Polym. Phys. 2013, 51, 1041–1050. [Google Scholar] [CrossRef]
- Williamson, B.R. Processing Polymers with Cyclodextrins. Ph.D. Thesis, North Carolina State University, Raleigh, NC, USA, 2010; p. 1332. [Google Scholar]
- Joijode, A.S. Nano-Structuring Polymers by Processing Them with Small Molecule Hosts. Ph.D. Thesis, North Carolina State University, Raleigh, NC, USA, 2014. Chapter 3. [Google Scholar]
- Gurarslan, A.; Caydamli, Y.; Shen, J.; Tse, S.; Yetukuri, M.; Tonelli, A.E. Coalesced poly (ε-caprolactone) fibers are stronger. Biomacromolecules 2015, 16, 890–893. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, A.E. Restructuring polymers via nanoconfinement and subsequent release. Beilstein J. Org. Chem. 2012, 8, 1318. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, S.; Lippits, D.R.; Peters, G.W.M.; Graf, R.; Yao, Y.; Spiess, H.W. Heterogeneity in polymer melts from melting of polymer crystals. Nat. Mater. 2005, 4, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Gurarslan, A.; Joijode, A.S.; Tonelli, A.E. Polymers coalesced from their cyclodextrin inclusion complexes: What can they tell us about the morphology of melt-crystallized polymers? J. Polym. Sci. Part B Polym. Phys. 2012, 50, 813–823. [Google Scholar] [CrossRef]
- Beers, D.E.; Ramirez, J.E. Vectran high-performance fibre. J. Text. Inst. 1990, 81, 561–574. [Google Scholar] [CrossRef]
- McLeish, T.C.B. A theory for heterogeneous states of polymer melts produced by single chain crystal melting. Soft Matter 2007, 3, 83–87. [Google Scholar] [CrossRef]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Density at 25 °C (g/cm3) (below Tg) | Density at 58 °C (g/cm3) (above Tg) |
|---|---|---|
| asr-PVAc | 1.093 | 1.040 |
| c-PVAc (γ-CD) | 1.156 | 1.077 |
| c-PVAc (urea) | 1.154 | 1.076 |
| Physical properties | asr-PCL fiber | c-PCL fiber | Drawn asr-PCL fiber | Drawn c-PCL fiber |
|---|---|---|---|---|
| modulus (MPa) | 41 ± 6 | 271 ± 20 | 465 ± 12 | 770 ± 32 |
| elongation at break (mm) | 197 ± 19 | 110 ± 8 | 32 ± 2 | 14 ± 2 |
| % crystallinity | 40.6 | 50.1 | 50.8 | 53.3 |
| PET samples | Sample density at 25 °C (g·cm−3) | Permeability (P × 1014) (cm3·s−1·Pa−1) |
|---|---|---|
| asr-PET | 1.368 | 1.64 |
| nuc-PET | 1.386 | 0.57 |
| Annealing time (days) | Tg (°C) |
|---|---|
| 0 | 41.5 |
| 2 | 41.7 |
| 8 | 41.5 |
| 14 | 41.2 |
| Sample * | Density (g/cm3) (above Tg) at 58 °C |
|---|---|
| asr-PVAc | 1.044 |
| c-PVAc | 1.077 |
| 50/50 = asr/c-PVAc blend | 1.076 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gurarslan, A.; Joijode, A.; Shen, J.; Narayanan, G.; Antony, G.J.; Li, S.; Caydamli, Y.; Tonelli, A.E. Reorganizing Polymer Chains with Cyclodextrins. Polymers 2017, 9, 673. https://doi.org/10.3390/polym9120673
Gurarslan A, Joijode A, Shen J, Narayanan G, Antony GJ, Li S, Caydamli Y, Tonelli AE. Reorganizing Polymer Chains with Cyclodextrins. Polymers. 2017; 9(12):673. https://doi.org/10.3390/polym9120673
Chicago/Turabian StyleGurarslan, Alper, Abhay Joijode, Jialong Shen, Ganesh Narayanan, Gerry J. Antony, Shanshan Li, Yavuz Caydamli, and Alan E. Tonelli. 2017. "Reorganizing Polymer Chains with Cyclodextrins" Polymers 9, no. 12: 673. https://doi.org/10.3390/polym9120673
APA StyleGurarslan, A., Joijode, A., Shen, J., Narayanan, G., Antony, G. J., Li, S., Caydamli, Y., & Tonelli, A. E. (2017). Reorganizing Polymer Chains with Cyclodextrins. Polymers, 9(12), 673. https://doi.org/10.3390/polym9120673