3. Results and Discussion
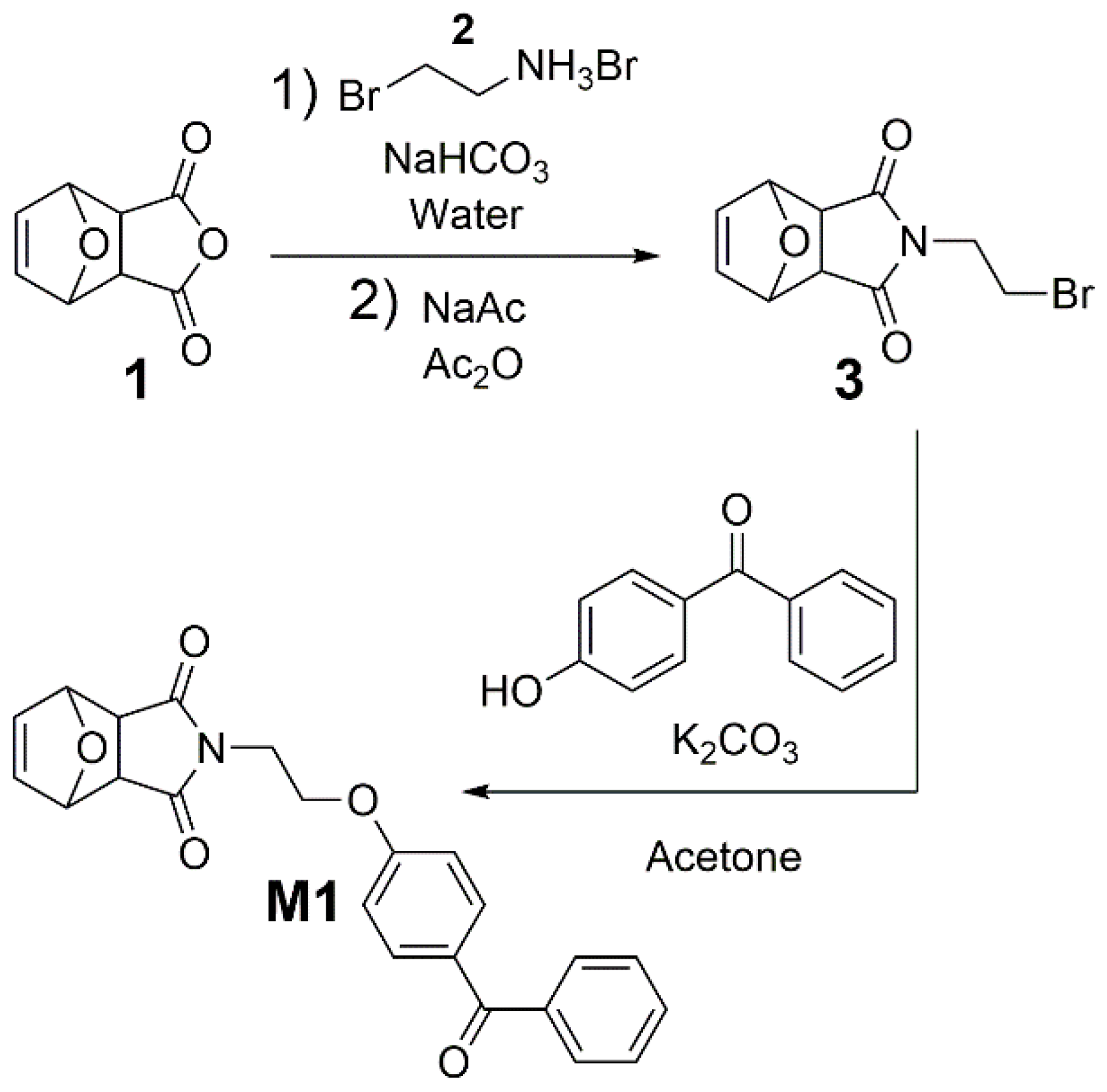
The benzophenone-containing poly(alkenylnorbornenes)
P(2-3-BP) (
Figure 1c) and poly(oxonorbornenes)
P(4-5-BP) (
Figure 1d) used in this study were obtained by copolymerizing the benzophenone-containing monomer
M1 (
Scheme 1) with the respective functional monomers
M2 to
M5 (
Scheme 2). To obtain
M1, first oxonorbornene anhydride
1 was reacted with the brominated amine
2 to give
N-(2-bromoethyl)-3,6-tetrahydrophthalimide (
3,
Scheme 1). The latter was then reacted with hydroxybenzophenone and K
2CO
3 in dry acetone for 24 h under reflux, yielding monomer
M1 (
Scheme 1). Details are given in the Experimental.
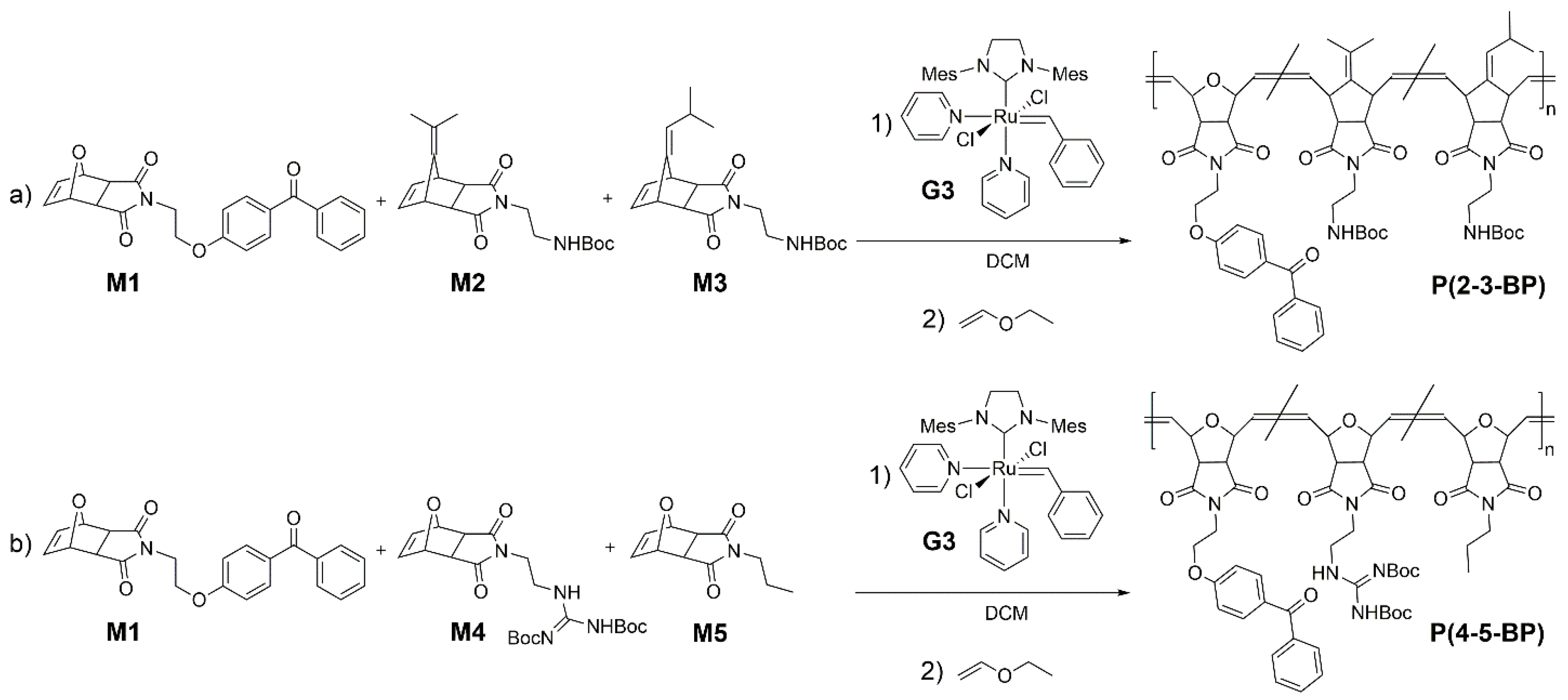
To synthesize different variants of the target poly(alkenylnorbornene)
P(2-3-BP), monomer
M1 and the alkenylnorbornene derivatives
M2 and
M3 were mixed in various monomer feed ratios and polymerized via ring opening metathesis polymerization (ROMP) using the Grubbs’ third generation catalyst (
G3) (
Scheme 2a). The catalyst-to-monomer ratio was adjusted to obtain copolymers with the desired molecular weight (target molecular weight 100,000 g·mol
−1). The target content of BP repeat units was 2.5% to 5%. Since the polymerization rates of the monomers used varied significantly (
M2 and
M3 being much less reactive than the oxonorbornene imide
M1) [
26,
27], the BP-containing monomer
M1 was added dropwise over 120 min to the reaction mixture containing
M2,
M3 and the polymerization catalyst. That way, a more homogeneous distribution of the BP-monomer units along the polymer chain was obtained. The molar composition of the polymers thus obtained, and their molar mass distribution parameters (determined by gel permeation chromatography) are summarized in
Table 1. It was confirmed by
1H NMR spectroscopy via a comparison of the integrated peaks of the benzophenone group (6.85 to 7.85 ppm, 9H) and the
N-Boc protective group (1.41 ppm, 9H) that the actual BP content of the polymers matched the target content.
To obtain different variants of the polymers
P(4-5-BP), monomer
M1 was copolymerized with different amounts of the oxonorbornene derivatives
M4 and
M5 (
Scheme 2b). These three monomers had similar polymerizable groups and therefore similar polymerization rates, and were thus mixed and added at the beginning of the reaction. The target BP content was 5 to 15 mol %. As before, the comonomer feed ratio and catalyst-to-monomer ratio were adjusted to obtain the desired copolymer molecular weights and compositions (
Table 1). For some of the network formation studies presented below, the
N-
tert-butyloxycarbonyl (
N-Boc) protective groups of the polymers were cleaved with trifluoroacetic acid before cross-linking. Unless otherwise specified, the reactions and measurements were performed on the protected polymers.
To synthesize surface-attached polymer networks from the
P(4-5-BP) polymers, they were dissolved in dichloromethane (DCM) and spin-coated onto silicon wafers that had been previously functionalized with benzophenone using the anchor group triethoxy benzophenone silane (
3EBP) [
25].
3EBP contains a benzophenone moiety and a silane group, which links it covalently to the silicon surface. Upon UV irradiation, the BP moiety of the
3EBP anchor group formed a covalent bond to
P(4-5-BP) and thus attached it to the surface, while the BP moieties that were part of
P(4-5-BP) simultaneously formed cross-links between the polymer chains, so that overall a surface-attached polymer network was obtained (
Figure 1b).
The degree of cross-linking of a network or hydrogel can be quantified by determining its gel content. The gel content of the here presented polymer films was determined by first measuring the overall dry film thickness
d0 of the networks just after UV irradiation via ellipsometry (i.e., the thickness of the gel plus the sol). The networks were then extracted in DCM overnight to remove unbound polymer chains. After this treatment, the remaining dry layer thickness
dextract (i.e., the gel thickness) was also measured via ellipsometry. From this, the gel content of each layer was calculated using Equation (1).
When different polymers are cross-linked using comparable conditions (e.g., the same irradiation doses and wavelengths), the gel content of the networks or hydrogels formed can be considered as a quantitative measure for the relative propensity of each polymer to cross-link. With this in mind, the gel content of networks made from different members of the
P(2-3-BP) and the
P(4-5-BP) families was determined after UV irradiation at different wavelengths (λ = 254 or 365 nm, respectively) and different energy doses. In
Figure 2, the gel content of the
P(4-5-BP) polymer networks thus obtained is shown. (For these polymers, the subscripts indicate the relative amount of each repeat unit, i.e.,
P(485-510-BP5) refers to a polymer made from 85%
M4, 10%
M5, and 5%
M1, and
P(495-BP5) to a polymer made from 95%
M4, 0%
M5, and 5%
M1.) The thickness data onto which the gel content curves are based are listed in the
Supporting Information (Tables S3–S5).
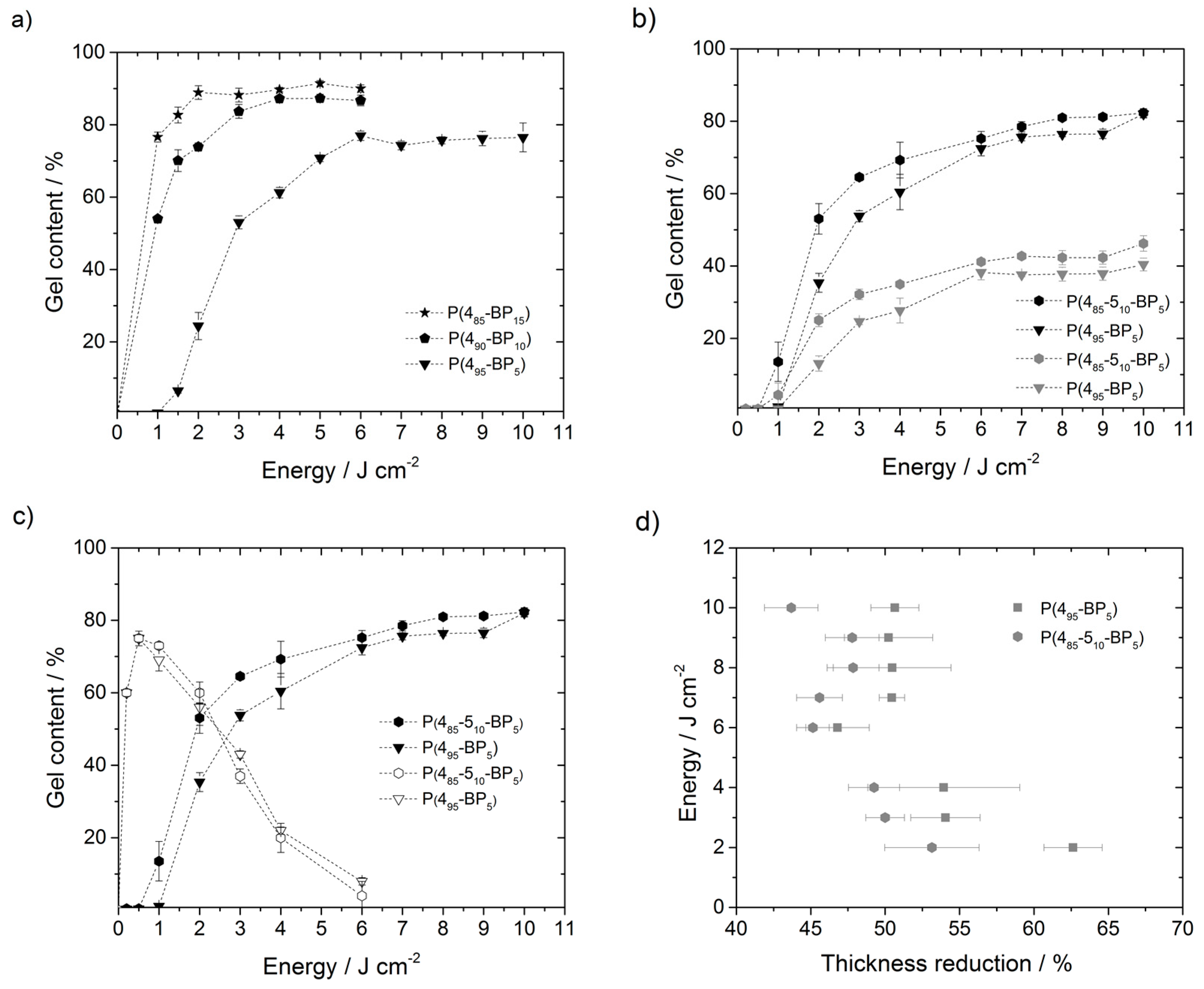
Figure 2a shows the gel content of
P(4-BP) networks with 5–15 mol % of BP for various irradiation energy doses at λ = 365 nm. In this graph, the gel content first increases steeply to the optimum dose, and then levels off into a plateau at higher energy doses. This is the usual behavior described for BP cross-linked polymers in the literature [
15], even though the energy doses needed to obtain a substantial amount of cross-linking in this polymer were quite high. Also in line with theory [
12], the data shows that layers made from polymers with a higher BP content also had a higher gel content when the same irradiation energy dose was used. For example,
P(485-BP15) irradiated with 2 J·cm
−2 had a gel content of around 90%, while the gel content of
P(490-BP10) was only 74%, and that of
P(495-BP5) was only 24%. It is noteworthy, though, that surprisingly high BP contents and energy doses were needed to obtain significant amounts of cross-linking.
When the
P(4-5-BP) polymer networks were treated with acid to remove their
tert-butyloxycarbonyl (Boc) protective groups (to obtain the corresponding polymer hydrogels), a layer thickness decrease of 46% to 63% was observed. For example, the layer thickness of
P(495-BP5) (energy dose 10 J·cm
−2) decreased from 229 to 113 nm after deprotection. This was mostly caused by the high mass loss occurring when the Boc groups were removed (42 mass %). At the same time, though, some polymer chains that are cross-linked predominantly via bonds to these Boc groups might also become detached, which would further increase the mass loss. To investigate if such chains formed was a significant fraction of the polymer networks obtained, the gel content of networks made from protected and the deprotected polymers
P(495-BP5) (irradiated with different energy doses at λ = 365 nm) were determined and compared to the gel contents of the corresponding
P(485-510-BP5) polymers.
P(485-510-BP5) had the same benzophenone content as
P(495-BP5), but 10 mol % repeat units containing propyl substituents, and thus more C–H bonds available for cross-linking that would not be cleaved by deprotection (
Figure 1d). When irradiated at 365 nm,
P(485-510-BP5) consistently had higher gel contents than
P(495-BP5) throughout the energy dose range investigated (
Figure 2b), even though it had a lower absolute number of C–H bonds compared to
P(495-BP5). For example,
P(485-510-BP5) had a gel content of around 81% after illumination with 8 J·cm
−2, whereas that of
P(495-BP5) was only 76% when irradiated with the same energy dose. This indicates that the propyl C–H groups per se were more easily available for cross-linking than the C–H bonds of the two CH
2CH
2N-Boc fragments. The same trend was observed for the corresponding deprotected polymers of
P(485-510-BP5) and
P(495-BP5) (
Figure 2b). In
Figure 2d, the relative thickness loss due to deprotection (in %) of the two polymers is compared. When all Boc groups are cleaved, the theoretical mass loss of deprotected
P(495-BP5) should be 42 mass %, corresponding to a thickness reduction of roughly the same percentage, while that of
P(485-510-BP5) should be only slightly lower (40 mass %).
Figure 2d shows that the mass loss for the deprotected
P(495-BP5) polymers was considerably larger than for the
P(485-510-BP5) polymers, indicating that the former contained a larger fraction of polymer chains that were exclusively cross-linked via bonds to the Boc groups. In line with this interpretation, the data also shows that for both polymers, the thickness decrease becomes smaller with increasing irradiation energy, i.e., in the strongly irradiated polymer networks (and the corresponding deprotected hydrogels), more cross-links to polymer parts other than the Boc group could be formed.
To look more closely into the aspect of the surprisingly high-energy doses and cross-linker contents needed for substantial cross-linking of the
P(4-5-BP) polymers,
P(485-510-BP5) and
P(495-BP5) were both irradiated with higher energy photons (λ = 254 nm) and different energy doses (
Figure 2c). Under these conditions, a significant amount of cross-linking was achieved even with an energy dose as low as 0.5 J·cm
−2 (
Figure 2c). For example, polymer
P(495-BP5) had a layer thickness of 211 nm and a gel content of 75% when irradiated with 0.5 J·cm
−2. However, when this polymer was irradiated with higher doses (up to 6 J·cm
−2), the layer thickness decreased to 20 nm and the gel content to only 8%. This effect was also observed for the copolymer
P(485-510-BP5) (
Figure 2c). This indicates that higher energy photons could, in principle, more efficiently cross-link these polymers. However, when too many photons of that energy were provided, unfavorable side reactions such as polymer main chain scissions, leading to lower gel content, become dominant. To confirm this, UV absorption spectra of
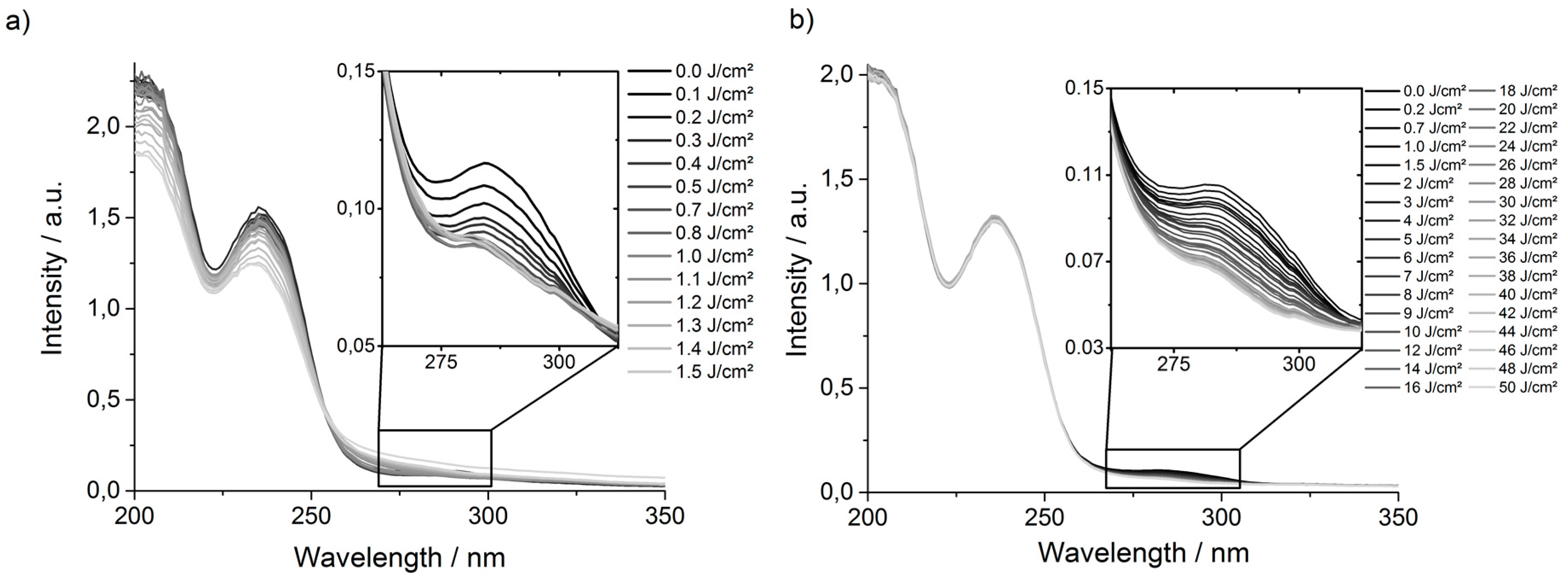
P(495-BP5) films that had been irradiated at 254 nm with different energy doses were measured (
Figure 3a).
These spectra showed that the UV absorption band of the BP group (at ~280 nm) decreased continuously when irradiated with energy doses from 0 to 0.5 J·cm
−2 at λ = 254 nm (
Figure 3a), i.e., the BP groups were quantitatively consumed in that energy range. When the samples were exposed to higher energy doses at that wavelength, the whole shape of the curve started to alter, particularly the peaks at 200 and 235 nm decreased significantly. This confirms that other reactions besides BP activation also occurred (
Figure 2c). It can be speculated that when the BP groups were used up, the 254 nm photons could interact with other functional groups, which could have led to cleavage of the polymer backbone. When the
P(495-BP5) films were irradiated with less energetic photons (λ = 365 nm), the UV absorption spectra did not show a decrease in the region of 200 to ~260 nm, even for energy doses as high as 50 J·cm
−2 (
Figure 3b). Instead, a steady decrease of the UV absorption band of the BP group at 280 nm with increasing energy dose was observed (
Figure 3b). Thus, these lower energy photons had a lower probability to trigger cross-linking via the BP group, and it therefore took higher energy doses to obtain similarly high gel contents as for the 254 nm photons. On the other hand, the 365 nm photons did not UV-activate the other functional groups in the polymer, so that the desired polymer networks were obtained.
A similar analysis was performed with the
P(2-3-BP) polymers (
Figure 1c). Polymer films were also obtained by spin-coating the respective polymers from solution onto
3EBP-functionalized substrates as described above for the
P(4-5-BP) samples. Again, the polymer films were cross-linked at wavelengths of 254 or 365 nm with defined energy doses. They were extracted in dichloromethane overnight to remove unattached polymer chains, and their gel content was determined. The thickness data obtained by ellipsometry for the different surface-attached
P(2-3-BP) networks and hydrogels are listed in the
supporting information (Tables S6 and S7) and summarized in
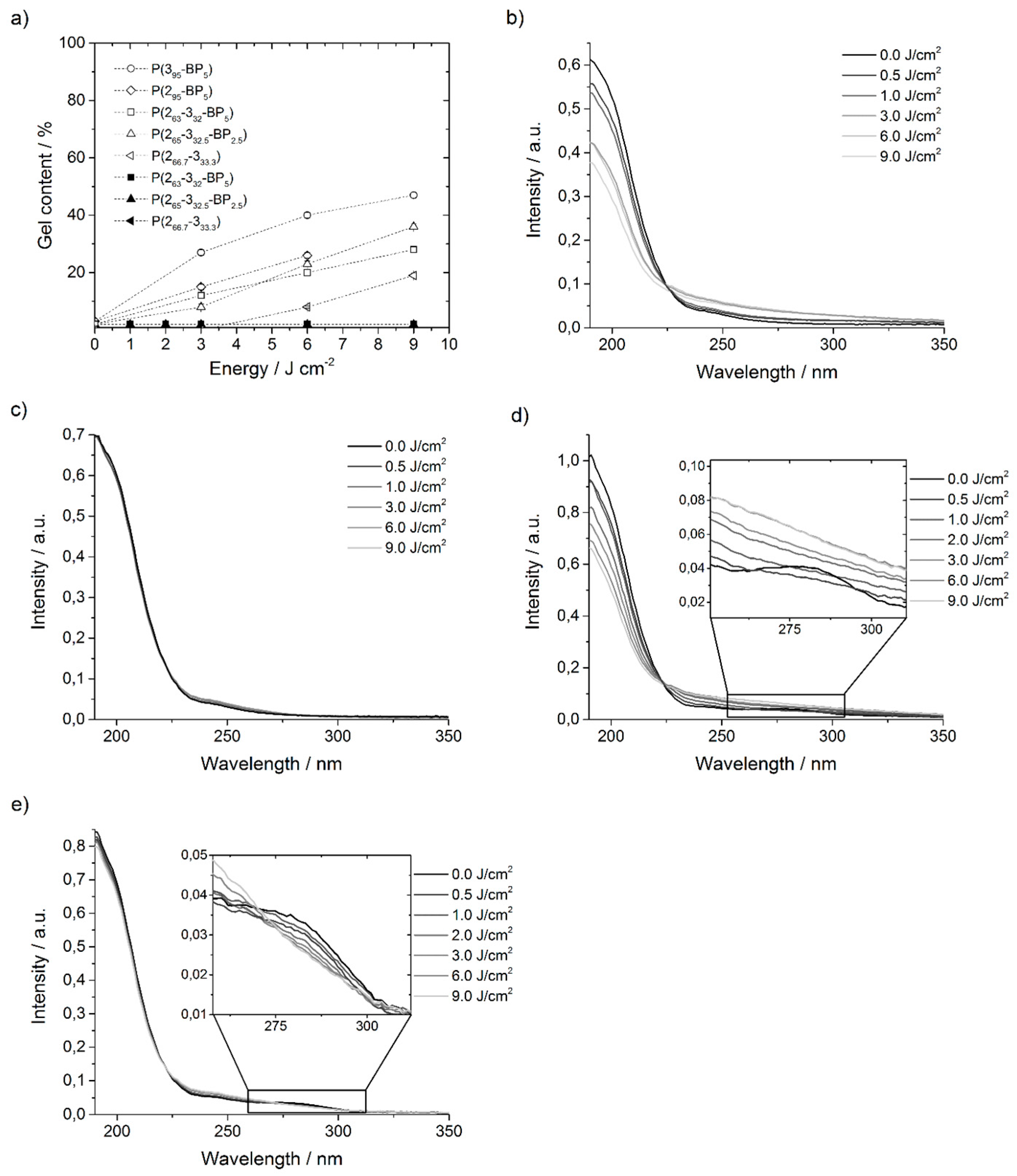
Figure 4a. (As before, the subscripts of these polymers indicate the relative amount of each repeat unit in the polymer, i.e.,
P(265-332.5-BP2.5) is a polymer made from 65%
M2, 32.5%
M3, and 2.5%
M1).
In contrast to the
P(4-5-BP) polymers, no networks formed when different
P(2-3-BP) polymers were irradiated at λ = 365 nm, regardless of the energy dose used (up to 9 J·cm
−2) or the amount of BP units in the polymer chain (
Figure 4a and
Table S6 in the Supporting Information). On the other hand, when the same experiments were repeated with higher energy photons at λ = 254 nm, gel contents of up to 47% were reached (
Figure 4a). Networks made from polymer
P(263-332-BP5) with 5% BP repeat units had a gel content that was similar to that of networks made from
P(265-332.5-BP2.5) with 2.5% BP repeat units, i.e., the number of BP units did not significantly affect the gel content. Interestingly, polymer
P(266.7-333.3) with no benzophenone groups self-crosslinked to a significant extent (19% gel content when irradiated with 9 J·cm
−2) at 254 nm, but not when irradiated at the same dose at 365 nm (
Figure 4a). To understand this, the network formation of
P(266.7-333.3) was also studied by recording UV spectra of films that had been irradiated at 254 nm (
Figure 4b) and 365 nm, respectively (
Figure 4c), and by comparing them to UV spectra of
P(265-332.5-BP2.5) that had been likewise treated (
Figure 4d,e). The spectra of
P(266.7-333.3) showed that the UV absorption bands at ~200 nm decreased from 0 to 9 J·cm
−2 when the sample was irradiated at 254 nm (
Figure 4b) and that the whole shape of the curve started to alter. Since at the same time a network formation was observed (
Figure 4a,
Table S7), this indicates a structural change of the polymer, which led to cross-linking. UV absorption at wavelengths around 200 nm are characteristic for an electron transition from the π-orbital to an antibonding π*-orbital of a C=C bond [
28]. Since there are many double bonds in the
P(2-3) polymer backbone, cross-linking can apparently occur also via those groups, presumably via a [2 + 2] cycloaddition involving the alkenyl group (which is not present in the corresponding poly(oxonorbornenes) that undergo chain cleavage when irradiated at 254 nm). When the same investigation was repeated with
P(266.7-333.3)-films irradiated at λ = 365 nm, the UV absorption spectra did not show any decrease of the peak at 200 nm (
Figure 4c), which matches the finding that no network formation was observed (
Figure 4a). This verifies the assumption that cross-linking occurred also via a reaction involving the alkenyl double bonds, for which the activation energy is provided by photons at λ = 254 nm, while irradiation at 365 nm is insufficient. The same set of experiments was performed with
P(265-332.5-BP2.5) films, which had a similar ratio of functional repeat units as
P(266.7-333.3), but additionally a small amount of BP groups. When irradiated at 365 nm, the UV absorption spectra of
P(265-332.5-BP2.5) only show a slight decrease of the peak at 200 nm, but a steady decrease of the UV absorption band of BP at ~280 nm with increasing energy dose (
Figure 4e). This indicates that the energy at λ = 365 nm was enough for the activation of the BP groups. However, thickness measurements showed that no network formed. When the
P(265-332.5-BP2.5) films were irradiated at 254 nm, the UV absorption band of the BP group (~280 nm) vanished already at 0.5 J·cm
−2 (
Figure 4d). Again, when the sample was irradiated with higher energy doses, the whole shape of the curve started to alter. Those results are consistent with the ones of the pure
P(266.7-333.3)-films irradiated at 254 nm, only the gel content and thus the degree of cross-linking was higher for
P(265-332.5-BP2.5). Thus, for the poly(alkenylnorbornenes) here investigated, network formation at λ = 254 nm occurs via a synergistic effect of BP-induced cross-linking and [2 + 2] cycloaddition. This finding also explains why
P(395-BP5) cross-linked more efficiently than
P(295-BP5) (
Figure 4a): the fully substituted double bond of
P(295-BP5) is sterically more hindered than the only triply substituted one of
P(395-BP5), and thus
P(295-BP5) has a lower propensity to undergo a [2 + 2] cycloaddition.
When the Boc protective groups of the
P(2-3-BP) polymers were removed with hydrochloric acid to obtain surface-attached hydrogels, this treatment near-quantitatively destroyed the network (see
Table S7 of the supporting information). For example, the polymer network consisting of
P(395-BP5) had a layer thickness of 49 nm after irradiation with 9 J·cm
−2 and extraction, which decreased after deprotection to a thickness of only 5 nm. This suggested that in the
P(2-3-BP) networks, most of the cross-links formed via the C–H groups in the Boc groups. To confirm this, polymer
P(265-332.5-BP2.5) was first deprotected and then spin coated on a
3EBP-functionalized wafer. After illumination at λ = 245 nm with 9 J·cm
−2, a surface-attached hydrogel with a gel content of 60% was formed, while the polymer coating made from the protected version of this polymer, treated under the same conditions, yielded only a gel content of 36%. This indicated that the Boc protective groups sterically shielded the other C–H bonds of the polymer, so that BP cross-linking mainly occurred with the C–H bonds in these protective group.
In general, the ability of a polymer to effectively attach to a substrate by cross-linking depends on the exact circumstances of the network or hydrogel formation process. Körner et al. used a simple percolation model to describe the formation of polymer networks as a function of the reaction conditions and the reaction time [
29]. In this model, polymers first form gel clusters inside the polymer layer, and a network is obtained once these clusters percolate through the entire polymer film. For UV irradiation of polymer films from above (e.g., using poly(styrene) containing additional BP repeat units, cross-linked at λ = 365 nm), it was observed that there was a long induction period before any macroscopic network formation was detected, as the formation of the clusters started at the air-polymer interface, and slowly percolated down to the polymer-substrate interface. In this particular case, a significant amount of network formation occurred only at an energy dose of >10 J·cm
−2 (corresponding to 100 min irradiation time at 2.2 mW·cm
−2) [
29]. Thus, it is possible that for polymers of the
P(2-3-BP) family, such a gel cluster formation is also very slow, so that no network percolation is observed at 365 nm in the energy range investigated. When irradiated with 254 nm, however, these polymers formed surface-attached polymer networks, albeit with a low gel content, and with synergistic BP cross-linking and double-bond-induced cross-linking. The poly(oxonorbornene) family
P(4-5-BP), on the other hand, degraded when irradiated at 254 nm at energy doses higher than 0.5 J·cm
−2. Unlike
P(2-3-BP), those polymers did not have the additional alkenyl substituent on the backbone. This double bond, however, seems to be a protection against main chain cleavage at λ = 254 nm, since it can absorb the high-energy photons via a π → π* transition. Additionally, this double bond assists network formation by its ability to self-crosslink at that wavelength. At 365 nm, on the other hand, the cross-linking behavior of the
P(4-5-BP) copolymers was textbook-like, and with higher energy doses higher gel contents were reached. The finding that the
P(4-5-BP) polymers undergo chain scission by photons having a wavelength of 254 nm is in line with the results of Christensen et al., who previously observed that highly abstractable hydrogens especially on the main chain tend to cause chain scission [
19].
Another factor affecting the reactivity of a certain C–H bond in C–H insertion reactions, besides their electronic activation, is their steric availability. C–H abstraction can be sterically hindered by flexible, large side groups that can shield the C–H bonds near the polymer backbone [
15]. For the polymers used in this study, the C–H bonds in the Boc group and the C–H bonds in the alkyl or alkenyl substituents were the sterically most easily accessible ones. In the norbornene polymers of the
P(2-3-BP) family, the Boc groups effectively shielded the C–H bonds near the backbone, so that cross-linking via the BP units mainly occurred via the Boc group. As a result, these networks disintegrated when the Boc groups were cleaved off. Only when the Boc groups were removed before the network formation, the C–H groups nearer to the backbone were significantly involved in the cross-linking process. In case of the poly(oxanorbornenes)
P(4-5-BP) family, on the other hand, BP could abstract hydrogen atoms from either the Boc groups or from the backbone. While the C–H bonds on the Boc group had less steric hindrance, the C–H bonds next to the oxygen and nitrogen atoms could be readily abstracted due to their lower bond energies, despite the higher steric hindrance.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}