The Effect of Hydrogen Bonding on Radical Semi-Batch Copolymerization of Butyl Acrylate and 2-Hydroxyethyl Acrylate


Abstract
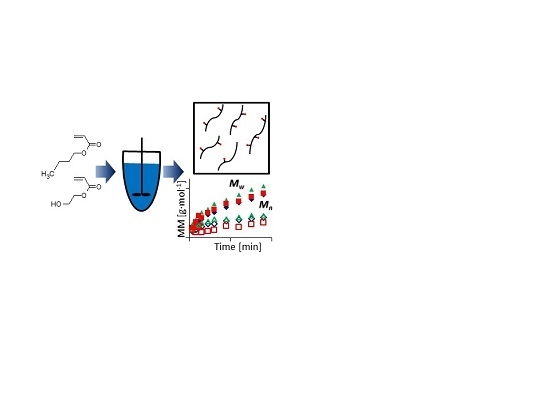
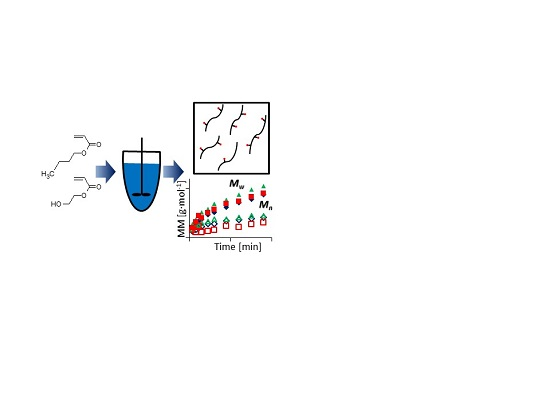
:
1. Introduction
2. Materials and Methods
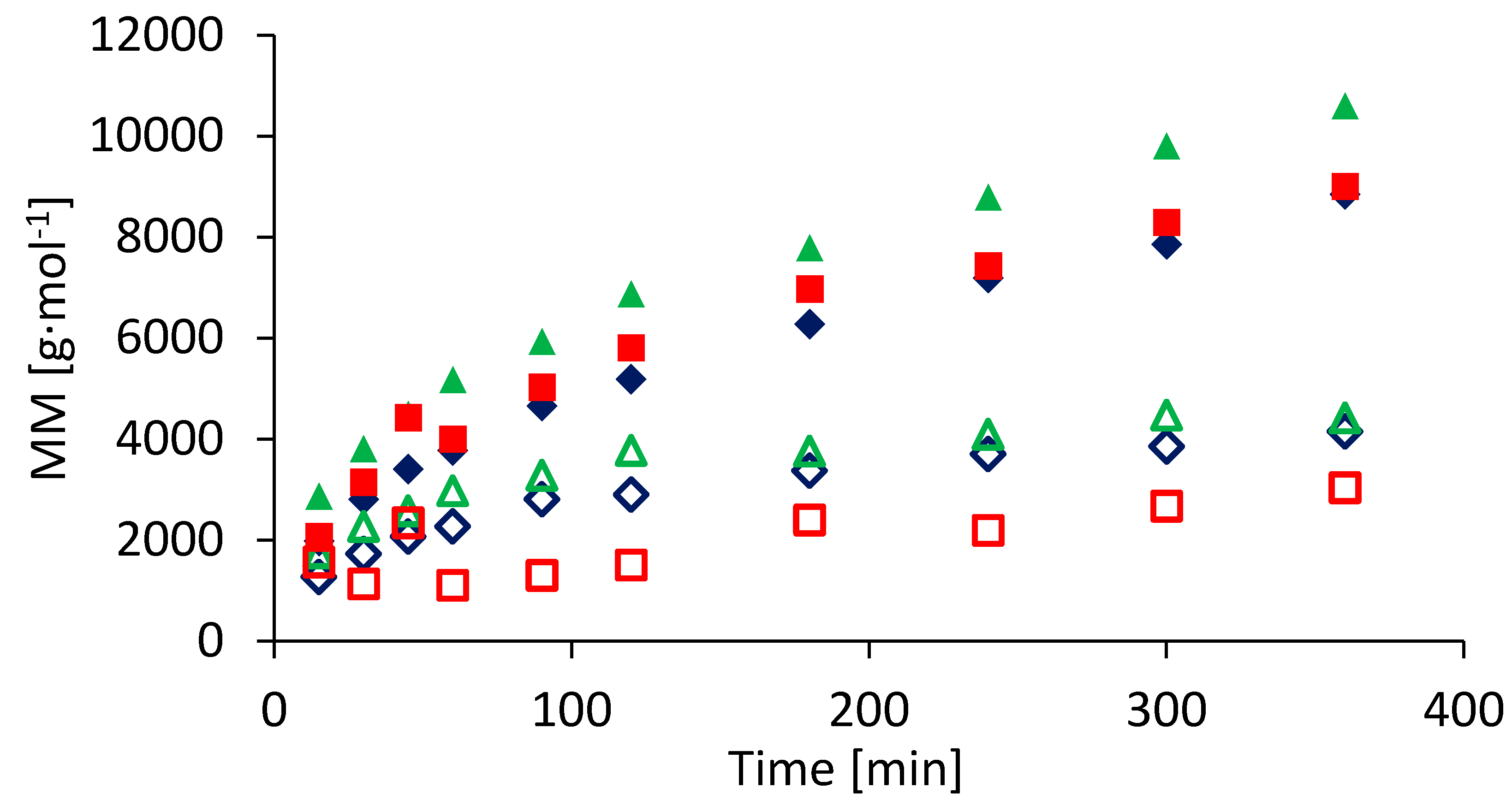
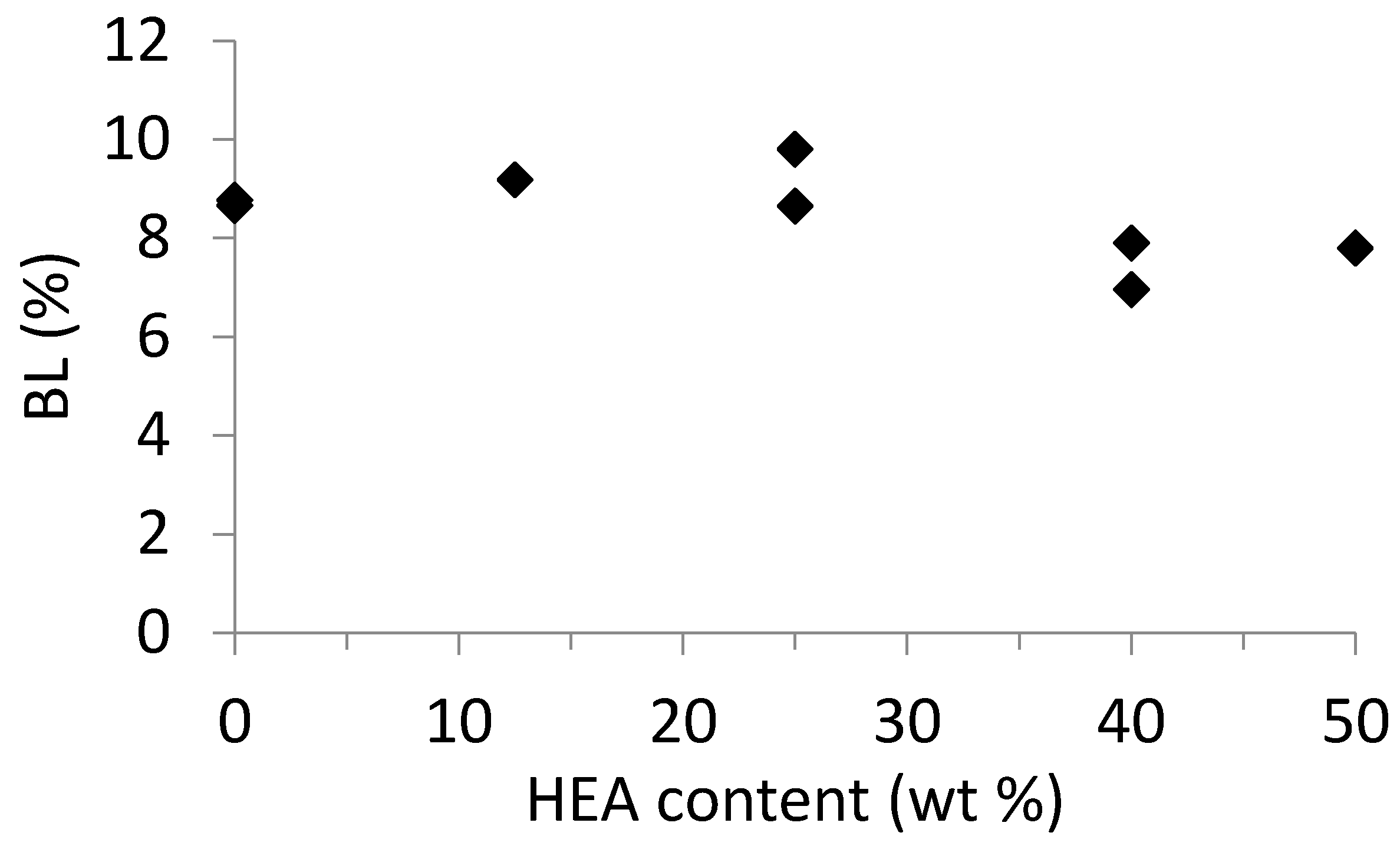
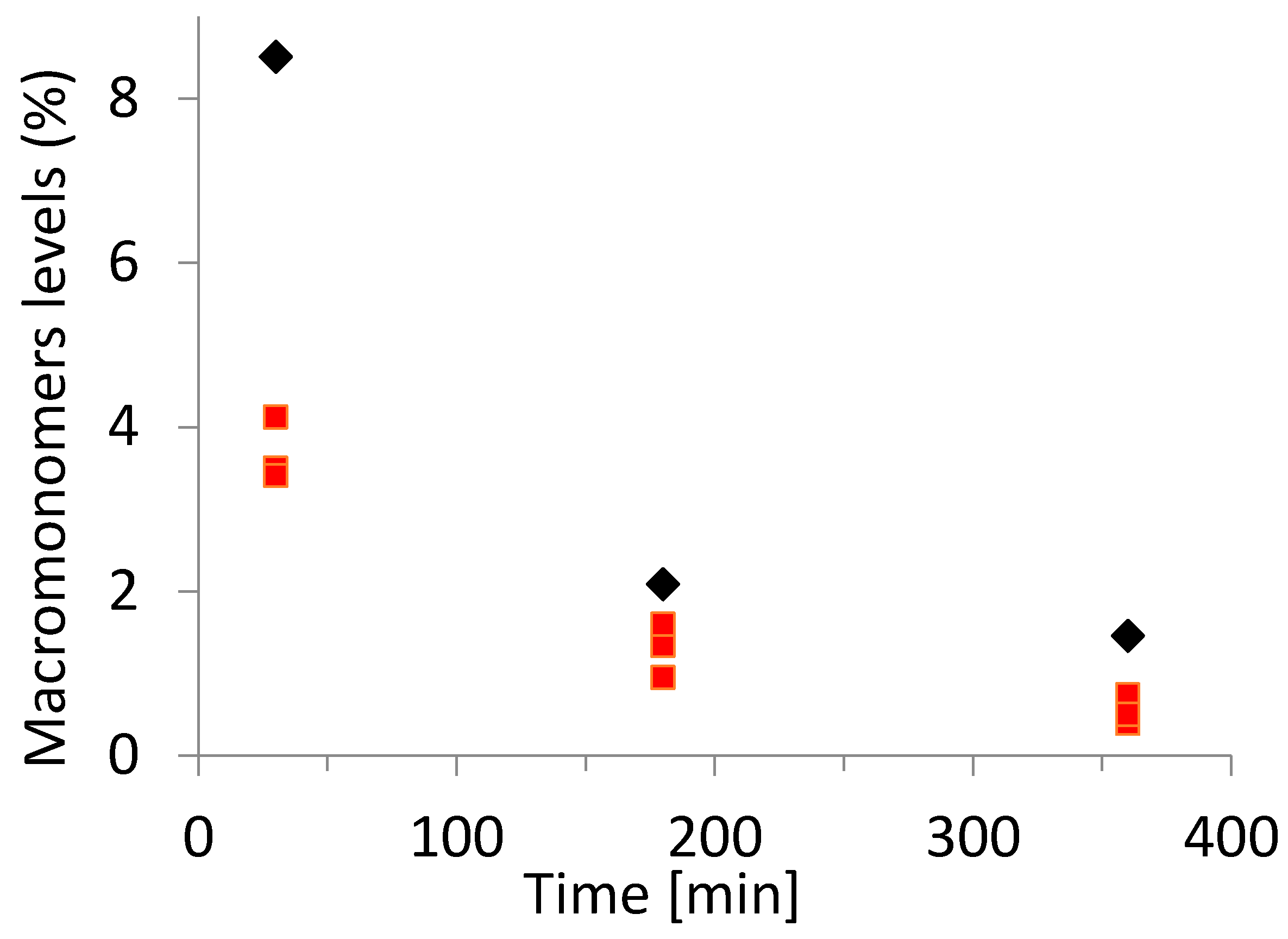
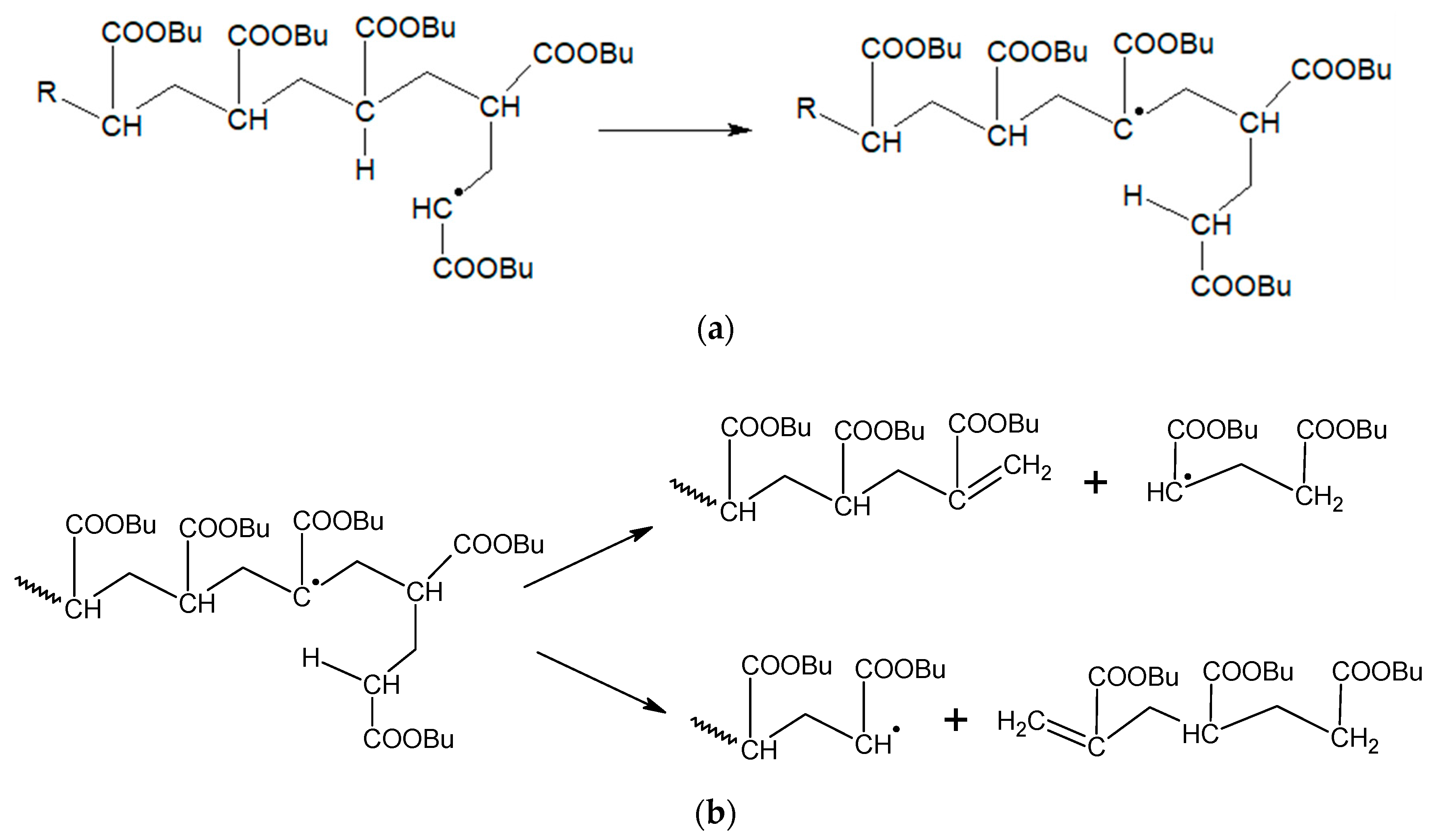
3. Results and Discussion
3.1. Batch Experiments
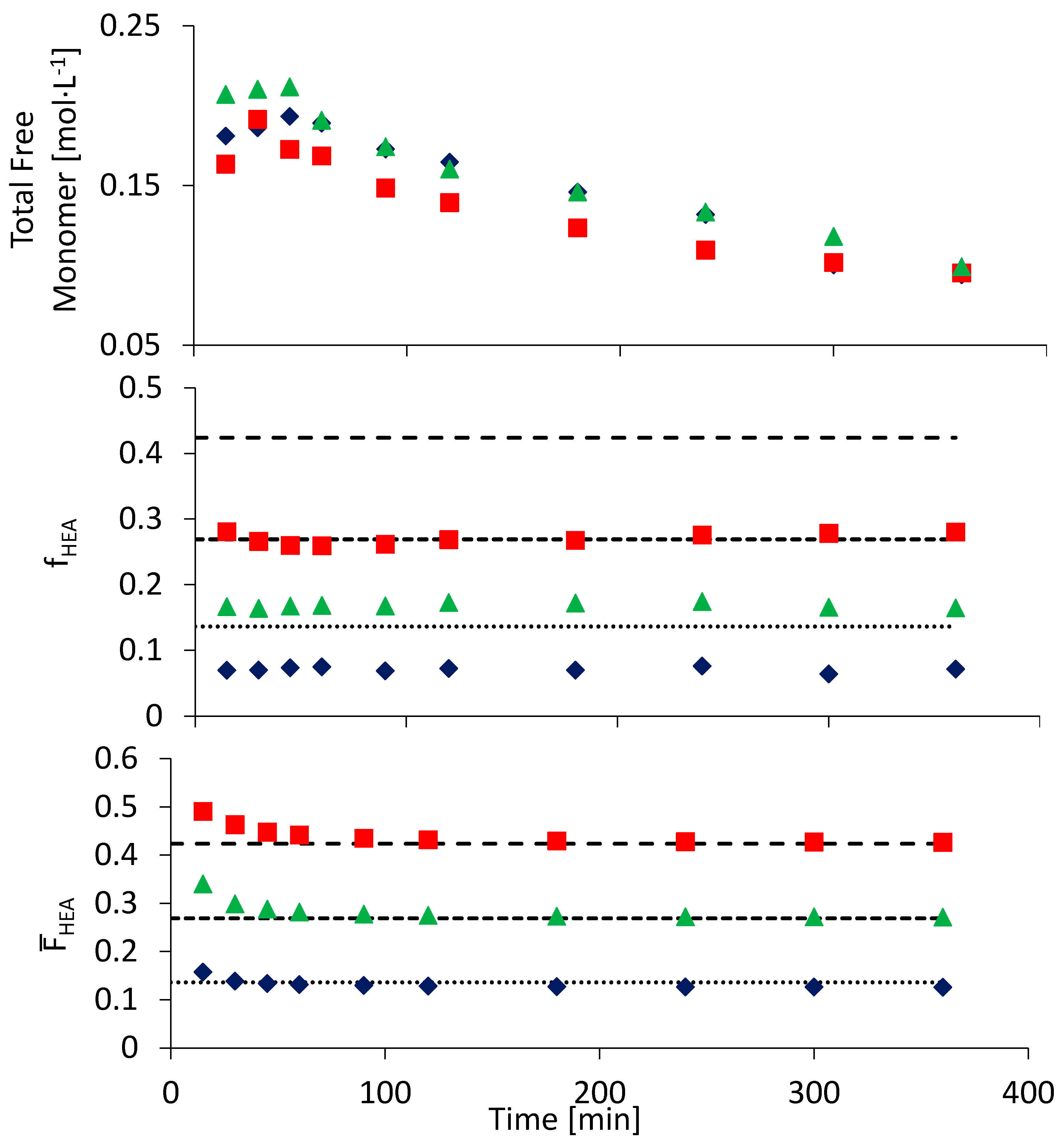
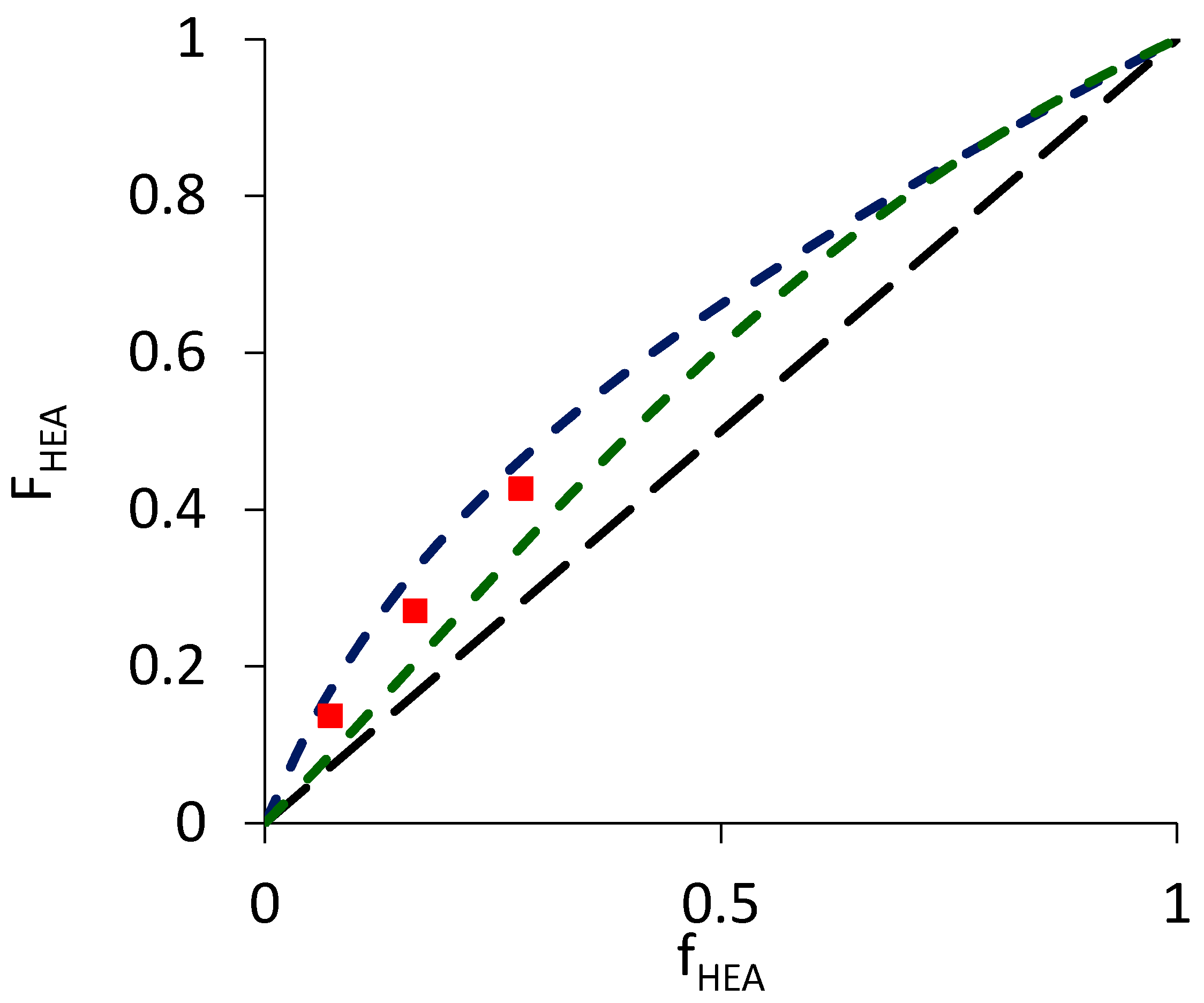
3.2. Semi-Batch Copolymerization of BA and HEA in Various Solvents
3.3. Acrylate Backbiting under Semi-Batch Conditions
4. Conclusions and Outlook
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Noble, B.B.; Coote, M.L. First principles modelling of free-radical polymerisation kinetics. Int. Rev. Phys. Chem. 2013, 32, 467–513. [Google Scholar] [CrossRef]
- Wang, J.S.; Matyjaszewski, K. Controlled “living” Radical Polymerization. Atom Transfer Radical Polzmerization in the Presenece of Transition-Metal Complexes. J. Am. Chem. Soc. 1995, 117, 5614–5615. [Google Scholar] [CrossRef]
- Chiefari, J.; Chong, Y.K.; Ercole, F.; Krstina, J.; Jeffery, J.; Le, T.P.T.; Mayadunne, R.T.A.; Meijs, G.F.; Moad, C.L.; Moad, G.; et al. Living Free-Radical Polymerization by Reversible Addition-Fragmentation Chain Transfer: The RAFT Process. Macromolecules 1998, 31, 5559–5562. [Google Scholar] [CrossRef]
- Hawker, C.J.; Barclay, G.G.; Orellana, A.; Dao, J.; Devonport, W. Initiating Systems for Nitroxide-Mediated “Living” Free Radical Polymerizations: Synthesis and Evaluation. Macromolecules 1996, 29, 5245–5254. [Google Scholar] [CrossRef]
- Szwarc, M. “Living” Polymers. Nature 1956, 178, 1168–1169. [Google Scholar] [CrossRef]
- Mülhaupt, R. Catalytic Polymerization and Post Polymerization Catalysis Fifty Years Ater the Discovery of Ziegler’s Catalyst. Macromol. Chem. Phys. 2003, 204, 289–327. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Davis, T.P. Handbook of Radical Polymerization; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2002. [Google Scholar]
- Grady, M.C.; Simonsick, W.J.; Hutchinson, R.A. Studies of higher temperature polymerization of n-butyl methacrylate and n-butyl arylate. Macromol. Symp. 2002, 182, 149–168. [Google Scholar] [CrossRef]
- Uhelská, L.; Chorvát, D.; Hutchinson, R.A.; Santanakrishnan, S.; Buback, M.; Lacík, I. Radical Propagation Kinetics of N-Vinylpyrrolidone in Organic Solvents Studied by Pulsed-Laser Polymerization-Size-Exclusion Chromatography (PLP-SEC). Macromol. Chem. Phys. 2014, 215, 2327–2336. [Google Scholar] [CrossRef]
- Lebduška, J.; Šnupárek, J.; Kašpar, K.; Čermák, V. Solution copolymerization of styrene and 2-hydroxyethyl mechacrylate. J. Polym. Sci. A 1986, 24, 777–791. [Google Scholar] [CrossRef]
- O’Driscoll, K.F.; Davis, T.P.; Klumperman, B.; Madruga, E.L. Solvent effects in copolymerization. Macromol. Rapid Commun. 1995, 16, 207–210. [Google Scholar] [CrossRef]
- Liang, K.; Rooney, T.R.; Hutchinson, R.A. Solvent Effects on Kinetics of 2-Hydroxyethyl Methacrylate Semibatch Radical Copolymerization. Ind. Eng. Chem. Res. 2014, 53, 7296–7304. [Google Scholar] [CrossRef]
- Schier, J.E.S.; Hutchinson, R.A. The influence of hydrogen bonding on radical chain-growth parameters for butyl methacrylate/2-hydroxyethyl acrylate solution copolymerization. Polym. Chem. 2016, 7, 4567–4574. [Google Scholar] [CrossRef]
- Schier, J.E.S.; Cohen-Sacal, D.; Hutchinson, R.A. Hydrogen bonding in radical solution copolymerization kinetics of acrylates and methacrylates: A comparison of hydroxy- and methoxy- functionality. Polym. Chem. 2017, 8, 1943–1952. [Google Scholar] [CrossRef]
- Ahmad, N.M.; Heatley, F.; Lovell, P.A. Chain Transfer to Polymer in Free-Radical Solution Polymerization of n-Butyl Acrylate Studied by NMR Spectroscopy. Macromolecules 1998, 31, 2822–2827. [Google Scholar] [CrossRef]
- González, I.; Asua, J.M.; Leiza, J.R. The role of methyl methacrylate on branching and gel formation in the emulsion copolymerization of BA/MMA. Polymer 2007, 48, 2542–2547. [Google Scholar] [CrossRef]
- Peck, A.N.F.; Hutchinson, R.A. Secondary reactions in the high-temperature free radical polymerization of butyl acrylate. Macromolecules 2004, 37, 5944–5951. [Google Scholar] [CrossRef]
- Junkers, T.; Barner-Kowollik, C. The Role of Mid-Chain Radicals in Acrylate Free Radical Polymerization: Branching and Scission. J. Polym. Sci. A 2008, 46, 7585–7605. [Google Scholar] [CrossRef]
- Nikitin, A.N.; Hutchinson, R.A.; Buback, M.; Hesse, P. Determination of Intramolecular Chain Transfer and Midchain Radical Propagation Rate Coefficients for Butyl Acrylate by Pulsed Laser Polymerization. Macromolecules 2007, 40, 8631–8641. [Google Scholar] [CrossRef]
- Barth, J.; Buback, M.; Hesse, P.; Sergeeva, T. Termination and Transfer Kinetics of Butyl Acrylate Radical Polymerization Studied via SP-PLP-EPR. Macromolecules 2010, 43, 4023–4031. [Google Scholar] [CrossRef]
- Nikitin, A.N.; Hutchinson, R.A. The Effect of Intramolecular Transfer to Polymer on Stationary Free Radical Polymerization of Alkyl Acrylates. Macromolecules 2005, 38, 1581–1590. [Google Scholar] [CrossRef]
- Wang, W.; Nikitin, A.N.; Hutchinson, R.A. Consideration of Macromonomer Reactions in n-Butyl Acrylate Free Radical Polymerization. Macromol. Rapid Commun. 2009, 30, 2022–2027. [Google Scholar] [CrossRef] [PubMed]
- Plessis, C.; Arzamendi, G.; Leiza, J.R.; Schoonbrood, H.A.S.; Charmot, D.; Asua, J.M. Seeded Semibatch Emulsion Polymerization of n-Butyl Acrylate. Kinetics and Structural Properties. Macromolecules 2000, 33, 5041–5047. [Google Scholar] [CrossRef]
- Asua, J.M.; Beuermann, S.; Buback, M.; Castignolles, P.; Charleux, B.; Gilbert, R.G.; Hutchinson, R.A.; Leiza, J.R.; Nikitin, A.N.; Vairon, J.-P.; et al. Critically Evaluated Rate Coefficients for Free-Radical Polymerization: 5. Propagation Rate Coefficient for Butyl Acrylate. Macromol. Chem. Phys. 2004, 205, 2151–2160. [Google Scholar]
- Nikitin, A.N.; Hutchinson, R.A.; Kalfas, G.A.; Richards, J.R.; Bruni, C. The Effect of Intramolecular Transfer to Polymer on Stationary Free-Radical Polymerization of Alkyl Acrylates, 3—Consideration of Solution Polymerization up to High Conversions. Macromol. Theory Simul. 2009, 18, 247–258. [Google Scholar] [CrossRef]
- Nikitin, A.N.; Hutchinson, R.A.; Wang, W.; Kalfas, G.A.; Richards, J.R.; Bruni, C. Effect of Intramolecular Transfer to Polymer on Stationary Free-Radical Polymerization of Alkyl Acrylates, 5—Consideration of Solution Polymerization up to High Temperatures. Macromol. React. Eng. 2010, 4, 691–706. [Google Scholar] [CrossRef]
- Wang, W.; Hutchinson, R.A. A comprehensive kinetic model for high-temperature free radical production of styrene/methacrylate/acrylate resins. AIChE J. 2011, 57, 227–238. [Google Scholar] [CrossRef]
- Hamzehlou, S.; Ballard, N.; Reyes, Y.; Aguirre, A.; Asua, J.M.; Leiza, J.R. Analyzing the discrepancies in the activation energies of the backbiting and β-scission reactions in the radical polymerization of n-butyl acrylate. Polym. Chem. 2016, 7, 2069–2077. [Google Scholar] [CrossRef]
- Wagner, M.H. The Rheology of Linear and Long-chain Branched Polymer Melts. Macromol. Symp. 2006, 236, 219–227. [Google Scholar] [CrossRef]
- Kontopoulou, M. Applied Polymer Rheology: Polymeric Fluids with Industrial Applications; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011. [Google Scholar] [CrossRef]
- Stadler, F.J.; Mahmoudi, T. Understanding the effect of short-chain branches by analyzing viscosity functions of linear and short-chain branched polyethylenes. Korea Aust. Rheol. J. 2011, 23, 185–193. [Google Scholar] [CrossRef]
- Liang, K.; Hutchinson, R.A.; Barth, J.; Samrock, S.; Buback, M. Reduced Branching in Poly(butyl acrylate) via Solution Radical Polymerization in n-Butanol. Macromolecules 2011, 44, 5843–5845. [Google Scholar] [CrossRef]
- Liang, K.; Hutchinson, R.A. The effect of hydrogen bonding on intramolecular chain transfer in polymerization of acrylates. Macromol. Rapid Commun. 2011, 32, 1090–1095. [Google Scholar] [CrossRef] [PubMed]
- Mavroudakis, E.; Liang, K.; Moscatelli, D.; Hutchinson, R.A. A combined computational and experimental study on the free-radical copolymerization of styrene and hydroxyethyl acrylate. Macromol. Chem. Phys. 2012, 213, 1706–1716. [Google Scholar] [CrossRef]
- Schier, J.E.S. Hydrogen Bonding in Radical Copolymerization: A Kinetic Investigation under Industrially Relevant Conditions. Ph.D. Thesis, Queen’s University, Kingston, ON, Canada, 2017. [Google Scholar]
- Beuermann, S. Impact of Hydrogen Bonding on Propagation Kinetics in Butyl Methacrylate Radical Polymerizations. Macromolecules 2004, 37, 1037–1041. [Google Scholar] [CrossRef]
- Liang, K. Free Radical Copolymerization of Hydroxy-Functional Monomers: Kinetic and Semibatch Studies. Ph.D. Thesis, Queen’s University, Kingston, ON, Canada, 2013. [Google Scholar]
- Liang, K.; Hutchinson, R.A. Solvent Effects on Free-Radical Copolymerization Propagation Kinetics of Styrene and Methacrylates. Macromolecules 2010, 43, 6311–6320. [Google Scholar] [CrossRef]
- Buback, M.; Schroeder, H.; Kattner, H. Detailed Kinetic and Mechanistic Insight into Radical Polymerization by Spectroscopic Techniques. Macromolecules 2016, 49, 3193–3213. [Google Scholar] [CrossRef]
- Barner-Kowollik, C.; Russell, G.T. Chain-length-dependent termination in radical polymerization: Subtle revolution in tackling a long-standing challenge. Prog. Polym. Sci. 2009, 34, 1211–1259. [Google Scholar] [CrossRef]
- Hansen, C.M. Hansen Solubility Parameters: A User’s Handbook; CRC Press: Boca Raton, FL, USA, 2007; ISBN 9780849372483. [Google Scholar]
- Jeličič, A.; Köhler, F.; Winter, A.; Beuermann, S. On the origin of ionic liquid-induced variations in hydroxypropyl methacrylate propagation rate coefficients. J. Polym. Sci. A 2010, 48, 3188–3199. [Google Scholar] [CrossRef]
- Moghadam, N.; Srinivasan, S.; Grady, M.C.; Rappe, A.M.; Soroush, M. Theoretical Study of Chain Transfer to Solvent Reactions of Alkyl Acrylates. J. Phys. Chem. A 2014, 118, 5474–5487. [Google Scholar] [CrossRef] [PubMed]
- Magee, C.; Sugihara, Y.; Zetterlund, P.B.; Aldabbagh, F. Chain transfer to solvent in radical polymerization of structurally diverse acrylamide monomers using straight-chain and branched alcohols as solvents. Polym. Chem. 2014, 5, 2259–2265. [Google Scholar] [CrossRef]
- Sugihara, Y.; O’Connor, P.; Zetterlund, P.B.; Aldabbagh, F. Chain Transfer to Solvent in the Radical Polymerization of N-Isopropylacrylamide. J. Polym. Sci. A 2011, 49, 1856–1864. [Google Scholar] [CrossRef]
- Mendrek, B.; Trzebicka, B.; Walach, W.; Dworak, A. Solution behaviour of 4-arm Poly(tert-butyl acrylate)star polymers. Eur. Polym. J. 2010, 46, 2341–2351. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Monomer | dn/dc (mL⋅g−1) | Mark-Houwink Parameters in THF | |
|---|---|---|---|
| K (dL⋅g−1) | a | ||
| HEA [34] | 0.066 | 0.000322 | 0.602 |
| BA [24] | 0.064 | 0.000122 | 0.700 |
| Styrene [34] | 0.184 | 0.000114 | 0.716 |
| Monomer | Solvent | [M]ini (mol∙L−1) | Conversion (%) | Mn/Mw (g∙mol−1) | Branching Level b (% per Acrylate Unit) |
|---|---|---|---|---|---|
| BA | Xylenes | 1.65 | 80.7 | 21,100/41,100 | 3.1 |
| BA | Toluene | 1.65 | 75.9 | 17,100/39,500 | 3.0 |
| BA | n-Butanol | 1.65 | 84.2 | 36,800/87,700 | 2.4 |
| HEA | DMF | 2.25 | 84.8 | n.a. | 1.8 |
| HEA | n-Butanol | 2.25 | 90.0 | n.a | 1.2 |
| BA/HEAa | Xylenes | 1.90 | Phase separation/precipitation occurred before 30 min; FHEA >> fHEA at conv. ≈ 50% | 1.2 | |
| BA/HEAa | MIBK | 1.90 | 1.8 | ||
| Comonomer Feed (wt %) and Solvent | FHEA a | fHEA a | Mw b (g∙mol−1) | BL b | ∫[M] c (mol∙L−1) | kbb/kpcop (L∙mol−1) |
|---|---|---|---|---|---|---|
| BA (100) BPi | 0 | 0 | 8600 | 0.088 | 0.203 | 1.95 × 10−2 |
| BA/HEA (87.5/12.5) BPi | 0.13 | 0.07 | 8900 | 0.092 | 0.141 | 1.43 × 10−2 |
| BA/HEA (75/25) BPi | 0.27 | 0.17 | 9000 | 0.087 | 0.146 | 1.38 × 10−2 |
| BA/HEA (60/40) BPi | 0.43 | 0.28 | 10,600 | 0.092 | 0.125 | 1.27 × 10−2 |
| BA/HEA (75/25) MIAK | 0.27 | 0.16 | 7100 | 0.098 | 0.151 | 1.64 × 10−2 |
| BA/HEA (75/25) xylenes | 0.27 | 0.15 | 7600 | 0.072 | 0.193 | 1.49 × 10−2 |
| BA PeOH | 0 | 0 | 3900 | 0.064 | 0.113 | 0.78 × 10−2 |
| BA/HEA (75/25) PeOH | 0.27 | 0.14 | 4400 | 0.067 | 0.152 | 1.09 × 10−2 |
| BA/HEA (50/50) PeOH | 0.53 | 0.32 | 3400 | 0.071 | 0.110 | 0.84 × 10−2 |
| BA/HEA (75/25) DMF | 0.27 | 0.20 | 4000 | 0.095 | 0.111 | 1.17 × 10−2 |
| BA/HEA (50/50) DMF | 0.53 | 0.41 | 3800 | 0.093 | 0.099 | 1.01 × 10−2 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schier, J.E.S.; Cohen-Sacal, D.; Larsen, O.R.; Hutchinson, R.A. The Effect of Hydrogen Bonding on Radical Semi-Batch Copolymerization of Butyl Acrylate and 2-Hydroxyethyl Acrylate. Polymers 2017, 9, 368. https://doi.org/10.3390/polym9080368
Schier JES, Cohen-Sacal D, Larsen OR, Hutchinson RA. The Effect of Hydrogen Bonding on Radical Semi-Batch Copolymerization of Butyl Acrylate and 2-Hydroxyethyl Acrylate. Polymers. 2017; 9(8):368. https://doi.org/10.3390/polym9080368
Chicago/Turabian StyleSchier, Jan E. S., David Cohen-Sacal, Owen R. Larsen, and Robin A. Hutchinson. 2017. "The Effect of Hydrogen Bonding on Radical Semi-Batch Copolymerization of Butyl Acrylate and 2-Hydroxyethyl Acrylate" Polymers 9, no. 8: 368. https://doi.org/10.3390/polym9080368
APA StyleSchier, J. E. S., Cohen-Sacal, D., Larsen, O. R., & Hutchinson, R. A. (2017). The Effect of Hydrogen Bonding on Radical Semi-Batch Copolymerization of Butyl Acrylate and 2-Hydroxyethyl Acrylate. Polymers, 9(8), 368. https://doi.org/10.3390/polym9080368