
Transcriptome Sequencing Reveals Key Genes for Sunflower Unsaturated Fatty Acid Synthesis


Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Design and Seed Sampling
2.2. FA Content Determination
2.3. RNA Isolation and Library Construction
2.4. Transcriptome Sequencing and Principal Component Analysis (PCA)
2.5. Reference Transcriptome, Quantification of Gene Expression Levels and Screening of DEGs
2.6. GO and KEGG Enrichment Analysis of DEGs
2.7. Quantitative Real-Time PCR (qRT-PCR) Analysis
3. Results
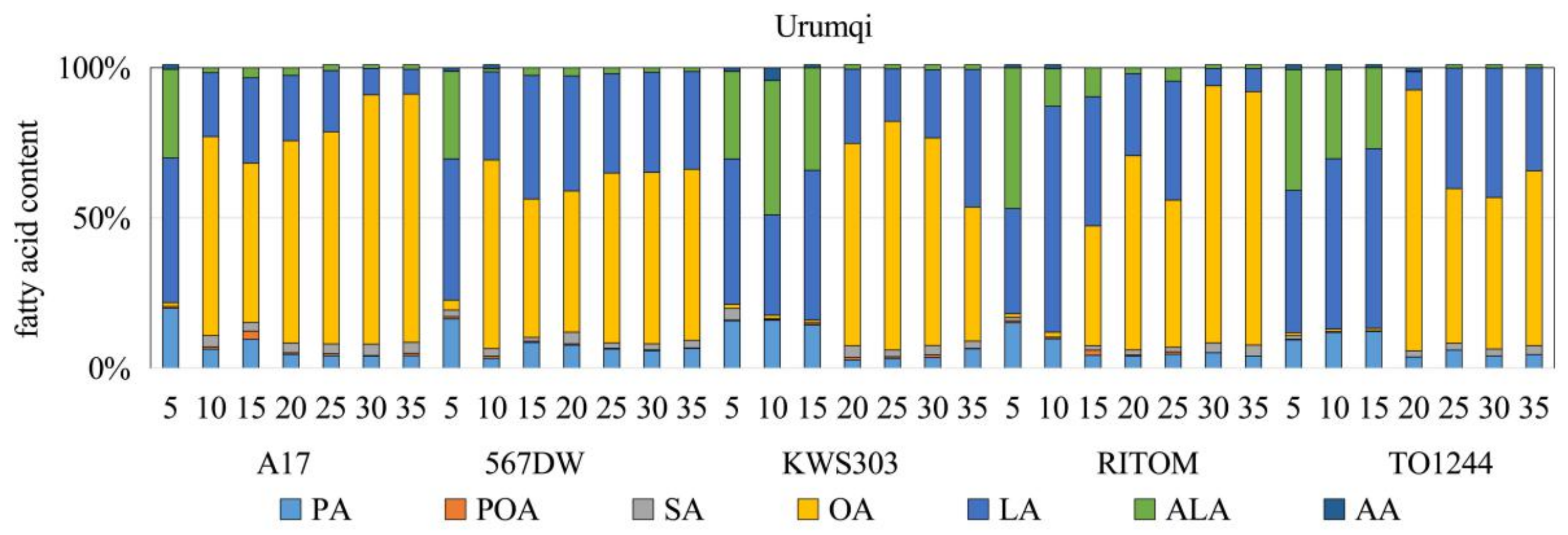
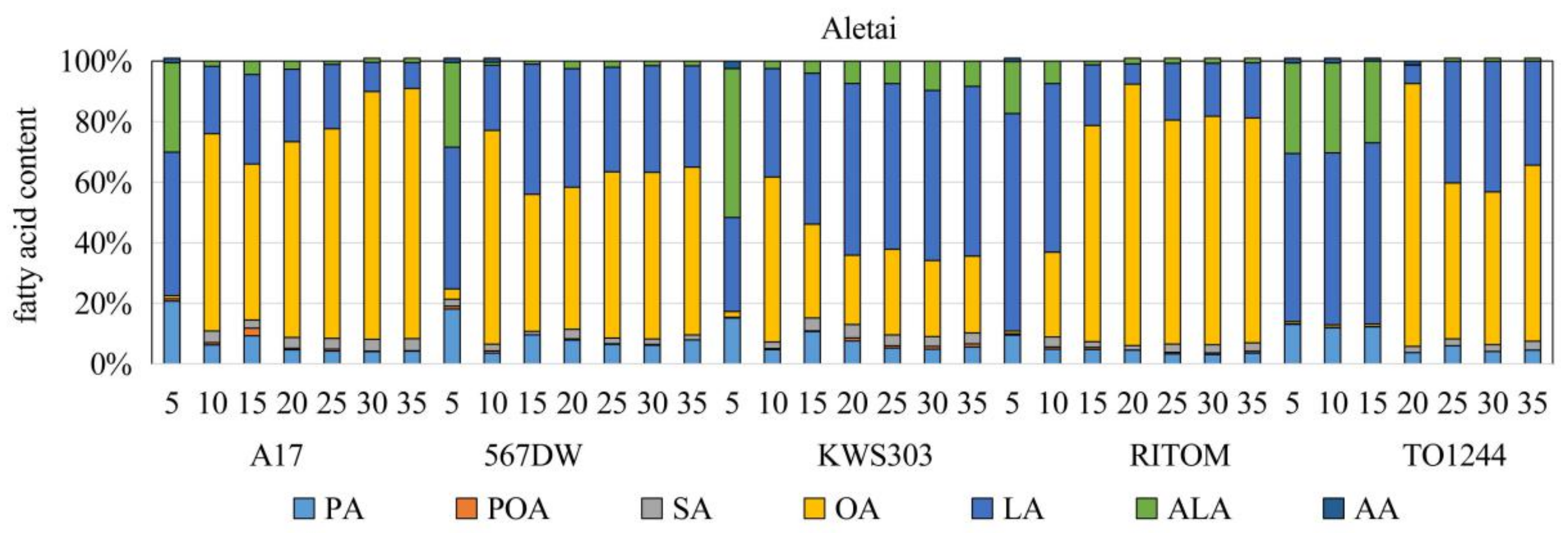
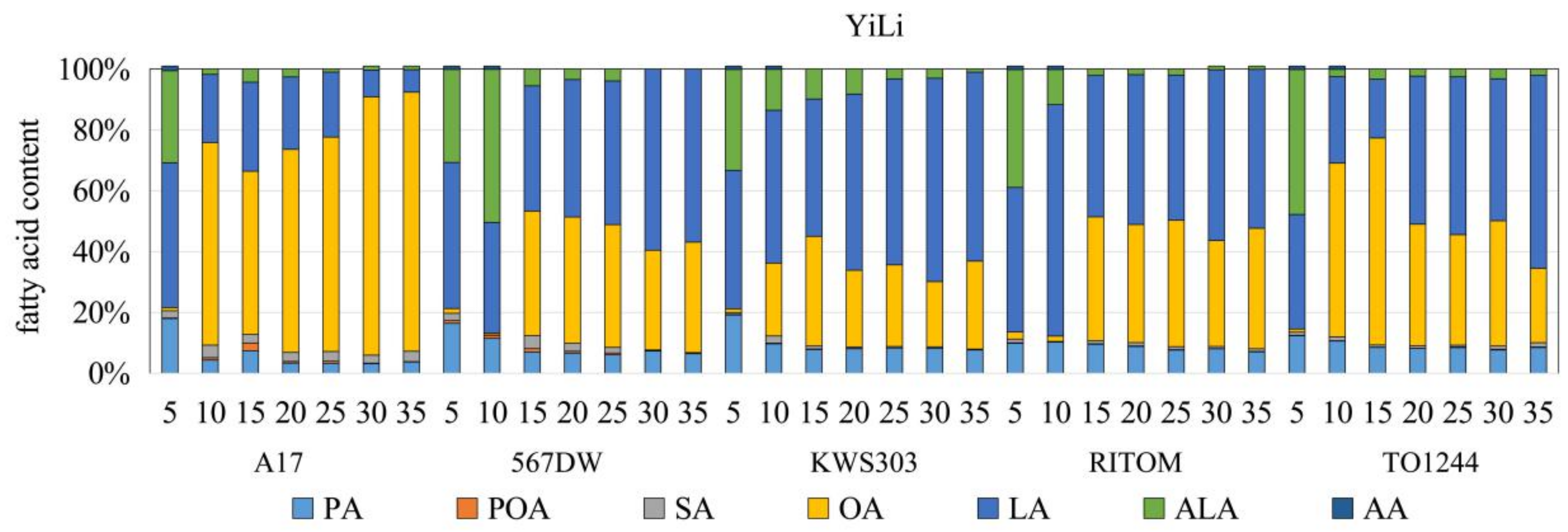
3.1. Analysis of the FA Composition and Contents
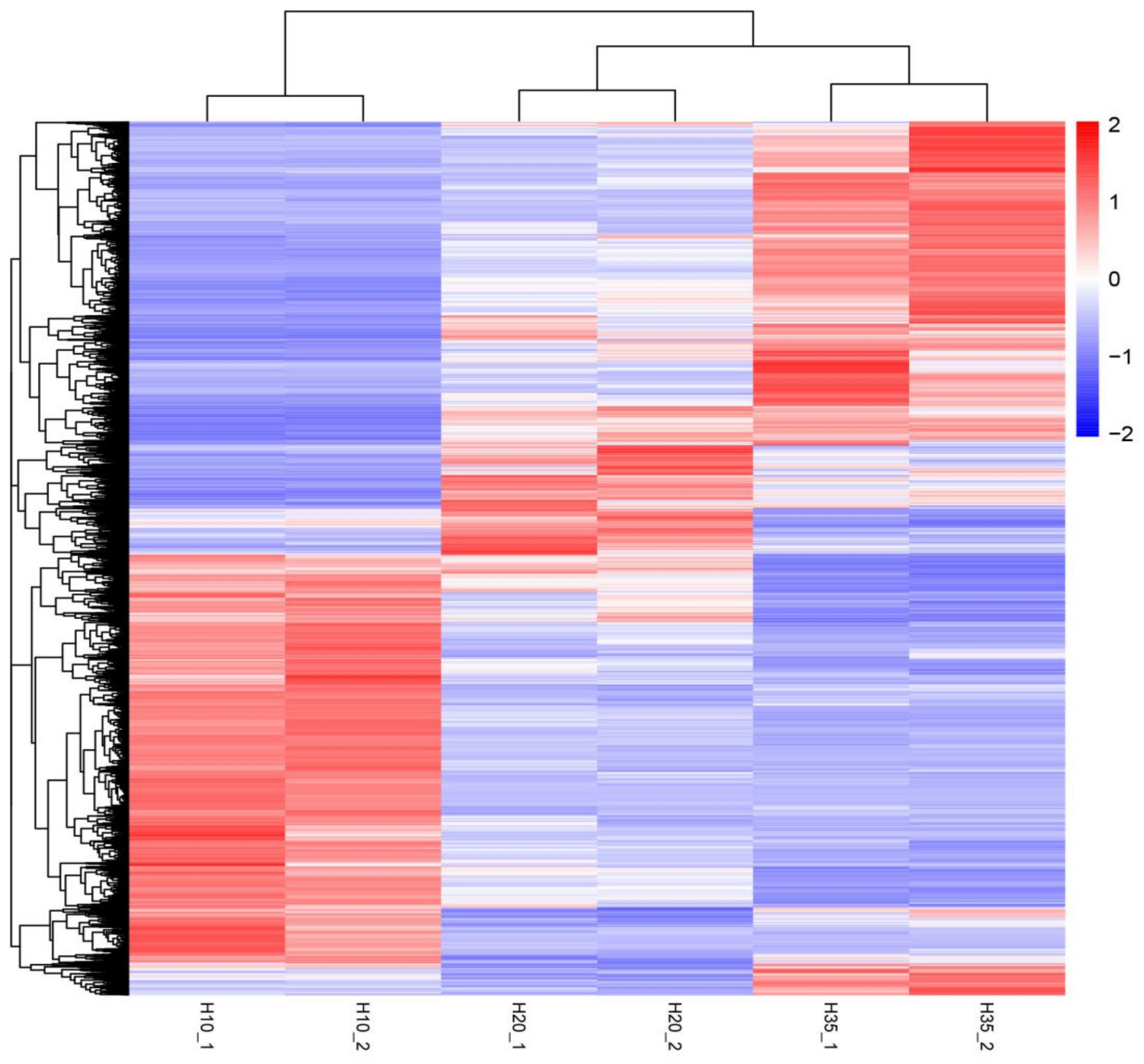
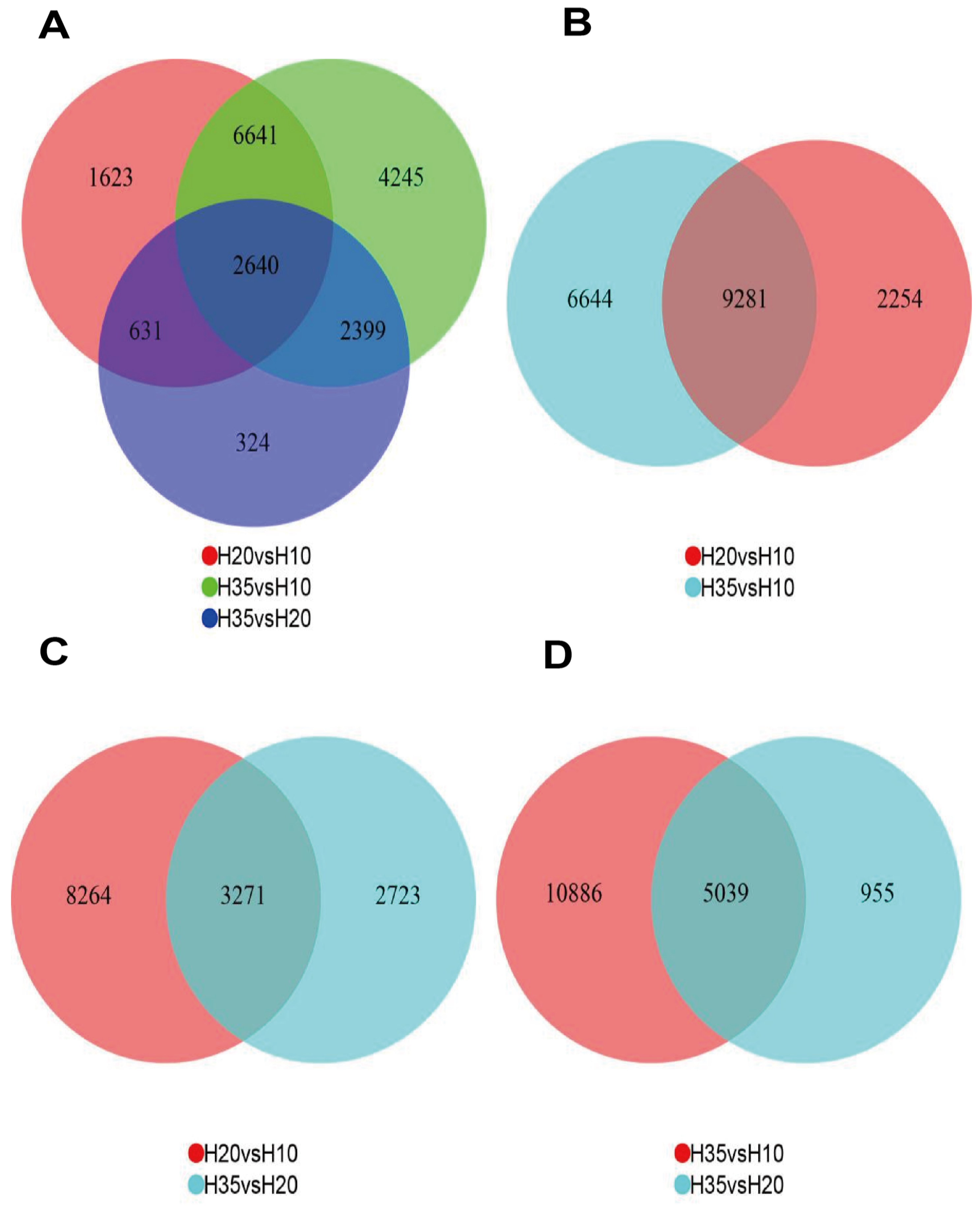
3.2. PCA and DEG Analysis at 10 d, 20 d and 35 d after Flowering
3.3. Co-Expression of the DEGs and KEGG Enrichment Analysis of Common DEGs
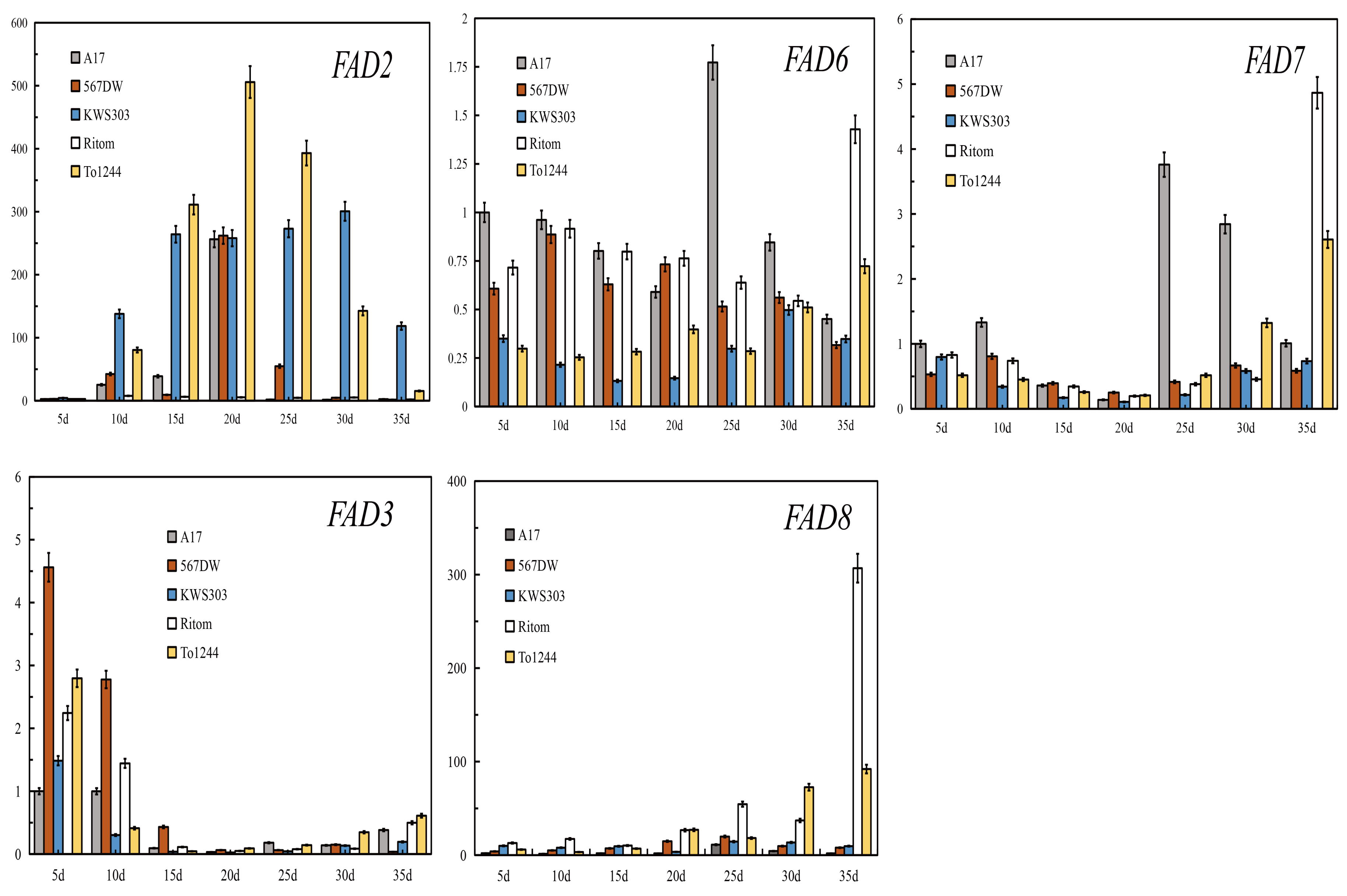
3.4. Expression Analysis of the Genes Related to the UFA Synthesis Pathways
3.5. Correlation Analysis between the UFA Synthesis Pathway Genes
3.6. Correlation Analysis between the Expression Levels of the UFA Synthesis Genes and Final Contents of the FA Components
4. Discussion
4.1. Accumulation Patterns of FAs and OAs at Different Stages of Seed Development
4.2. Transcriptional Expression of Key Genes during Oil Accumulation
4.3. Correlation of Key Genes during Oil Accumulation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kane, N.C.; Burke, J.M.; Marek, L.; Seiler, G.; Vear, F.; Baute, G.; Knapp, S.J.; Vincourt, P.; Rieseberg, L.H. Sunflower genetic, genomic and ecological resources. Mol. Ecol. Resour. 2013, 13, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Seiler, G.J.; Qi, L.L.; Marek, L.F. Utilization of sunflower crop wild relatives for cultivated sunflower improvement. Crop Sci. 2017, 57, 1083–1101. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Zhang, Z.; Li, J.; Zhu, Y.; Yin, Y.; Zhang, X.; Dai, Y.; Zhang, A.; Li, C.; Zhu, Y.; et al. Genome-wide identification and characterization of lncRNAs in sunflower endosperm. BMC Plant Biol. 2022, 22, 494. [Google Scholar] [CrossRef] [PubMed]
- Rial, C.; Varela, R.M.; Molinillo, J.M.G.; Peralta, S.; Macías, F.A. Sunflower metabolites involved in resistance mechanisms against broomrape. Agronomy 2021, 11, 501. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, Y.; Shi, H.; Hu, H.; Yi, L.; Hou, J. Time-course transcriptome and WGCNA analysis revealed the drought response mechanism of two sunflower inbred lines. PLoS ONE 2022, 17, e0265447. [Google Scholar] [CrossRef] [PubMed]
- Gogna, M.; Bhatla, S.C. Biochemical mechanisms regulating salt tolerance in sunflower. Plant Signal Behav. 2019, 14, 1670597. [Google Scholar] [CrossRef]
- Ferretti, C.A.; Spotti, M.L.; Di Cosimo, J.I. Diglyceride-rich oils from glycerolysis of edible vegetable oils. Catal. Today 2018, 302, 233–241. [Google Scholar] [CrossRef]
- Li, Z.; Dong, F.; Sun, Y.; Sun, Z.; Song, X.; Dong, Y.; Huang, X.; Zhong, J.; Zhang, R.; Wang, M.; et al. Qualitative and quantitative analysis of six fatty acid amides in 11 edible vegetable oils using liquid chromatography-mass spectrometry. Front. Nutr. 2022, 9, 857858. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Hou, H.-Y.; Huyen, N.T.; Yang, I.-S.; Chen, X.-B. Raman spectroscopy and 2DCOS analysis of unsaturated fatty acid in edible vegetable oils. Appl. Sci. 2019, 9, 2807. [Google Scholar] [CrossRef] [Green Version]
- Danov, S.M.; Kazantsev, O.A.; Esipovich, A.L.; Belousov, A.S.; Rogozhin, A.E.; Kanakov, E.A. Recent advances in the field of selective epoxidation of vegetable oils and their derivatives: A review and perspective. Catal. Sci. Technol. 2017, 7, 3659–3675. [Google Scholar] [CrossRef]
- Yang, R.; Zhang, L.; Li, P.; Yu, L.; Mao, J.; Wang, X.; Zhang, Q. A review of chemical composition and nutritional properties of minor vegetable oils in China. Trends Food Sci. Tech. 2018, 74, 26–32. [Google Scholar] [CrossRef]
- Hosni, T.; Abbes, Z.; Abaza, L.; Medimagh, S.; Ben Salah, H.; Kharrat, M.; Al-Farga, A. Biochemical characterization of seed oil of tunisian sunflower (Helianthus annuus L.) accessions with special reference to its fatty acid composition and oil content. J. Food Qual. 2022, 2022, 2875072. [Google Scholar] [CrossRef]
- Yelchuri, V.; Srikanth, K.; Prasad, R.B.N.; Karuna, M.S.L. Olefin metathesis of fatty acids and vegetable oils. J. Chem. Sci. 2019, 131, 39. [Google Scholar] [CrossRef] [Green Version]
- Czumaj, A.; Sledzinski, T. Biological role of unsaturated fatty acid desaturases in health and disease. Nutrients 2020, 12, 356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petermann, A.B.; Reyna-Jeldes, M.; Ortega, L.; Coddou, C.; Yevenes, G.E. Roles of the unsaturated fatty acid docosahexaenoic acid in the central nervous system: Molecular and cellular insights. Int. J. Mol. Sci. 2022, 23, 5390. [Google Scholar] [CrossRef]
- Akkaya, M.R. Prediction of fatty acid composition of sunflower seeds by near-infrared reflectance spectroscopy. J. Food Sci. Technol. 2018, 55, 2318–2325. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Olofsson, K.; Linares-Pasten, J.A.; Nordberg Karlsson, E. Investigation of structural features of two related lipases and the impact on fatty acid specificity in vegetable fats. Int. J. Mol. Sci. 2022, 23, 7072. [Google Scholar] [CrossRef] [PubMed]
- Zare, T.; Rupasinghe, T.W.T.; Boughton, B.A.; Roessner, U. The changes in the release level of polyunsaturated fatty acids (ω-3 and ω-6) and lipids in the untreated and water-soaked chia seed. Food Res. Int. 2019, 126, 108665. [Google Scholar] [CrossRef] [PubMed]
- Taipale, S.; Peltomaa, E.; Salmi, P. Variation in ω-3 and ω-6 polyunsaturated fatty acids produced by different phytoplankton taxa at early and late growth phase. Biomolecules 2020, 10, 559. [Google Scholar] [CrossRef] [Green Version]
- Gladyshev, M.I. Fatty Acids: Essential nutrients and important biomarkers. Biomolecules 2022, 12, 1250. [Google Scholar] [CrossRef]
- Premnath, A.; Narayana, M.; Ramakrishnan, C.; Kuppusamy, S.; Chockalingam, V. Mapping quantitative trait loci controlling oil content, oleic acid and linoleic acid content in sunflower (Helianthus annuus L.). Mol. Breed. 2016, 36, 106. [Google Scholar] [CrossRef]
- Chernova, A.I.; Gubaev, R.F.; Singh, A.; Sherbina, K.; Goryunova, S.V.; Martynova, E.U.; Goryunov, D.V.; Boldyrev, S.V.; Vanyushkina, A.A.; Anikanov, N.A.; et al. Genotyping and lipid profiling of 601 cultivated sunflower lines reveals novel genetic determinants of oil fatty acid content. BMC Genom. 2021, 22, 505. [Google Scholar] [CrossRef] [PubMed]
- Badouin, H.; Gouzy, J.; Grassa, C.J.; Murat, F.; Staton, S.E.; Cottret, L.; Lelandais-Brière, C.; Owens, G.L.; Carrère, S.; Mayjonade, B.; et al. The sunflower genome provides insights into oil metabolism, flowering and Asterid evolution. Nature 2017, 546, 148–152. [Google Scholar] [CrossRef] [Green Version]
- Jarecki, W. Effect of varying nitrogen and micronutrient fertilization on yield quantity and quality of sunflower (Helianthus annuus L.) achenes. Agronomy 2022, 12, 2352. [Google Scholar] [CrossRef]
- Nguyen, L.N.; Nosanchuk, J.D. The inhibitory effect of cerulenin to yeasts is fungicidal. Commun. Integr. Biol. 2011, 4, 631–632. [Google Scholar] [CrossRef] [PubMed]
- Lynch, J.H.; Roje, S. A higher plant FAD synthetase is fused to an inactivated FAD pyrophosphatase. J. Biol. Chem. 2022, 10, 2626. [Google Scholar] [CrossRef] [PubMed]
- An, J.U.; Lee, I.G.; Ko, Y.J.; Oh, D.K. Microbial synthesis of linoleate 9 S-lipoxygenase derived plant C18 oxylipins from C18 polyunsaturated fatty acids. J. Agric. Food Chem. 2019, 67, 3209–3219. [Google Scholar] [CrossRef]
- Hashempour, A.; Ghasemnezhad, M.; Sohani, M.M.; Ghazvini, R.F.; Abedi, A. Effects of freezing stress on the expression of fatty acid desaturase (FAD2, FAD6 and FAD7) and beta-glucosidase (BGLC) genes in tolerant and sensitive olive cultivars. Russ. J. Plant Physl. 2019, 66, 214–222. [Google Scholar] [CrossRef]
- Ben Ayed, R.; Chirmade, T.; Hanana, M.; Khamassi, K.; Ercisli, S.; Choudhary, R.; Kadoo, N.; Karunakaran, R. Comparative analysis and structural modeling of Elaeis oleifera FAD2, a fatty acid desaturase involved in unsaturated fatty acid composition of american oil palm. Biology 2022, 11, 529. [Google Scholar] [CrossRef] [PubMed]
- Wen, S.; Liu, H.; Li, X.; Chen, X.; Hong, Y.; Li, H.; Lu, Q.; Liang, X. TALEN-mediated targeted mutagenesis of fatty acid desaturase 2 (FAD2) in peanut (Arachis hypogaea L.) promotes the accumulation of oleic acid. Plant Mol. Biol. 2018, 97, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Banilas, G.; Moressis, A.; Nikoloudakis, N.; Hatzopoulos, P. Spatial and temporal expressions of two distinct oleate desaturases from olive (Olea europaea L.). Plant Sci. 2005, 168, 547–555. [Google Scholar] [CrossRef]
- Salimonti, A.; Carbone, F.; Romano, E.; Pellegrino, M.; Benincasa, C.; Micali, S.; Tondelli, A.; Conforti, F.L.; Perri, E.; Ienco, A.; et al. Association study of the 5′UTR intron of the FAD2-2 gene with oleic and linoleic acid content in Olea europaea L. Front. Plant Sci. 2020, 11, 66. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Wang, W.; Li, M.; Wan, S.; Sui, N. Antioxidants and unsaturated fatty acids are involved in salt tolerance in peanut. Acta Physiol. Plant. 2017, 39, 207. [Google Scholar] [CrossRef]
- Chen, Y.; Tian, L.; Wan, L.; Xia, J.; Liu, F.; Zhao, R.; Wang, T.; Han, C. The cold resistance of cottonseed during germination is related to the content of unsaturated fatty acids. Crop Sci. 2021, 61, 3598–3609. [Google Scholar] [CrossRef]
- Sui, N.; Wang, Y.; Liu, S.; Yang, Z.; Wang, F.; Wan, S. Transcriptomic and physiological evidence for the relationship between unsaturated fatty acid and salt stress in peanut. Front. Plant Sci. 2018, 9, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unamba, C.I.; Nag, A.; Sharma, R.K. Next generation sequencing technologies: The doorway to the unexplored genomics of non-model plants. Front. Plant Sci. 2015, 6, 1074. [Google Scholar] [CrossRef] [Green Version]
- Dimitrijevic, A.; Horn, R. Sunflower hybrid breeding: From markers to genomic selection. Front. Plant Sci. 2017, 8, 2238. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.; Zuo, Y.; Zhang, Y.; Wu, C.; Su, W.; Jin, W.; Yu, H.; An, Y.; Li, Q. Large-scale transcriptome comparison of sunflower genes responsive to Verticillium dahliae. BMC Genom. 2017, 18, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Ma, L.; Li, Y.; Wang, S.a.; Li, L.; Yang, R. Transcriptome analysis reveals sunflower cytochrome P450 CYP93A1 responses to high salinity treatment at the seedling stage. Genes Genom. 2017, 39, 581–591. [Google Scholar] [CrossRef]
- Sarazin, V.; Duclercq, J.; Guillot, X.; Sangwan, B.; Sangwan, R.S. Water-stressed sunflower transcriptome analysis revealed important molecular markers involved in drought stress response and tolerance. Environ. Exp. Bot. 2017, 142, 45–53. [Google Scholar] [CrossRef]
- Wu, Y.; Shi, H.; Yu, H.; Ma, Y.; Hu, H.; Han, Z.; Zhang, Y.; Zhen, Z.; Yi, L.; Hou, J. Combined GWAS and transcriptome analyses provide new insights into the response mechanisms of sunflower against drought stress. Front. Plant Sci. 2022, 13, 847435. [Google Scholar] [CrossRef] [PubMed]
- Roche, J.; Mouloungui, Z.; Cerny, M.; Merah, O. Fatty acid and phytosterol accumulation during seed development in three oilseed species. Int. J. Food Sci. Technol. 2016, 51, 1820–1826. [Google Scholar] [CrossRef] [Green Version]
- Seo, S.H.; Jo, S.M.; Kim, J.; Lee, M.; Lee, Y.; Kang, I. Peanut sprout extracts attenuate triglyceride accumulation by promoting mitochondrial fatty acid oxidation in adipocytes. Int. J. Mol. Sci. 2019, 20, 1216. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.H.; Cahoon, R.E.; Horn, P.J.; Shi, H.; Prakash, R.R.; Cai, Y.; Hearney, M.; Chapman, K.D.; Cahoon, E.B.; Schwender, J.; et al. Identification of bottlenecks in the accumulation of cyclic fatty acids in camelina seed oil. Plant Biotechnol. J. 2018, 16, 926–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, Q.; Wang, J.; Sun, P.; Cai, F.; Ran, B.; Deng, J.; Shi, T.; Chen, Q.; Li, H. Evaluation of the composition and accumulation pattern of fatty acids in tartary buckwheat seed at the germplasm level. Agronomy 2022, 12, 2447. [Google Scholar] [CrossRef]
- Azarkovich, M.I. Dehydrins in orthodox and recalcitrant seeds. Russ. J. Plant Physl. 2020, 67, 221–230. [Google Scholar] [CrossRef]
- Abdul Rahman, S.M.; Ellis, R.H. Seed quality in rice is most sensitive to drought and high temperature in early seed development. Seed Sci. Res. 2019, 29, 238–249. [Google Scholar] [CrossRef] [Green Version]
- Abou Chehade, L.; Angelini, L.G.; Tavarini, S. Genotype and seasonal variation affect yield and oil quality of safflower (Carthamus tinctorius L.) under mediterranean conditions. Agronomy 2022, 12, 122. [Google Scholar] [CrossRef]
- He, M.; Qin, C.X.; Wang, X.; Ding, N.Z. Plant unsaturated fatty acids: Biosynthesis and regulation. Front. Plant Sci. 2020, 11, 390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, B.; Hao, Y.; Lu, J.; Bai, H.; Guan, L.; Zhang, T. Transcriptomic analysis of Perilla frutescens seed to insight into the biosynthesis and metabolic of unsaturated fatty acids. BMC Genom. 2018, 19, 213. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.Y.; Yu, R.; Xie, L.H.; Rahman, M.M.; Kilaru, A.; Niu, L.X.; Zhang, Y.L. Fatty acid and associated gene expression analyses of three tree peony species reveal key genes for α-linolenic acid synthesis in seeds. Front. Plant Sci. 2018, 9, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.-R.; Kim, E.-H.; Roh, K.H.; Kim, J.-B.; Kang, H.-C.; Go, Y.S.; Suh, M.C.; Kim, H.U. High-oleic oilseed rapes developed with seed-specific suppression of FAD2 gene expression. Appl. Biol. Chem. 2016, 59, 669–676. [Google Scholar] [CrossRef]
- Yuan, M.; Zhu, J.; Gong, L.; He, L.; Lee, C.; Han, S.; Chen, C.; He, G. Mutagenesis of FAD2 genes in peanut with CRISPR/Cas9 based gene editing. BMC Biotechnol. 2019, 19, 24. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Zhang, X.; Wang, R.; Chen, B.; Jiang, J.; Win, A.N.; Chai, Y. Cloning and expression of Perilla frutescens FAD2 gene and polymorphism analysis among cultivars. Acta Physiol. Plant. 2017, 39, 84. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Experimental Groups |
|---|---|
| W1 | H20vsH10-H35vsH10 |
| W2 | H20vsH10-H35vsH20 |
| W3 | H35vsH10-H35vsH20 |
| g-W1 | G- H20vsH10-H35vsH10 |
| g-W2 | G- H20vsH10-H35vsH20 |
| g-W3 | G- H35vsH10-H35vsH20 |
| W0 | H20vsH10-H35vsH10- H35vsH20 |
| Experimental Group | UFA Enrichment Ranking | Related Enriched Gene |
|---|---|---|
| w1 | 104 | 110894736 |
| w2 | 69 | 110894736 |
| w3 | 34 | 110894736 |
| g-w3 | 21 | 110894736 |
| w0 | 64 | 110894736 |
| ID | H10_1 | H10_2 | H20_1 | H20_2 | log2FoldChange | Expression Trend |
|---|---|---|---|---|---|---|
| 110894736 | 269.4909 | 340.185 | 287.9888 | 293.8598 | 0.067055 | Down-regulated |
| ID | H20_1 | H20_2 | H35_1 | H35_2 | log2FoldChange | Expression trend |
| 110894736 | 200.6852 | 205.976 | 0.995999 | 3 | 6.670405 | Down-regulated |
| FAD2 (LOC110894736) | FAD6 (LOC110886589) | FAD3 (LOC110872089) | FAD7 (LOC110885030) | FAD8 (LOC110868972) | |
|---|---|---|---|---|---|
| FAD2 (LOC110894736) | 1.000 | ||||
| FAD6 (LOC110886589) | −0.463 ** | 1.000 | |||
| FAD3 (LOC110872089) | −0.347 ** | 0.118 | 1.000 | ||
| FAD7 (LOC110885030) | −0.354 ** | 0.704 ** | 0.006 | 1.000 | |
| FAD8 (LOC110868972) | −0.124 | 0.348 ** | −0.084 | 0.678 ** | 1.000 |
| FAs | Gene | 5 d | 10 d | 15 d | 20 d | 25 d | 30 d | 35 d |
|---|---|---|---|---|---|---|---|---|
| OA | FAD2 (LOC110894736) | −0.693 ** | −0.888 ** | −0.667 ** | −0.539 ** | −0.709 ** | −0.781 ** | −0.704 ** |
| FAD6 (LOC110886589) | 0.782 ** | 0.692 ** | 0.879 ** | 0.702 * | 0.694 ** | 0.587 * | 0.617 * | |
| FAD3 (LOC110872089) | −0.297 | 0.172 | −0.084 | −0.106 | 0.409 | −0.343 | 0.447 | |
| FAD7 (LOC110885030) | 0.533 ** | 0.721 ** | 0.628 * | 0.081 | 0.567 * | 0.432 | 0.567 * | |
| FAD8 (LOC110868972) | −0.901 * | 0.269 | −0.318 | 0.154 | 0.498 | −0.055 | 0.475 | |
| LA | FAD2 (LOC110894736) | 0.689 * | 0.899 ** | 0.709 ** | 0.557 * | 0.753 ** | 0.802 ** | 0.706 * |
| FAD6 (LOC110886589) | −0.823 ** | −0.716 ** | −0.903 ** | −0.720 ** | −0.725 ** | −0.618 * | −0.575 * | |
| FAD3 (LOC110872089) | 0.284 | −0.216 | 0.037 | 0.141 | 0.398 | 0.384 | 0.387 | |
| FAD7 (LOC110885030) | −0.548 * | −0.758 ** | −0.660 ** | −0.08 | −0.592 * | −0.444 | −0.518 * | |
| FAD8 (LOC110868972) | 0.909 ** | −0.248 | 0.344 | −0.107 | −0.474 | 0.117 | −0.427 | |
| ALA | FAD2 (LOC110894736) | 0.056 | −0.094 | −0.534 * | −0.200 | −0.470 | −0.260 | −0.054 |
| FAD6 (LOC110886589) | 0.319 | 0.280 | 0.214 | 0.313 | 0.050 | 0.075 | 0.536 | |
| FAD3 (LOC110872089) | 0.556 * | 0.800 ** | 0.884 ** | −0.210 | −0.382 | −0.454 | −0.917 ** | |
| FAD7 (LOC110885030) | −0.16 | 0.241 | 0.452 | 0.291 | −0.041 | −0.180 | −0.597 | |
| FAD8 (LOC110868972) | 0.009 | −0.168 | −0.070 | −0.377 | −0.205 | −0.684 ** | −0.539 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Q.; Xiang, L.; Zhang, L.; Maimaiti, Y.; Luo, W.; Lei, Z. Transcriptome Sequencing Reveals Key Genes for Sunflower Unsaturated Fatty Acid Synthesis. Agronomy 2023, 13, 885. https://doi.org/10.3390/agronomy13030885
Huang Q, Xiang L, Zhang L, Maimaiti Y, Luo W, Lei Z. Transcriptome Sequencing Reveals Key Genes for Sunflower Unsaturated Fatty Acid Synthesis. Agronomy. 2023; 13(3):885. https://doi.org/10.3390/agronomy13030885
Chicago/Turabian StyleHuang, Qixiu, Lijun Xiang, Li Zhang, Yushanjiang Maimaiti, Wenfang Luo, and Zhonghua Lei. 2023. "Transcriptome Sequencing Reveals Key Genes for Sunflower Unsaturated Fatty Acid Synthesis" Agronomy 13, no. 3: 885. https://doi.org/10.3390/agronomy13030885
APA StyleHuang, Q., Xiang, L., Zhang, L., Maimaiti, Y., Luo, W., & Lei, Z. (2023). Transcriptome Sequencing Reveals Key Genes for Sunflower Unsaturated Fatty Acid Synthesis. Agronomy, 13(3), 885. https://doi.org/10.3390/agronomy13030885