
Genome-Wide Identification, Characterization, and Expression Profiling of TaDUF668 Gene Family in Triticum aestivum


 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Identification and Phylogenetic Analysis of TaDUF668
2.2. Chromosome Localization and Interspecific Evolutionary Analysis of TaDUF668
2.3. TaDUF668 Gene Structure and Conserved Motif Analysis
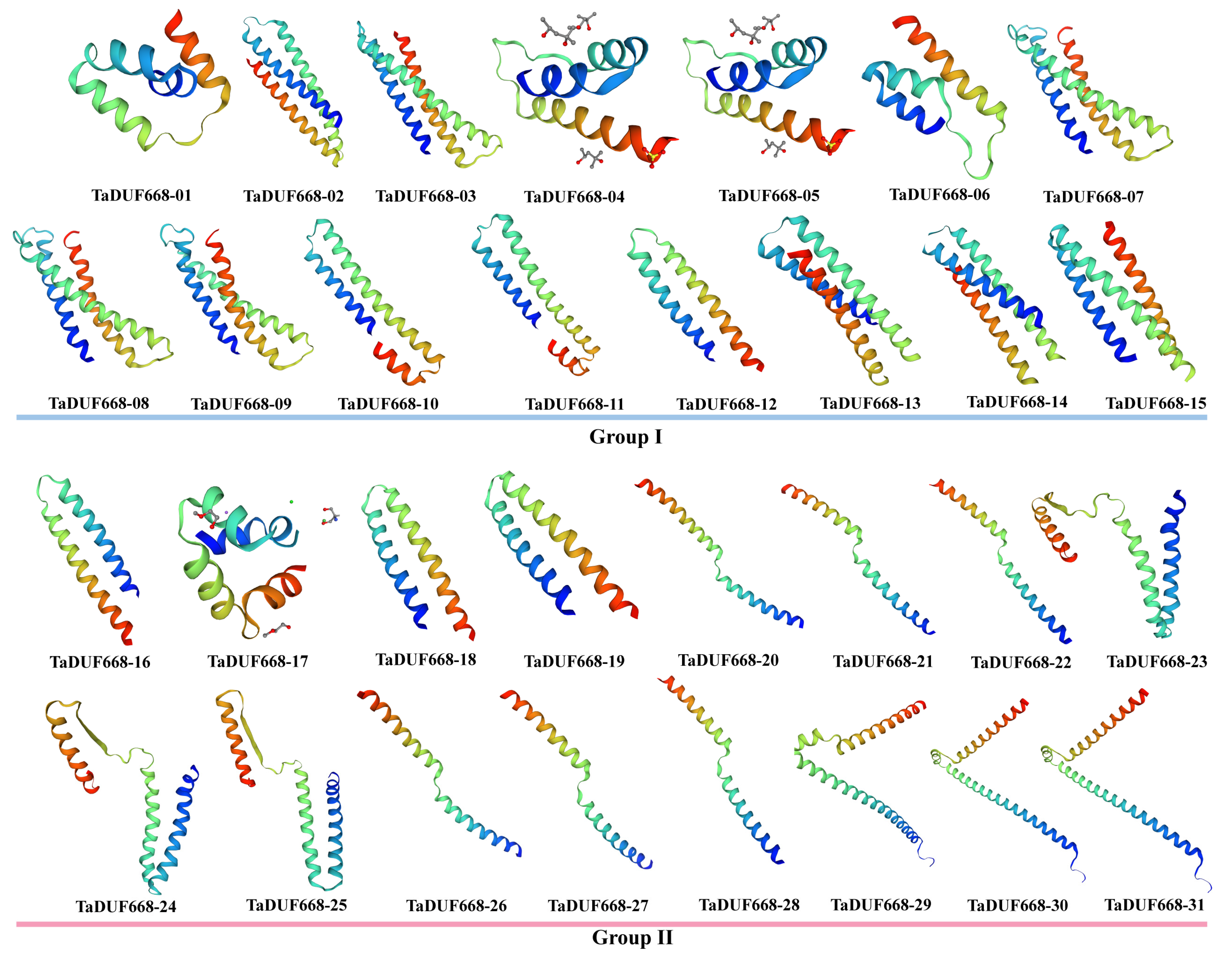
2.4. Characteristics, Three-Dimensional Structure Prediction, and Subcellular Localization of TaDUF668 Proteins
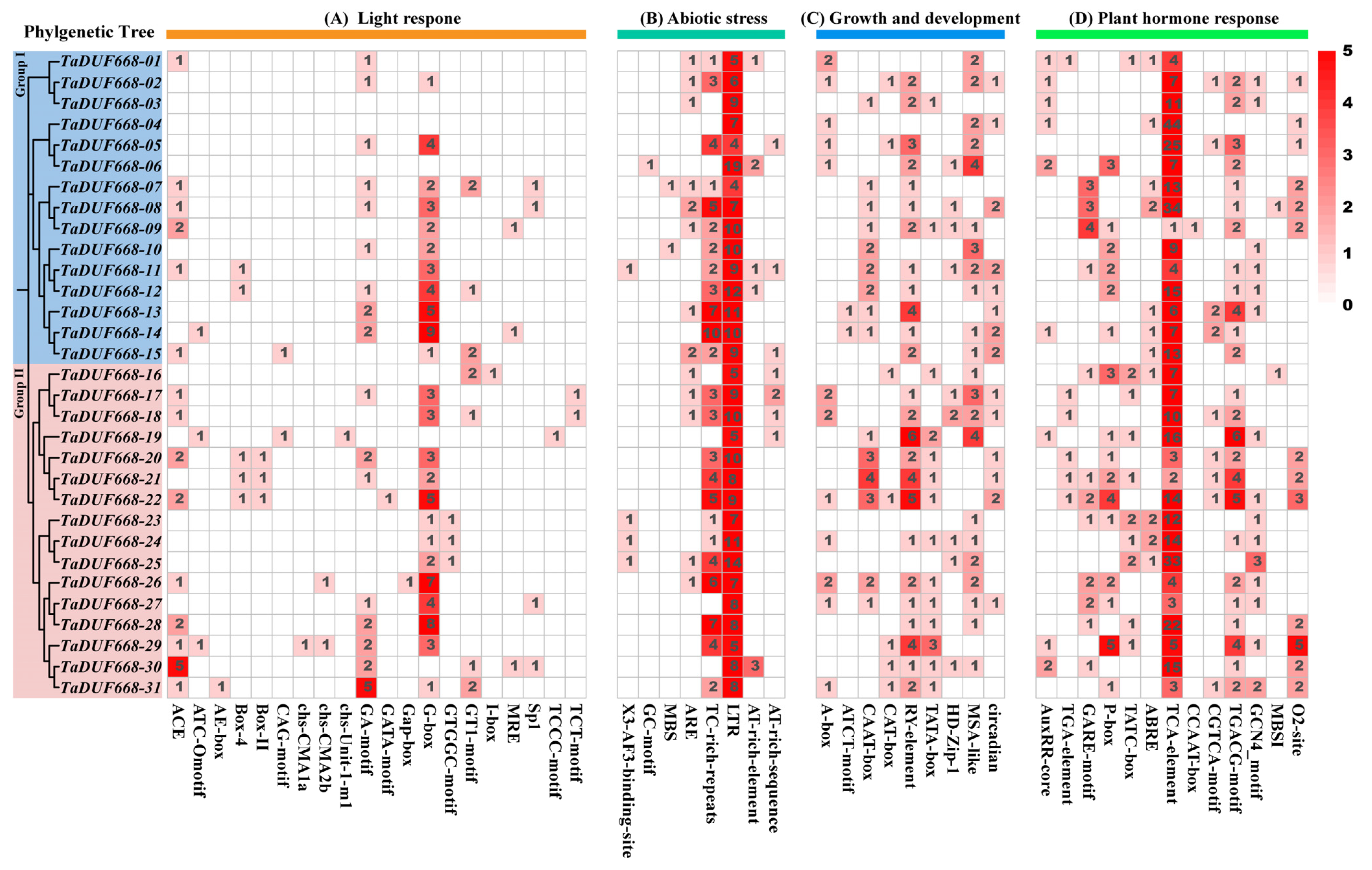
2.5. Cis-Regulatory Element Analysis of TaDUF668
2.6. Expression Profiling of TaDUF668s
2.7. Prediction of the Targeting Relationship between miRNAs and TaDUF668
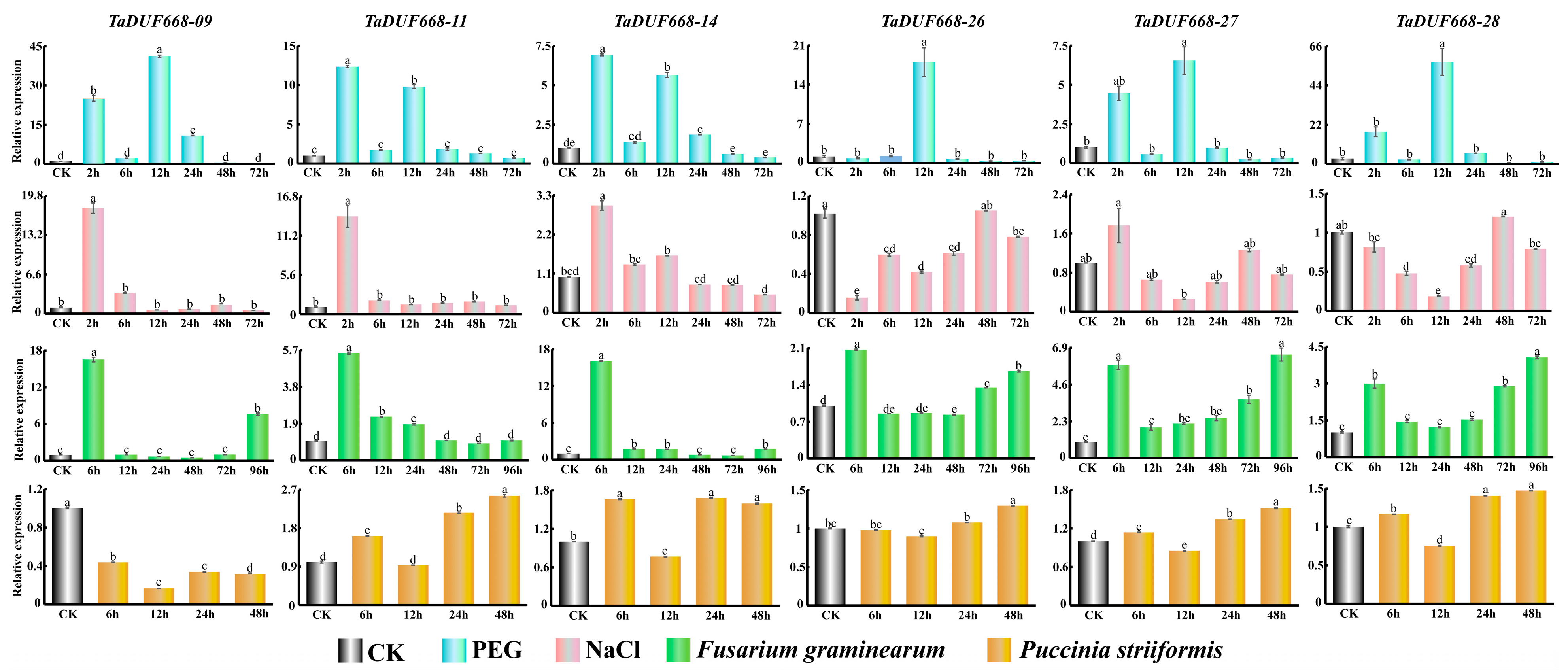
2.8. Stress Treatment of Wheat
2.9. Real-Time Quantitative PCR
3. Results
3.1. Identification and Phylogenetic Analysis of TaDUF668s
3.2. Chromosome Localization and Evolution Analysis of TaDUF668s
3.3. TaDUF668 Gene Structure and Conserved Motifs
3.4. TaDUF668 Protein Characteristics, Subcellular Localization, and 3D Structure Prediction
3.5. TaDUF668 cis-Element Analysis
3.6. TaDUF668 Expression Profiling Analysis
3.7. Post-Transcriptional Regulation of TaDUF668 by miRNAs
3.8. Real-Time Quantitative PCR Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Li, Y.T.; Liu, X.; Xiao, Y.X.; Wen, Y.; Li, K.K.; Ma, Z.L.; Yang, L.J.; Zhu, Y.X.; Yin, J.L. Genome-wide characterization and function analysis uncovered roles of wheat LIMs in responding to adverse stresses and TaLIM8-4D function as a susceptible gene. Plant Genome 2022, 15, 103. [Google Scholar] [CrossRef]
- Yang, F.; Zhang, J.J.; Liu, Q.R.; Liu, H.; Zhou, Y.H.; Yang, W.Y.; Ma, W.J. Improvement and Re-Evolution of Tetraploid Wheat for Global Environmental Challenge and Diversity Consumption Demand. Int. J. Mol. Sci. 2022, 23, 2206. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.X.; Yang, L.; Liu, N.; Yang, J.; Zhou, X.K.; Xia, Y.-C.; He, Y.; He, Y.Q.; Gong, H.J.; Ma, D.F.; et al. Genome-wide identification, structure characterization, and expression pattern profiling of aquaporin gene family in cucumber. BMC Plant Biol. 2019, 19, 497. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Kang, Z. Fighting wheat rusts in China: A look back and into the future. Phytopathol. Res. 2023, 5, 938. [Google Scholar] [CrossRef]
- Bateman, A.; Coggill, P.; Finn, R.D. DUFs: Families in search of function. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2010, 66, 1148–1152. [Google Scholar] [CrossRef] [PubMed]
- Lv, P.Y.; Wan, J.L.; Zhang, C.T.; Hina, A.; Al Amin, G.M.; Begum, N.; Zhao, T.J. Unraveling the Diverse Roles of Neglected Genes Containing Domains of Unknown Function (DUFs): Progress and Perspective. Int. J. Mol. Sci. 2023, 24, 4187. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.Y.; Zhu, X.G.; Shao, W.N.; Song, J.H.; Jiang, W.Q.; He, Y.Q.; Yin, J.L.; Ma, D.F.; Qiao, Y.L. Genome-Wide Mining of Wheat DUF966 Gene Family Provides New Insights into Salt Stress Responses. Sci. Lett. 2020, 118, 569838. [Google Scholar] [CrossRef]
- Zhao, J.Y.; Wang, P.; Gao, W.J.; Long, Y.L.; Wang, Y.X.; Geng, S.W.; Su, X.N.; Jiao, Y.; Chen, Q.J.; Qu, Y.Y. Genome-wide identification of the DUF668 gene family in cotton and expression profiling analysis of GhDUF668 in Gossypium hirsutum under adverse stress. BMC Genom. 2021, 22, 395. [Google Scholar] [CrossRef]
- Ying, S. Genome-Wide Identification and Transcriptional Analysis of Arabidopsis DUF506 Gene Family. Int. J. Mol. Sci. 2021, 22, 11442. [Google Scholar] [CrossRef]
- Qin, D.D.; Xie, S.; Liu, G.; Ni, Z.; Yao, Y.; Sun, Q.; Peng, H. Isolation and Functional Characterization of Heat-stressresponsive Gene TaWTF1 from Wheat. Chin. Bull. Bot. 2013, 48, 34–41. [Google Scholar]
- Guo, F.L.; Shan, Z.L.; Yu, J.F.; Xu, G.B.; Zhang, Z.Y. The Cysteine-Rich Repeat Protein TaCRR1 Participates in Defense against Both Rhizoctonia cerealis and Bipolaris sorokinianain in Wheat. Int. J. Mol. Sci. 2020, 21, 5698. [Google Scholar] [CrossRef] [PubMed]
- Ding, P.Y.; Ma, J.; Yang, Y.J.; Luo, W.; Zou, Y.Y.; Li, T.; Mu, Y.; Tang, H.P.; Lan, X.J. Structure and expression analysis of TaGW5 in common wheat. Indian J. Genet. Plant Breed. 2019, 79, 383–393. [Google Scholar] [CrossRef]
- Zhong, H.; Zhang, H.Y.; Guo, R.; Wang, Q.; Huang, X.P.; Liao, J.L.; Li, Y.S.; Huang, Y.J.; Wang, Z.H. Characterization and Functional Divergence of a Novel DUF668 Gene Family in Rice Based on Comprehensive Expression Patterns. Genes 2019, 10, 980. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.L.; Li, Z.Q.; Luo, Z.Q.; Xu, L.L.; Jin, P.; Ji, S.; Zhou, G.H.; Wang, Z.Y.; Zhou, Z.L.; Zhang, H. Genome-Wide Identification of DUF668 Gene Family and Expression Analysis under Drought and Salt Stresses in Sweet Potato [Ipomoea batatas (L.) Lam]. Genes 2023, 14, 217. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.W.; Jiang, W.Q.; He, Y.Q.; Ma, D.F.; Liu, Y.K.; Wang, S.P.; Zhang, Y.X.; Yin, J.L. Genome-Wide Identification, Structure Characterization, and Expression Profiling of Dof Transcription Factor Gene Family in Wheat (Triticum aestivum L.). Agronomy 2020, 10, 294. [Google Scholar] [CrossRef]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Liu, S. Genome-wide identification and phylogenetic analysis of the ERF gene family in cucumbers. Genet. Mol. Biol. 2011, 34, 624–633. [Google Scholar] [CrossRef]
- Hao, L.D.; Zhang, J.S.; Shi, S.B.; Li, P.; Li, D.D.; Zhang, T.J.; Guo, H.B. Identification and expression profiles of the YABBY transcription factors in wheat. Peerj. 2022, 10, 941. [Google Scholar] [CrossRef]
- Jiang, W.Q.; Geng, Y.P.; Liu, Y.K.; Chen, S.H.; Cao, S.L.; Li, W.; Chen, H.G.; Ma, D.F.; Yin, J.L. Genome-wide identification and characterization of SRO gene family in wheat: Molecular evolution and expression profiles during different stresses. Plant Physiol. Biochem. 2020, 154, 106. [Google Scholar] [CrossRef]
- Chen, C.J.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.H.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 114. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef] [PubMed]
- Duvaud, S.; Gabella, C.; Lisacek, F.; Stockinger, H.; Ioannidis, V.; Durinx, C. Expasy, the Swiss Bioinformatics Resource Portal, as designed by its users. Nucleic Acids Res. 2021, 49, W216–W227. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.-C.; Shen, H.-B. Plant-mPLoc: A Top-Down Strategy to Augment the Power for Predicting Plant Protein Subcellular Localization. PLoS ONE 2010, 5, 511. [Google Scholar] [CrossRef]
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Kiefer, F.; Cassarino, T.G.; Bertoni, M.; Bordoli, L.; et al. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014, 42, W252–W258. [Google Scholar] [CrossRef]
- Zan, T.; Zhang, L.; Xie, T.T.; Li, L.Q. Genome-Wide Identification and Analysis of the Growth-Regulating Factor (GRF) Gene Family and GRF-Interacting Factor Family in Triticum aestivum L. Biochem. Genet. 2020, 58, 705–724. [Google Scholar] [CrossRef]
- Wang, W.P.; Cui, H.; Xiao, X.F.; Wu, B.J.; Sun, J.L.; Zhang, Y.X.; Yang, Q.Y.; Zhao, Y.P.; Liu, G.X.; Qin, T.F. Genome-Wide Identification of Cotton (Gossypium spp.) Trehalose-6-Phosphate Phosphatase (TPP) Gene Family Members and the Role of GhTPP22 in the Response to Drought Stress. Plants 2022, 11, 1079. [Google Scholar] [CrossRef]
- Yin, J.L.; Yan, J.H.; Hou, L.; Jiang, L.L.; Xian, W.R.; Guo, Q.Y. Identification and functional deciphering suggested the regulatory roles of long intergenic ncRNAs (lincRNAs) in increasing grafting pepper resistance to Phytophthora capsici. BMC Genom. 2021, 22, 122. [Google Scholar] [CrossRef]
- Zhang, P.; Zhu, Y.; Ma, D.; Xu, W.; Zhou, J.; Yan, H.; Yang, L.; Yin, J. Screening, Identification, and Optimization of Fermentation Conditions of an Antagonistic Endophyte to Wheat Head Blight. Agronomy 2019, 9, 476. [Google Scholar] [CrossRef]
- Yang, X.M.; Ma, J.; Li, H.B.; Ma, H.X.; Yao, J.B.; Liu, C.J. Different genes can be responsible for crown rot resistance at different developmental stages of wheat and barley. Eur. J. Plant Pathol. 2010, 128, 495–502.891. [Google Scholar] [CrossRef]
- Zhan, C.; Li, Y.T.; Li, H.; Wang, M.R.; Gong, S.J.; Ma, D.F.; Li, Y. Phylogenomic analysis of phenylalanine ammonia-lyase (PAL) multigene family and their differential expression analysis in wheat (Triticum aestivum L.) suggested their roles during different stress responses. Front. Plant Sci. 2022, 13, 889. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Ekinci, M.; Kiziloglu, F.M.; Yildirim, E.; Turan, M.; Kotan, R.; Ors, S. Ameliorative Effects of Plant Growth Promoting Bacteria on Water-yield Relationships, Growth, and Nutrient Uptake of Lettuce Plants under Different Irrigation Levels. Hortscience 2015, 50, 1379–1386. [Google Scholar] [CrossRef]
- Hurst, L.D. The Ka/Ks ratio: Diagnosing the form of sequence evolution. Trends Genet. TIG 2002, 18, 486. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.R.; Tian, B.B.; Gao, C.; Gao, W.; NanYan, S.; Yao, H.; Wang, X.Y.; Jiang, Y.T.; Hu, L.X.; Pan, X.; et al. Identification and expression analysis of candidate genes related to seed dormancy and germination in the wheat GATA family. Plant Physiol. Biochem. PPB 2021, 169, 150. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.S.; Liu, Y.L.; Deng, X.; Liu, D.M.; Liu, Y.; Hu, Y.K.; Yan, Y.M. Genome-wide identification and expression analysis of expansin gene family in common wheat (Triticum aestivum L.). BMC Genom. 2019, 20, 528. [Google Scholar] [CrossRef] [PubMed]
- Avina-Padilla, K.; Ramirez-Rafael, J.A.; Herrera-Oropeza, G.E.; Muley, V.Y.; Valdivia, D.I.; Diaz-Valenzuela, E.; Garcia-Garcia, A.; Varela-Echavarria, A.; Hernandez-Rosales, M. Evolutionary Perspective and Expression Analysis of Intronless Genes Highlight the Conservation of Their Regulatory Role. Front. Genet. 2021, 12, 654256. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, S. Dof Domain Proteins: Plant-Specific Transcription Factors Associated with Diverse Phenomena Unique to Plants. Plant Cell Physiol. 2004, 45, 386–391.531. [Google Scholar] [CrossRef] [PubMed]
- Soltani, B.M.; Ehlting, J.; Hamberger, B.; Douglas, C.J. Multiple cis-regulatory elements regulate distinct and complex patterns of developmental and wound-induced expression of Arabidopsis thaliana 4CL gene family members. Planta 2006, 224, 1226–1238. [Google Scholar] [CrossRef]
- Walther, D.; Brunnemann, R.; Selbig, J. The regulatory code for transcriptional response diversity and its relation to genome structural properties in A. thaliana. PLoS Genet. 2007, 3, e11. [Google Scholar] [CrossRef]
- Nakashima, K.; Yamaguchi-Shinozaki, K.; Shinozaki, K. The transcriptional regulatory network in the drought response and its crosstalk in abiotic stress responses including drought, cold, and heat. Front. Plant Sci. 2014, 5, 530. [Google Scholar] [CrossRef]
- Xie, D.W.; Wang, X.N.; Fu, L.S.; Sun, J.; Zheng, W.; Li, Z.F. Identification of the trehalose-6-phosphate synthase gene family in winter wheat and expression analysis under conditions of freezing stress. J. Genet. 2015, 94, 55–65. [Google Scholar] [CrossRef]
- Han, R.; Jian, C.; Lv, J.Y.; Yan, Y.; Chi, Q.; Li, Z.J.; Wang, Q.; Zhang, J.; Liu, X.L.; Zhao, H.X. Identification and characterization of microRNAs in the flag leaf and developing seed of wheat (Triticum aestivum L.). BMC Genom. 2014, 15, 289. [Google Scholar] [CrossRef]
- Jones-Rhoades, M.W.; Bartel, D.P. Computational identification of plant microRNAs and their targets, including a stress-induced miRNA. Mol. Cell 2004, 14, 787–799. [Google Scholar] [CrossRef]
- Shi, G.Q.; Fu, J.Y.; Rong, L.J.; Zhang, P.Y.; Guo, C.J.; Xiao, K. TaMIR1119, a miRNA family member of wheat (Triticum aestivum),is essential in the regulation of plant drought tolerance. J. Integr. Agric. 2018, 17, 2369–2378. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Forward Primer | Reverse Primer |
|---|---|---|
| TaDUF668-09 | GGGAACCACCCGAGGAAATA | TATCCGCACGCCATCTGAAT |
| TaDUF668-11 | GCAGGAAGTGAAGAGCCAAAGT | ATGACCAAAGGCGTCGTAAATC |
| TaDUF668-14 | TTCTCGCTCATCTTCTGTTCCT | CAAGCCACCGTAATGTCTTCTC |
| TaDUF668-26 | GCGGAAGCAACGGTTCAG | CAACGAGCACCTGGACGAT |
| TaDUF668-27 | GGACGAGCCAAGCGAGAAG | CTGTCAACGGAGGTGGCAAT |
| TaDUF668-28 | GGACGAGCCAAGCGAGAAG | CTGTCAACGGAGGTGGCAAT |
| Name | Gene ID | Len. | MW. | pI. | Ins. | GRAVY | Sig. | Sub. |
|---|---|---|---|---|---|---|---|---|
| TaDUF668-01 | TraesCS5A03G0714700 | 156 | 17.45 | 6.43 | 44.15 | −0.10 | No | Nuc |
| TaDUF668-02 | TraesCS4B03G0220500 | 492 | 56.11 | 9.48 | 62.02 | −0.59 | No | Nuc |
| TaDUF668-03 | TraesCS4A03G0577300 | 552 | 62.15 | 8.87 | 55.17 | −0.56 | No | Chl/Nuc/Cyt |
| TaDUF668-04 | TraesCS2B03G0837000 | 538 | 59.50 | 8.66 | 43.69 | −0.33 | No | Chl |
| TaDUF668-05 | TraesCS2D03G0700600 | 538 | 59.74 | 8.73 | 44.08 | −0.37 | No | Chl |
| TaDUF668-06 | TraesCS2A03G0759400 | 534 | 59.33 | 8.80 | 41.33 | −0.32 | No | Chl |
| TaDUF668-07 | TraesCS7A03G1285000 | 624 | 70.07 | 8.98 | 57.30 | −0.70 | No | Nuc |
| TaDUF668-08 | TraesCS7D03G1219600 | 622 | 69.85 | 8.97 | 59.03 | −0.70 | No | Nuc |
| TaDUF668-09 | TraesCS7B03G1202200 | 622 | 69.66 | 8.95 | 58.49 | −0.70 | No | Nuc |
| TaDUF668-10 | TraesCS1A03G0682400 | 645 | 72.40 | 7.18 | 52.38 | −0.52 | No | Nuc |
| TaDUF668-11 | TraesCS1D03G0646400 | 644 | 72.05 | 7.44 | 52.28 | −0.50 | No | Nuc |
| TaDUF668-12 | TraesCS1B03G0776500 | 647 | 72.53 | 7.74 | 52.66 | −0.52 | No | Nuc |
| TaDUF668-13 | TraesCS3A03G0907200 | 667 | 74.07 | 9.17 | 54.52 | −0.68 | No | Nuc |
| TaDUF668-14 | TraesCS3D03G0833600 | 666 | 73.96 | 9.16 | 55.08 | −0.68 | No | Nuc |
| TaDUF668-15 | TraesCS3B03G1028600 | 667 | 74.06 | 9.16 | 54.80 | −0.68 | No | Nuc |
| TaDUF668-16 | TraesCS5B03G1360500 | 537 | 57.58 | 9.61 | 48.32 | −0.18 | No | Nuc |
| TaDUF668-17 | TraesCS5D03G1204500 | 542 | 57.93 | 9.67 | 49.08 | −0.20 | No | Nuc |
| TaDUF668-18 | TraesCS4A03G0791000 | 541 | 57.95 | 9.67 | 48.51 | −0.21 | No | Nuc |
| TaDUF668-19 | TraesCS5B03G0355900 | 240 | 26.25 | 6.50 | 39.64 | −0.40 | No | Nuc |
| TaDUF668-20 | TraesCS5B03G0360900 | 545 | 59.58 | 9.65 | 47.45 | −0.25 | No | Chl |
| TaDUF668-21 | TraesCS5D03G0347700 | 542 | 59.32 | 9.62 | 47.93 | −0.22 | No | Chl |
| TaDUF668-22 | TraesCS5A03G0365500 | 545 | 59.49 | 9.53 | 47.11 | −0.23 | No | Chl |
| TaDUF668-23 | TraesCS5B03G1143800 | 573 | 62.51 | 7.03 | 60.36 | −0.27 | No | Nuc |
| TaDUF668-24 | TraesCS5D03G1034300 | 569 | 62.12 | 7.63 | 59.34 | −0.24 | No | Nuc |
| TaDUF668-25 | TraesCS5A03G1081600 | 571 | 62.29 | 7.30 | 58.04 | −0.23 | No | Nuc |
| TaDUF668-26 | TraesCS4B03G0579600 | 464 | 50.95 | 9.96 | 55.67 | −0.33 | No | Chl |
| TaDUF668-27 | TraesCS4D03G0514500 | 465 | 51.17 | 9.84 | 57.11 | −0.35 | No | Chl |
| TaDUF668-28 | TraesCS4A03G0195500 | 465 | 51.19 | 9.92 | 55.40 | −0.35 | No | Chl |
| TaDUF668-29 | TraesCS3D03G0788700 | 487 | 53.13 | 9.37 | 50.59 | −0.22 | No | Cem/Chl/Nuc |
| TaDUF668-30 | TraesCS3B03G0984500 | 487 | 53.13 | 9.37 | 49.99 | −0.20 | No | Cem/Chl/Nuc |
| TaDUF668-31 | TraesCS3A03G0859800 | 487 | 53.13 | 9.37 | 50.04 | −0.21 | No | Cem/Chl/Nuc |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yin, X.; Yuan, Y.; Han, X.; Han, S.; Li, Y.; Ma, D.; Fang, Z.; Gong, S.; Yin, J. Genome-Wide Identification, Characterization, and Expression Profiling of TaDUF668 Gene Family in Triticum aestivum. Agronomy 2023, 13, 2178. https://doi.org/10.3390/agronomy13082178
Yin X, Yuan Y, Han X, Han S, Li Y, Ma D, Fang Z, Gong S, Yin J. Genome-Wide Identification, Characterization, and Expression Profiling of TaDUF668 Gene Family in Triticum aestivum. Agronomy. 2023; 13(8):2178. https://doi.org/10.3390/agronomy13082178
Chicago/Turabian StyleYin, Xiaohui, Yi Yuan, Xiaowen Han, Shuo Han, Yiting Li, Dongfang Ma, Zhengwu Fang, Shuangjun Gong, and Junliang Yin. 2023. "Genome-Wide Identification, Characterization, and Expression Profiling of TaDUF668 Gene Family in Triticum aestivum" Agronomy 13, no. 8: 2178. https://doi.org/10.3390/agronomy13082178
APA StyleYin, X., Yuan, Y., Han, X., Han, S., Li, Y., Ma, D., Fang, Z., Gong, S., & Yin, J. (2023). Genome-Wide Identification, Characterization, and Expression Profiling of TaDUF668 Gene Family in Triticum aestivum. Agronomy, 13(8), 2178. https://doi.org/10.3390/agronomy13082178