Cellular and Molecular Mechanisms of R/S-Roscovitine and CDKs Related Inhibition under Both Focal and Global Cerebral Ischemia: A Focus on Neurovascular Unit and Immune Cells



Abstract
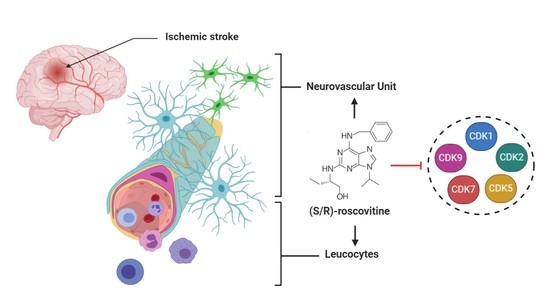
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Effects on Clinical Neuroscore, Infarct Size, and Edema: Role of Roscovitine and CDKs Specific Inhibition
2.1. Neurological Recovery
2.1.1. Roscovitine
(R)-Roscovitine and Neurological Recovery after Focal and Global Ischemia
(S)-Roscovitine and Neurological Recovery after Focal Ischemia
2.1.2. Specific CDKs INHIBITION on Neurological Recovery
2.1.3. Conclusions
2.2. Infarct Size
2.2.1. Roscovitine
(R)-Roscovitine and Infarct Size in Focal Ischemia
(S)-Roscovitine and Infarct Size in Focal Ischemia
2.2.2. Specific CDKs Inhibition on Infarct Size
CDK1 and Infarct Size
CDK5 and Infarct Size
2.2.3. Conclusions
2.3. Edema
2.3.1. Roscovitine and Brain Edema
2.3.2. CDKs Inhibition and Brain Edema
3. Effects of Roscovitine on Neurovascular Unit
3.1. Neurons
3.1.1. Pathological Processes
3.1.2. Roscovitine in Ischemic Stroke Models In Vitro and In Vivo
(R)-Roscovitine and Neurons
(S)-Roscovitine and Neurons
3.1.3. Specific CDKs Inhibition on Neurons
CDK1 and Neurons
CDK2 and Neurons
CDK5 and Neurons
CDK7 and Neurons
3.1.4. Conclusions
3.2. Microglia
3.2.1. Pathological Processes
3.2.2. Roscovitine in Ischemic Stroke Models
(R)-Roscovitine and Microglia
(S)-Roscovitine and Microglia
3.2.3. Roscovitine in Other Non-Ischemic Models
3.2.4. Specific CDKs Inhibition in Microglia
CDK1 and Microglia
CDK2 and Microglia
CDK5 and Microglia
3.2.5. Conclusions
3.3. Astrocytes
3.3.1. Pathological Processes
3.3.2. Roscovitine in Ischemic Stroke Models
(R)-Roscovitine and Astrocytes
(S)-Roscovitine and Astrocytes
3.3.3. Roscovitine in Other Non-Ischemic Models
3.3.4. Specific CDKs Inhibition in Astrocytes
CDK2 and Astrocytes
CDK5 and Astrocytes
CDK9 and Astrocytes
3.3.5. Conclusions
3.4. Oligodendrocytes
3.4.1. Pathological Processes
3.4.2. Roscovitine
3.4.3. Specific CDKs Inhibition in Oligodendrocytes
CDK1 and Oligodendrocytes
CDK2 and Oligodendrocytes
CDK5 and Oligodendrocytes
3.4.4. Conclusions
3.5. Endothelial Cells
3.5.1. Pathological Processes
3.5.2. Roscovitine in Ischemic Stroke Models
(R)-Roscovitine and Endothelial Cells
(S)-Roscovitine and Endothelial Cells
3.5.3. Roscovitine in Other Non-Ischemic Models
3.5.4. Specific CDKs Inhibition in Endothelial Cells
CDK1 and Endothelial Cells
CDK2 and Endothelial Cells
CDK5 and Endothelial Cells
CDK7 and Endothelial Cells
CDK9 and Endothelial Cells
3.5.5. Conclusions
4. Effects of Roscovitine on Leucocytes
4.1. Macrophages
4.1.1. Pathological Processes
4.1.2. Roscovitine and Macrophages
4.1.3. Specific CDKs Inhibition in Macrophages
CDK1 and Macrophages
CDK5 and Macrophages
CDK7 and Macrophages
4.1.4. Conclusions
4.2. Neutrophils
4.2.1. Pathological Processes
4.2.2. Roscovitine and Neutrophils
4.2.3. Specific CDKs Inhibition in Neutrophils
CDK1 and Neutrophils
CDK2 and Neutrophils
CDK5 and Neutrophils
CDK9 and Neutrophils
4.2.4. Conclusions
4.3. Eosinophils
4.3.1. Pathological Processes
4.3.2. Roscovitine and Eosinophils
4.3.3. Specific CDKs Inhibition in Eosinophils
4.3.4. Conclusions
4.4. T Lymphocytes
4.4.1. Pathological Processes
4.4.2. Roscovitine
4.4.3. Specific CDKs Inhibition in T Cells
CDK1 and T Lymphocytes
CDK2 and T Lymphocytes
CDK5 and T Lymphocytes
CDK9 and T Lymphocytes
4.4.4. Th17/Treg Lymphocytes Imbalance
Roscovitine in Th17/Treg Lymphocytes Imbalance
CDKs in Th17/Treg Lymphocytes Imbalance
4.4.5. Conclusions
4.5. B Lymphocytes
4.5.1. Pathological Processes
4.5.2. Roscovitine
4.5.3. Specific CDKs Inhibition in B-Lymphocytes
4.5.4. Conclusions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Moretti, A.; Ferrari, F.; Villa, R.F. Neuroprotection for Ischaemic Stroke: Current Status and Challenges. Pharmacol. Ther. 2015, 146, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.O.; Nguyen, M.; Roth, G.A.; Nichols, E.; Alam, T.; Abate, D.; Abd-Allah, F.; Abdelalim, A.; Abraha, H.N.; Abu-Rmeileh, N.M.; et al. Global, Regional, and National Burden of Stroke, 1990–2016: A Systematic Analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 439–458. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, A.M.; Firlik, A.D.; Fukui, M.B.; Wechsler, L.R.; Jungries, C.A.; Yonas, H. Ischemic Core and Penumbra in Human Stroke. Stroke 1999, 30, 93–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggoun-Zouaoui, D.; Margalli, I.; Borrega, F.; Represa, A.; Plotkine, M.; Ben-Ari, Y.; Charriaut-Marlangue, C. Ultrastructural Morphology of Neuronal Death Following Reversible Focal Ischemia in the Rat. Apoptosis 1998, 3, 133–141. [Google Scholar] [CrossRef]
- Astrup, J.; Symon, L.; Branston, N.M.; Lassen, N.A. Cortical Evoked Potential and Extracellular K+ and H+ at Critical Levels of Brain Ischemia. Stroke 1977, 8, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Rami, A.; Kögel, D. Apoptosis Meets Autophagy-like Cell Death in the Ischemic Penumbra: Two Sides of the Same Coin? Autophagy 2008, 4, 422–426. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J. Neuroprotective Strategies Targeting Apoptotic and Necrotic Cell Death for Stroke. Apoptosis Int. J. Program. Cell Death 2009, 14, 469–477. [Google Scholar] [CrossRef] [Green Version]
- Khaja, A.M.; Grotta, J.C. Established Treatments for Acute Ischaemic Stroke. Lancet Lond. Engl. 2007, 369, 319–330. [Google Scholar] [CrossRef]
- Aguiar de Sousa, D.; von Martial, R.; Abilleira, S.; Gattringer, T.; Kobayashi, A.; Gallofré, M.; Fazekas, F.; Szikora, I.; Feigin, V.; Caso, V.; et al. Access to and Delivery of Acute Ischaemic Stroke Treatments: A Survey of National Scientific Societies and Stroke Experts in 44 European Countries. Eur. Stroke J. 2019, 4, 13–28. [Google Scholar] [CrossRef] [Green Version]
- Shaw, C.M.; Alvord, E.C.; Berry, R.G. Swelling of the Brain Following Ischemic Infarction with Arterial Occlusion. Arch. Neurol. 1959, 1, 161–177. [Google Scholar] [CrossRef]
- Frank, J.I. Large Hemispheric Infarction, Deterioration, and Intracranial Pressure. Neurology 1995, 45, 1286–1290. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Tanaka, Y.; Tokushige, K.; Tanabe, A.; Kobayashi, S. Basilar Bifurcation Aneurysms Associated with Persistent Primitive Hypoglossal Artery. Neurosurg. Rev. 1998, 21, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Dostovic, Z.; Dostovic, E.; Smajlovic, D.; Ibrahimagic, O.C.; Avdic, L. Brain Edema After Ischaemic Stroke. Med. Arch. 2016, 70, 339–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hacke, W.; Schwab, S.; Horn, M.; Spranger, M.; De Georgia, M.; von Kummer, R. “Malignant” Middle Cerebral Artery Territory Infarction: Clinical Course and Prognostic Signs. Arch. Neurol. 1996, 53, 309–315. [Google Scholar] [CrossRef]
- Berrouschot, J.; Sterker, M.; Bettin, S.; Köster, J.; Schneider, D. Mortality of Space-Occupying (‘malignant’) Middle Cerebral Artery Infarction under Conservative Intensive Care. Intensive Care Med. 1998, 24, 620–623. [Google Scholar] [CrossRef] [PubMed]
- Michinaga, S.; Koyama, Y. Pathogenesis of Brain Edema and Investigation into Anti-Edema Drugs. Int. J. Mol. Sci. 2015, 16, 9949–9975. [Google Scholar] [CrossRef] [Green Version]
- Stokum, J.A.; Gerzanich, V.; Simard, J.M. Molecular Pathophysiology of Cerebral Edema. J. Cereb. Blood Flow Metab. 2016, 36, 513–538. [Google Scholar] [CrossRef] [Green Version]
- Betz, A.L.; Iannotti, F.; Hoff, J.T. Brain Edema: A Classification Based on Blood-Brain Barrier Integrity. Cerebrovasc. Brain Metab. Rev. 1989, 1, 133–154. [Google Scholar]
- Kahle, K.T.; Simard, J.M.; Staley, K.J.; Nahed, B.V.; Jones, P.S.; Sun, D. Molecular Mechanisms of Ischemic Cerebral Edema: Role of Electroneutral Ion Transport. Physiol. Bethesda Md 2009, 24, 257–265. [Google Scholar] [CrossRef] [Green Version]
- Heo, J.H.; Han, S.W.; Lee, S.K. Free Radicals as Triggers of Brain Edema Formation after Stroke. Free Radic. Biol. Med. 2005, 39, 51–70. [Google Scholar] [CrossRef]
- Mori, K.; Miyazaki, M.; Iwase, H.; Maeda, M. Temporal Profile of Changes in Brain Tissue Extracellular Space and Extracellular Ion (Na(+), K(+)) Concentrations after Cerebral Ischemia and the Effects of Mild Cerebral Hypothermia. J. Neurotrauma 2002, 19, 1261–1270. [Google Scholar] [CrossRef] [PubMed]
- Menzies, S.A.; Betz, A.L.; Hoff, J.T. Contributions of Ions and Albumin to the Formation and Resolution of Ischemic Brain Edema. J. Neurosurg. 1993, 78, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, O.; Asano, T.; Koide, T.; Takakura, K. Ischemic Brain Edema Following Occlusion of the Middle Cerebral Artery in the Rat. I: The Time Courses of the Brain Water, Sodium and Potassium Contents and Blood-Brain Barrier Permeability to 125I-Albumin. Stroke 1985, 16, 101–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vahedi, K.; Hofmeijer, J.; Juettler, E.; Vicaut, E.; George, B.; Algra, A.; Amelink, G.J.; Schmiedeck, P.; Schwab, S.; Rothwell, P.M.; et al. Early Decompressive Surgery in Malignant Infarction of the Middle Cerebral Artery: A Pooled Analysis of Three Randomised Controlled Trials. Lancet Neurol. 2007, 6, 215–222. [Google Scholar] [CrossRef]
- Hofmeijer, J.; Kappelle, L.J.; Algra, A.; Amelink, G.J.; van Gijn, J.; van der Worp, H.B. HAMLET investigators Surgical Decompression for Space-Occupying Cerebral Infarction (the Hemicraniectomy After Middle Cerebral Artery Infarction with Life-Threatening Edema Trial [HAMLET]): A Multicentre, Open, Randomised Trial. Lancet Neurol. 2009, 8, 326–333. [Google Scholar] [CrossRef]
- Timsit, S.; Menn, B. Cerebral Ischemia, Cell Cycle Elements and Cdk5. Biotechnol. J. 2007, 2, 958–966. [Google Scholar] [CrossRef]
- Ozaki, T.; Nakamura, H.; Kishima, H. Therapeutic Strategy against Ischemic Stroke with the Concept of Neurovascular Unit. Neurochem. Int. 2019, 126, 246–251. [Google Scholar] [CrossRef]
- Roy, C.S.; Sherrington, C.S. On the Regulation of the Blood-Supply of the Brain. J. Physiol. 1890, 11, 85–158. [Google Scholar] [CrossRef]
- del Zoppo, G.J. The Neurovascular Unit in the Setting of Stroke. J. Intern. Med. 2010, 267, 156–171. [Google Scholar] [CrossRef] [Green Version]
- Thurgur, H.; Pinteaux, E. Microglia in the Neurovascular Unit: Blood-Brain Barrier-Microglia Interactions After Central Nervous System Disorders. Neuroscience 2019, 405, 55–67. [Google Scholar] [CrossRef]
- Enzmann, G.; Mysiorek, C.; Gorina, R.; Cheng, Y.-J.; Ghavampour, S.; Hannocks, M.-J.; Prinz, V.; Dirnagl, U.; Endres, M.; Prinz, M.; et al. The Neurovascular Unit as a Selective Barrier to Polymorphonuclear Granulocyte (PMN) Infiltration into the Brain after Ischemic Injury. Acta Neuropathol. 2013, 125, 395–412. [Google Scholar] [CrossRef] [Green Version]
- Stanimirovic, D.B.; Friedman, A. Pathophysiology of the Neurovascular Unit: Disease Cause or Consequence? J. Cereb. Blood Flow Metab. 2012, 32, 1207–1221. [Google Scholar] [CrossRef] [Green Version]
- Jessen, N.A.; Munk, A.S.F.; Lundgaard, I.; Nedergaard, M. The Glymphatic System: A Beginner’s Guide. Neurochem. Res. 2015, 40, 2583–2599. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, M.K.; Mestre, H.; Nedergaard, M. The Glymphatic Pathway in Neurological Disorders. Lancet Neurol. 2018, 17, 1016–1024. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Nelson, A.R.; Betsholtz, C.; Zlokovic, B.V. Establishment and Dysfunction of the Blood-Brain Barrier. Cell 2015, 163, 1064–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muoio, V.; Persson, P.B.; Sendeski, M.M. The Neurovascular Unit—Concept Review. Acta Physiol. 2014, 210, 790–798. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, B.T.; Davis, T.P. The Blood-Brain Barrier/Neurovascular Unit in Health and Disease. Pharmacol. Rev. 2005, 57, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Knockaert, M.; Greengard, P.; Meijer, L. Pharmacological Inhibitors of Cyclin-Dependent Kinases. Trends Pharmacol. Sci. 2002, 23, 417–425. [Google Scholar] [CrossRef]
- Wang, F.; O’Hare, M.J.; Park, D.S. Cyclin-Dependent Kinases and Stroke. Expert Opin. Ther. Targets 2001, 5, 557–567. [Google Scholar] [CrossRef]
- Le Roy, L.; Amara, A.; Le Roux, C.; Bocher, O.; Létondor, A.; Benz, N.; Timsit, S. Principal Component Analysis, a Useful Tool to Study Cyclin-Dependent Kinase-Inhibitor’s Effect on Cerebral Ischaemia. Brain Commun. 2020, 2. [Google Scholar] [CrossRef]
- Roskoski, R. Modulation of Enzyme Activity. In xPharm: The Comprehensive Pharmacology Reference; Enna, S.J., Bylund, D.B., Eds.; Elsevier: New York, NY, USA, 2007; pp. 1–11. ISBN 978-0-08-055232-3. [Google Scholar]
- Roskoski, R. Cyclin-Dependent Protein Serine/Threonine Kinase Inhibitors as Anticancer Drugs. Pharmacol. Res. 2019, 139, 471–488. [Google Scholar] [CrossRef]
- Sherr, C.J. Cancer Cell Cycles. Science 1996, 274, 1672–1677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grison, A.; Atanasoski, S. Cyclins, Cyclin-Dependent Kinases, and Cyclin-Dependent Kinase Inhibitors in the Mouse Nervous System. Mol. Neurobiol. 2020, 57, 3206–3218. [Google Scholar] [CrossRef] [PubMed]
- Dhavan, R.; Tsai, L.H. A Decade of CDK5. Nat. Rev. Mol. Cell Biol. 2001, 2, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Samuels, B.A.; Tsai, L.-H. Neuronal Motility and Structure: Cdk5 Pathways. In Encyclopedia of Neuroscience; Squire, L.R., Ed.; Academic Press: Oxford, UK, 2009; pp. 703–710. ISBN 978-0-08-045046-9. [Google Scholar]
- Hellmich, M.R.; Pant, H.C.; Wada, E.; Battey, J.F. Neuronal Cdc2-like Kinase: A Cdc2-Related Protein Kinase with Predominantly Neuronal Expression. Proc. Natl. Acad. Sci. USA 1992, 89, 10867–10871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyerson, M.; Enders, G.H.; Wu, C.L.; Su, L.K.; Gorka, C.; Nelson, C.; Harlow, E.; Tsai, L.H. A Family of Human Cdc2-Related Protein Kinases. EMBO J. 1992, 11, 2909–2917. [Google Scholar] [CrossRef]
- Lee, M.H.; Nikolic, M.; Baptista, C.A.; Lai, E.; Tsai, L.H.; Massagué, J. The Brain-Specific Activator P35 Allows Cdk5 to Escape Inhibition by P27Kip1 in Neurons. Proc. Natl. Acad. Sci. USA 1996, 93, 3259–3263. [Google Scholar] [CrossRef] [Green Version]
- Humbert, S.; Dhavan, R.; Tsai, L. P39 Activates Cdk5 in Neurons, and Is Associated with the Actin Cytoskeleton. J. Cell Sci. 2000, 113 Pt 6, 975–983. [Google Scholar]
- Contreras-Vallejos, E.; Utreras, E.; Gonzalez-Billault, C. Going out of the Brain: Non-Nervous System Physiological and Pathological Functions of Cdk5. Cell. Signal. 2012, 24, 44–52. [Google Scholar] [CrossRef]
- Liebl, J.; Fürst, R.; Vollmar, A.M.; Zahler, S. Twice Switched at Birth: Cell Cycle-Independent Roles of the “Neuron-Specific” Cyclin-Dependent Kinase 5 (Cdk5) in Non-Neuronal Cells. Cell. Signal. 2011, 23, 1698–1707. [Google Scholar] [CrossRef] [PubMed]
- Stevens, J.L.; Aguanno, A. The Role of CDK5 in Non-Neuronal Tissues. FASEB J. 2007, 21, A992. [Google Scholar] [CrossRef]
- Meijer, L.; Borgne, A.; Mulner, O.; Chong, J.P.; Blow, J.J.; Inagaki, N.; Inagaki, M.; Delcros, J.G.; Moulinoux, J.P. Biochemical and Cellular Effects of Roscovitine, a Potent and Selective Inhibitor of the Cyclin-Dependent Kinases Cdc2, Cdk2 and Cdk5. Eur. J. Biochem. 1997, 243, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Khalil, H.S.; Mitev, V.; Vlaykova, T.; Cavicchi, L.; Zhelev, N. Discovery and Development of Seliciclib. How Systems Biology Approaches Can Lead to Better Drug Performance. J. Biotechnol. 2015, 202, 40–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; McClue, S.J.; Ferguson, J.R.; Hull, J.D.; Stokes, S.; Parsons, S.; Westwood, R.; Fischer, P.M. Synthesis and Configuration of the Cyclin-Dependent Kinase Inhibitor Roscovitine and Its Enantiomer. Tetrahedron Asymmetry 2001, 12, 2891–2894. [Google Scholar] [CrossRef]
- Bach, S.; Knockaert, M.; Reinhardt, J.; Lozach, O.; Schmitt, S.; Baratte, B.; Koken, M.; Coburn, S.P.; Tang, L.; Jiang, T.; et al. Roscovitine Targets, Protein Kinases and Pyridoxal Kinase. J. Biol. Chem. 2005, 280, 31208–31219. [Google Scholar] [CrossRef] [Green Version]
- Menn, B.; Bach, S.; Blevins, T.L.; Campbell, M.; Meijer, L.; Timsit, S. Delayed Treatment with Systemic (S)-Roscovitine Provides Neuroprotection and Inhibits in Vivo CDK5 Activity Increase in Animal Stroke Models. PLoS ONE 2010, 5, e12117. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Chen, C.; Lü, J.; Xie, M.; Pan, D.; Luo, X.; Yu, Z.; Dong, Q.; Wang, W. Cell Cycle Inhibition Attenuates Microglial Proliferation and Production of IL-1beta, MIP-1alpha, and NO after Focal Cerebral Ischemia in the Rat. Glia 2009, 57, 908–920. [Google Scholar] [CrossRef]
- Rousselet, E.; Létondor, A.; Menn, B.; Courbebaisse, Y.; Quillé, M.-L.; Timsit, S. Sustained (S)-Roscovitine Delivery Promotes Neuroprotection Associated with Functional Recovery and Decrease in Brain Edema in a Randomized Blind Focal Cerebral Ischemia Study. J. Cereb. Blood Flow Metab. 2018, 38, 1070–1084. [Google Scholar] [CrossRef]
- Marlier, Q.; Jibassia, F.; Verteneuil, S.; Linden, J.; Kaldis, P.; Meijer, L.; Nguyen, L.; Vandenbosch, R.; Malgrange, B. Genetic and Pharmacological Inhibition of Cdk1 Provides Neuroprotection towards Ischemic Neuronal Death. Cell Death Discov. 2018, 4. [Google Scholar] [CrossRef]
- Zhang, R.; Liu, C.; Ji, Y.; Teng, L.; Guo, Y. Neuregulin-1β Plays a Neuroprotective Role by Inhibiting the Cdk5 Signaling Pathway after Cerebral Ischemia-Reperfusion Injury in Rats. J. Mol. Neurosci. 2018, 66, 261–272. [Google Scholar] [CrossRef]
- Xu, L.; Di, Q.; Zhang, Y. Cell Cycle Proteins Preceded Neuronal Death after Chronic Cerebral Hypoperfusion in Rats. Neurol. Res. 2008, 30, 932–939. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Vargas, J.A.; Múnera, A.; Cardona-Gómez, G.P. CDK5 Knockdown Prevents Hippocampal Degeneration and Cognitive Dysfunction Produced by Cerebral Ischemia. J. Cereb. Blood Flow Metab. 2015, 35, 1937–1949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, Y.; Yang, S.; Liu, R.; Brun-Zinkernagel, A.M.; Koulen, P.; Simpkins, J.W. Transient Cerebral Ischemia Induces Aberrant Neuronal Cell Cycle Re-Entry and Alzheimer’s Disease-like Tauopathy in Female Rats. J. Biol. Chem. 2004, 279, 22684–22692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, S.L.; Kulp, K.S.; Vulliet, R. Cyclin-Dependent Protein Kinase 5 Activity Increases in Rat Brain Following Ischemia. Neurochem. Int. 1997, 31, 617–623. [Google Scholar] [CrossRef]
- Wen, Y.; Yang, S.-H.; Liu, R.; Perez, E.J.; Brun-Zinkernagel, A.M.; Koulen, P.; Simpkins, J.W. Cdk5 Is Involved in NFT-like Tauopathy Induced by Transient Cerebral Ischemia in Female Rats. Biochim. Biophys. Acta 2007, 1772, 473–483. [Google Scholar] [CrossRef]
- Meyer, D.A.; Torres-Altoro, M.I.; Tan, Z.; Tozzi, A.; Filippo, M.D.; DiNapoli, V.; Plattner, F.; Kansy, J.W.; Benkovic, S.A.; Huber, J.D.; et al. Ischemic Stroke Injury Is Mediated by Aberrant Cdk5. J. Neurosci. 2014, 34, 8259–8267. [Google Scholar] [CrossRef] [Green Version]
- Rashidian, J.; Rousseaux, M.W.; Venderova, K.; Qu, D.; Callaghan, S.M.; Phillips, M.; Bland, R.J.; During, M.J.; Mao, Z.; Slack, R.S.; et al. Essential Role of Cytoplasmic Cdk5 and Prx2 in Multiple Ischemic Injury Models, In Vivo. J. Neurosci. 2009, 29, 12497–12505. [Google Scholar] [CrossRef] [Green Version]
- Mitsios, N.; Pennucci, R.; Krupinski, J.; Sanfeliu, C.; Gaffney, J.; Kumar, P.; Kumar, S.; Juan-Babot, O.; Slevin, M. Expression of Cyclin-Dependent Kinase 5 MRNA and Protein in the Human Brain Following Acute Ischemic Stroke. Brain Pathol. 2007, 17, 11–23. [Google Scholar] [CrossRef]
- Gutiérrez-Vargas, J.A.; Moreno, H.; Cardona-Gómez, G.P. Targeting CDK5 Post-Stroke Provides Long-Term Neuroprotection and Rescues Synaptic Plasticity. J. Cereb. Blood Flow Metab. 2016. [Google Scholar] [CrossRef]
- Rashidian, J.; Iyirhiaro, G.; Aleyasin, H.; Rios, M.; Vincent, I.; Callaghan, S.; Bland, R.J.; Slack, R.S.; During, M.J.; Park, D.S. Multiple Cyclin-Dependent Kinases Signals Are Critical Mediators of Ischemia/Hypoxic Neuronal Death in Vitro and in Vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 14080–14085. [Google Scholar] [CrossRef] [Green Version]
- Puig, B.; Brenna, S.; Magnus, T. Molecular Communication of a Dying Neuron in Stroke. Int. J. Mol. Sci. 2018, 19, 2834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voll, R.E.; Herrmann, M.; Roth, E.A.; Stach, C.; Kalden, J.R.; Girkontaite, I. Immunosuppressive Effects of Apoptotic Cells. Nature 1997, 390, 350–351. [Google Scholar] [CrossRef] [PubMed]
- Henson, P.M. Dampening Inflammation. Nat. Immunol. 2005, 6, 1179–1181. [Google Scholar] [CrossRef] [PubMed]
- Rock, K.L.; Kono, H. The Inflammatory Response to Cell Death. Annu. Rev. Pathol. 2008, 3, 99–126. [Google Scholar] [CrossRef] [PubMed]
- Xing, C.; Lo, E.H. Help-Me Signaling: Non-Cell Autonomous Mechanisms of Neuroprotection and Neurorecovery. Prog. Neurobiol. 2017, 152, 181–199. [Google Scholar] [CrossRef] [Green Version]
- Hartings, J.A.; Rolli, M.L.; Lu, X.-C.M.; Tortella, F.C. Delayed Secondary Phase of Peri-Infarct Depolarizations after Focal Cerebral Ischemia: Relation to Infarct Growth and Neuroprotection. J. Neurosci. 2003, 23, 11602–11610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Ren, Q.-G.; Zhang, Z.-H.; Zhou, K.; Yu, Z.-Y.; Luo, X.; Wang, W. Phospho-Rb Mediating Cell Cycle Reentry Induces Early Apoptosis Following Oxygen-Glucose Deprivation in Rat Cortical Neurons. Neurochem. Res. 2012, 37, 503–511. [Google Scholar] [CrossRef]
- Lu, W.; Ai, H.; Peng, L.; Wang, J.; Zhang, B.; Liu, X.; Luo, J. A Novel Phosphorylation Site of N-Methyl-d-Aspartate Receptor GluN2B at S1284 Is Regulated by Cdk5 in Neuronal Ischemia. Exp. Neurol. 2015, 271, 251–258. [Google Scholar] [CrossRef]
- Shin, B.N.; Kim, D.W.; Kim, I.H.; Park, J.H.; Ahn, J.H.; Kang, I.J.; Lee, Y.L.; Lee, C.-H.; Hwang, I.K.; Kim, Y.-M.; et al. Down-Regulation of Cyclin-Dependent Kinase 5 Attenuates P53-Dependent Apoptosis of Hippocampal CA1 Pyramidal Neurons Following Transient Cerebral Ischemia. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef]
- Rashidian, J.; Iyirhiaro, G.O.; Park, D.S. Cell Cycle Machinery and Stroke. Biochim. Biophys. Acta BBA—Mol. Basis Dis. 2007, 1772, 484–493. [Google Scholar] [CrossRef] [Green Version]
- Timsit, S.; Menn, B. Cyclin-Dependent Kinase Inhibition with Roscovitine: Neuroprotection in Acute Ischemic Stroke. Clin. Pharmacol. Ther. 2012, 91, 327–332. [Google Scholar] [CrossRef]
- O’Hare, M.; Wang, F.; Park, D.S. Cyclin-Dependent Kinases as Potential Targets to Improve Stroke Outcome. Pharmacol. Ther. 2002, 93, 135–143. [Google Scholar] [CrossRef]
- Katchanov, J.; Harms, C.; Gertz, K.; Hauck, L.; Waeber, C.; Hirt, L.; Priller, J.; von Harsdorf, R.; Brück, W.; Hörtnagl, H.; et al. Mild Cerebral Ischemia Induces Loss of Cyclin-Dependent Kinase Inhibitors and Activation of Cell Cycle Machinery before Delayed Neuronal Cell Death. J. Neurosci. 2001, 21, 5045–5053. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Liu, S.; Fu, Y.; Wang, J.H.; Lu, Y. Cdk5 Activation Induces Hippocampal CA1 Cell Death by Directly Phosphorylating NMDA Receptors. Nat. Neurosci. 2003, 6, 1039–1047. [Google Scholar] [CrossRef] [PubMed]
- Timsit, S.; Rivera, S.; Ouaghi, P.; Guischard, F.; Tremblay, E.; Ben-Ari, Y.; Khrestchatisky, M. Increased Cyclin D1 in Vulnerable Neurons in the Hippocampus after Ischaemia and Epilepsy: A Modulator of in Vivo Programmed Cell Death? Eur. J. Neurosci. 1999, 11, 263–278. [Google Scholar] [CrossRef] [PubMed]
- Love, S. Neuronal Expression of Cell Cycle-Related Proteins after Brain Ischaemia in Man. Neurosci. Lett. 2003, 353, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Warita, H.; Abe, K.; Itoyama, Y. Expression of Cyclin-Dependent Kinase 5 and Its Activator P35 in Rat Brain after Middle Cerebral Artery Occlusion. Neurosci. Lett. 1999, 265, 37–40. [Google Scholar] [CrossRef]
- Jin, K.; Nagayama, T.; Chen, J.; Stetler, A.R.; Kawaguchi, K.; Simon, R.P.; Graham, S.H. Molecular Cloning of a Cell Cycle Regulation Gene Cyclin H from Ischemic Rat Brain. J. Neurochem. 1999, 73, 1598–1608. [Google Scholar] [CrossRef]
- Denes, A.; Vidyasagar, R.; Feng, J.; Narvainen, J.; McColl, B.W.; Kauppinen, R.A.; Allan, S.M. Proliferating Resident Microglia after Focal Cerebral Ischaemia in Mice. J. Cereb. Blood Flow Metab. 2007, 27, 1941–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lalancette-Hébert, M.; Gowing, G.; Simard, A.; Weng, Y.C.; Kriz, J. Selective Ablation of Proliferating Microglial Cells Exacerbates Ischemic Injury in the Brain. J. Neurosci. 2007, 27, 2596–2605. [Google Scholar] [CrossRef] [Green Version]
- Gelosa, P.; Lecca, D.; Fumagalli, M.; Wypych, D.; Pignieri, A.; Cimino, M.; Verderio, C.; Enerbäck, M.; Nikookhesal, E.; Tremoli, E.; et al. Microglia Is a Key Player in the Reduction of Stroke Damage Promoted by the New Antithrombotic Agent Ticagrelor. J. Cereb. Blood Flow Metab. 2014, 34, 979–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jolivel, V.; Bicker, F.; Binamé, F.; Ploen, R.; Keller, S.; Gollan, R.; Jurek, B.; Birkenstock, J.; Poisa-Beiro, L.; Bruttger, J.; et al. Perivascular Microglia Promote Blood Vessel Disintegration in the Ischemic Penumbra. Acta Neuropathol. (Berl.) 2015, 129, 279–295. [Google Scholar] [CrossRef] [PubMed]
- Hallenbeck, J.M. The Many Faces of Tumor Necrosis Factor in Stroke. Nat. Med. 2002, 8, 1363–1368. [Google Scholar] [CrossRef]
- Wu, M.-H.; Huang, C.-C.; Chio, C.-C.; Tsai, K.-J.; Chang, C.-P.; Lin, N.-K.; Lin, M.-T. Inhibition of Peripheral TNF-α and Downregulation of Microglial Activation by Alpha-Lipoic Acid and Etanercept Protect Rat Brain Against Ischemic Stroke. Mol. Neurobiol. 2016, 53, 4961–4971. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Gan, Y.; Liu, Q.; Yin, J.-X.; Liu, Q.; Shi, J.; Shi, F.-D. CX3CR1 Deficiency Suppresses Activation and Neurotoxicity of Microglia/Macrophage in Experimental Ischemic Stroke. J. Neuroinflammation 2014, 11, 26. [Google Scholar] [CrossRef] [Green Version]
- Yenari Midori, A.; Xu, L.; Tang, X.N.; Qiao, Y.; Giffard, R.G. Microglia Potentiate Damage to Blood–Brain Barrier Constituents. Stroke 2006, 37, 1087–1093. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Bao, J.; Zhao, X.; Shen, H.; Lv, J.; Ma, S.; Zhang, X.; Li, Z.; Wang, S.; Wang, Q.; et al. Activated Cyclin-Dependent Kinase 5 Promotes Microglial Phagocytosis of Fibrillar β-Amyloid by Up-Regulating Lipoprotein Lipase Expression. Mol. Cell. Proteomics MCP 2013, 12, 2833–2844. [Google Scholar] [CrossRef] [Green Version]
- Hilton, G.D.; Stoica, B.A.; Byrnes, K.R.; Faden, A.I. Roscovitine Reduces Neuronal Loss, Glial Activation, and Neurologic Deficits after Brain Trauma. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2008, 28, 1845–1859. [Google Scholar] [CrossRef] [Green Version]
- Kabadi, S.V.; Stoica, B.A.; Byrnes, K.R.; Hanscom, M.; Loane, D.J.; Faden, A.I. Selective CDK Inhibitor Limits Neuroinflammation and Progressive Neurodegeneration after Brain Trauma. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2012, 32, 137–149. [Google Scholar] [CrossRef] [Green Version]
- Kabadi, S.V.; Stoica, B.A.; Loane, D.J.; Luo, T.; Faden, A.I. CR8, a Novel Inhibitor of CDK, Limits Microglial Activation, Astrocytosis, Neuronal Loss, and Neurologic Dysfunction after Experimental Traumatic Brain Injury. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2014, 34, 502–513. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, S.; Kohsaka, S.; Nakajima, K. Role of Cell Cycle-Associated Proteins in Microglial Proliferation in the Axotomized Rat Facial Nucleus. Glia 2012, 60, 570–581. [Google Scholar] [CrossRef]
- Kim, J.-E.; Park, H.; Choi, S.-H.; Kong, M.-J.; Kang, T.-C. Roscovitine Attenuates Microglia Activation and Monocyte Infiltration via P38 MAPK Inhibition in the Rat Frontoparietal Cortex Following Status Epilepticus. Cells 2019, 8, 746. [Google Scholar] [CrossRef] [Green Version]
- Tomov, N.; Surchev, L.; Wiedenmann, C.; Döbrössy, M.; Nikkhah, G. Roscovitine, an Experimental CDK5 Inhibitor, Causes Delayed Suppression of Microglial, but Not Astroglial Recruitment around Intracerebral Dopaminergic Grafts. Exp. Neurol. 2019, 318, 135–144. [Google Scholar] [CrossRef]
- Zhang, H.H.; Yang, B.X.; Huang, J.L.; Shun, J.L.; Kong, F.J.; Chen, Z.G.; Lu, J.M. Cdk5 Contributes to Inflammation-Induced Thermal Hyperalgesia Mediated by the P38 MAPK Pathway in Microglia. Brain Res. 2015, 1619, 166–175. [Google Scholar] [CrossRef] [Green Version]
- Pepe, G.; De Maglie, M.; Minoli, L.; Villa, A.; Maggi, A.; Vegeto, E. Selective Proliferative Response of Microglia to Alternative Polarization Signals. J. Neuroinflammation 2017, 14, 236. [Google Scholar] [CrossRef] [Green Version]
- Zamanian, J.L.; Xu, L.; Foo, L.C.; Nouri, N.; Zhou, L.; Giffard, R.G.; Barres, B.A. Genomic Analysis of Reactive Astrogliosis. J. Neurosci. 2012, 32, 6391–6410. [Google Scholar] [CrossRef] [Green Version]
- Hol, E.M.; Pekny, M. Glial Fibrillary Acidic Protein (GFAP) and the Astrocyte Intermediate Filament System in Diseases of the Central Nervous System. Curr. Opin. Cell Biol. 2015, 32, 121–130. [Google Scholar] [CrossRef]
- Yao, X.; Derugin, N.; Manley, G.T.; Verkman, A.S. Reduced Brain Edema and Infarct Volume in Aquaporin-4 Deficient Mice after Transient Focal Cerebral Ischemia. Neurosci. Lett. 2015, 584, 368–372. [Google Scholar] [CrossRef] [Green Version]
- Colombo, E.; Farina, C. Astrocytes: Key Regulators of Neuroinflammation. Trends Immunol. 2016, 37, 608–620. [Google Scholar] [CrossRef]
- Tan, S.; Shan, Y.; Lin, Y.; Liao, S.; Zhang, B.; Zeng, Q.; Wang, Y.; Deng, Z.; Chen, C.; Hu, X.; et al. Neutralization of Interleukin-9 Ameliorates Experimental Stroke by Repairing the Blood-Brain Barrier via down-Regulation of Astrocyte-Derived Vascular Endothelial Growth Factor-A. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 4376–4387. [Google Scholar] [CrossRef]
- Li, Y.-N.; Pan, R.; Qin, X.-J.; Yang, W.-L.; Qi, Z.; Liu, W.; Liu, K.J. Ischemic Neurons Activate Astrocytes to Disrupt Endothelial Barrier via Increasing VEGF Expression. J. Neurochem. 2014, 129, 120–129. [Google Scholar] [CrossRef] [Green Version]
- Abeysinghe, H.C.S.; Phillips, E.L.; Chin-Cheng, H.; Beart, P.M.; Roulston, C.L. Modulating Astrocyte Transition after Stroke to Promote Brain Rescue and Functional Recovery: Emerging Targets Include Rho Kinase. Int. J. Mol. Sci. 2016, 17, 288. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Chopp, M. Astrocytes, Therapeutic Targets for Neuroprotection and Neurorestoration in Ischemic Stroke. Prog. Neurobiol. 2016, 144, 103–120. [Google Scholar] [CrossRef] [Green Version]
- Manley, G.T.; Fujimura, M.; Ma, T.; Noshita, N.; Filiz, F.; Bollen, A.W.; Chan, P.; Verkman, A.S. Aquaporin-4 Deletion in Mice Reduces Brain Edema after Acute Water Intoxication and Ischemic Stroke. Nat. Med. 2000, 6, 159–163. [Google Scholar] [CrossRef]
- Posada-Duque, R.A.; Palacio-Castañeda, V.; Cardona-Gómez, G.P. CDK5 Knockdown in Astrocytes Provide Neuroprotection as a Trophic Source via Rac1. Mol. Cell. Neurosci. 2015, 68, 151–166. [Google Scholar] [CrossRef]
- He, Y.; Li, H.-L.; Xie, W.-Y.; Yang, C.-Z.; Yu, A.C.H.; Wang, Y. The Presence of Active Cdk5 Associated with P35 in Astrocytes and Its Important Role in Process Elongation of Scratched Astrocyte. Glia 2007, 55, 573–583. [Google Scholar] [CrossRef]
- Zhu, Z.; Zhang, Q.; Yu, Z.; Zhang, L.; Tian, D.; Zhu, S.; Bu, B.; Xie, M.; Wang, W. Inhibiting Cell Cycle Progression Reduces Reactive Astrogliosis Initiated by Scratch Injury in Vitro and by Cerebral Ischemia in Vivo. Glia 2007, 55, 546–558. [Google Scholar] [CrossRef]
- Di Giovanni, S.; Movsesyan, V.; Ahmed, F.; Cernak, I.; Schinelli, S.; Stoica, B.; Faden, A.I. Cell Cycle Inhibition Provides Neuroprotection and Reduces Glial Proliferation and Scar Formation after Traumatic Brain Injury. Proc. Natl. Acad. Sci. USA 2005, 102, 8333–8338. [Google Scholar] [CrossRef] [Green Version]
- Hyun, H.-W.; Min, S.-J.; Kim, J.-E. CDK5 Inhibitors Prevent Astroglial Apoptosis and Reactive Astrogliosis by Regulating PKA and DRP1 Phosphorylations in the Rat Hippocampus. Neurosci. Res. 2017, 119, 24–37. [Google Scholar] [CrossRef]
- Zhong, Y.; Chen, J.; Chen, J.; Chen, Y.; Li, L.; Xie, Y. Crosstalk between Cdk5/P35 and ERK1/2 Signalling Mediates Spinal Astrocyte Activity via the PPARγ Pathway in a Rat Model of Chronic Constriction Injury. J. Neurochem. 2019, 151, 166–184. [Google Scholar] [CrossRef]
- Tanaka, T.; Tatsuno, I.; Noguchi, Y.; Uchida, D.; Oeda, T.; Narumiya, S.; Yasuda, T.; Higashi, H.; Kitagawa, M.; Nakayama, K.; et al. Activation of Cyclin-Dependent Kinase 2 (Cdk2) in Growth-Stimulated Rat Astrocytes. Geranylgeranylated Rho Small GTPase(s) Are Essential for the Induction of Cyclin E Gene Expression. J. Biol. Chem. 1998, 273, 26772–26778. [Google Scholar] [CrossRef] [Green Version]
- Tikoo, R.; Casaccia-Bonnefil, P.; Chao, M.V.; Koff, A. Changes in Cyclin-Dependent Kinase 2 and P27kip1 Accompany Glial Cell Differentiation of Central Glia-4 Cells. J. Biol. Chem. 1997, 272, 442–447. [Google Scholar] [CrossRef] [Green Version]
- Becerra-Calixto, A.; Posada-Duque, R.; Cardona-Gómez, G.P. Recovery of Neurovascular Unit Integrity by CDK5-KD Astrocyte Transplantation in a Global Cerebral Ischemia Model. Mol. Neurobiol. 2018, 55, 8563–8585. [Google Scholar] [CrossRef]
- Becerra-Calixto, A.; Cardona-Gómez, G.P. Neuroprotection Induced by Transplanted CDK5 Knockdown Astrocytes in Global Cerebral Ischemic Rats. Mol. Neurobiol. 2017, 54, 6681–6696. [Google Scholar] [CrossRef]
- Garriga, J.; Graña, X. CDK9 Inhibition Strategy Defines Distinct Sets of Target Genes. BMC Res. Notes 2014, 7, 301. [Google Scholar] [CrossRef] [Green Version]
- Mifsud, G.; Zammit, C.; Muscat, R.; Di Giovanni, G.; Valentino, M. Oligodendrocyte Pathophysiology and Treatment Strategies in Cerebral Ischemia. CNS Neurosci. Ther. 2014, 20, 603–612. [Google Scholar] [CrossRef]
- Dewar, D.; Underhill, S.M.; Goldberg, M.P. Oligodendrocytes and Ischemic Brain Injury. J. Cereb. Blood Flow Metab. 2016. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Lu, J.; Shao, A.; Zhang, J.H.; Zhang, J. Glial Cells: Role of the Immune Response in Ischemic Stroke. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Wang, L.; Geng, J.; Qu, M.; Yuan, F.; Wang, Y.; Pan, J.; Li, Y.; Ma, Y.; Zhou, P.; Zhang, Z.; et al. Oligodendrocyte Precursor Cells Transplantation Protects Blood–Brain Barrier in a Mouse Model of Brain Ischemia via Wnt/β-Catenin Signaling. Cell Death Dis. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Traiffort, E.; Kassoussi, A.; Zahaf, A.; Laouarem, Y. Astrocytes and Microglia as Major Players of Myelin Production in Normal and Pathological Conditions. Front. Cell. Neurosci. 2020, 14. [Google Scholar] [CrossRef] [Green Version]
- Marin, M.A.; Carmichael, S.T. Mechanisms of Demyelination and Remyelination in the Young and Aged Brain Following White Matter Stroke. Neurobiol. Dis. 2019, 126, 5–12. [Google Scholar] [CrossRef]
- Zhang, R.; Chopp, M.; Zhang, Z.G. Oligodendrogenesis after Cerebral Ischemia. Front. Cell. Neurosci. 2013, 7. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.; Malgrange, B.; Rocher, V.; Hans, G.; Moonen, G.; Rigo, J.-M.; Belachew, S. Chemical Inhibitors of Cyclin-Dependent Kinases Control Proliferation, Apoptosis and Differentiation of Oligodendroglial Cells. Int. J. Dev. Neurosci. 2003, 21, 321–326. [Google Scholar] [CrossRef]
- Miyamoto, Y.; Yamauchi, J.; Chan, J.R.; Okada, A.; Tomooka, Y.; Hisanaga, S.; Tanoue, A. Cdk5 Regulates Differentiation of Oligodendrocyte Precursor Cells through the Direct Phosphorylation of Paxillin. J. Cell Sci. 2007, 120, 4355–4366. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, Y.; Yamauchi, J.; Tanoue, A. Cdk5 Phosphorylation of WAVE2 Regulates Oligodendrocyte Precursor Cell Migration through Nonreceptor Tyrosine Kinase Fyn. J. Neurosci. 2008, 28, 8326–8337. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Wang, H.; Zhang, J.; Luo, F.; Herrup, K.; Bibb, J.A.; Lu, R.; Miller, R.H. Cyclin Dependent Kinase 5 Is Required for the Normal Development of Oligodendrocytes and Myelin Formation. Dev. Biol. 2013, 378, 94–106. [Google Scholar] [CrossRef] [Green Version]
- Luo, F.; Burke, K.; Kantor, C.; Miller, R.H.; Yang, Y. Cyclin-Dependent Kinase 5 Mediates Adult OPC Maturation and Myelin Repair through Modulation of Akt and GsK-3β Signaling. J. Neurosci. 2014, 34, 10415–10429. [Google Scholar] [CrossRef]
- Akundi, R.S.; Rivkees, S.A. Hypoxia Alters Cell Cycle Regulatory Protein Expression and Induces Premature Maturation of Oligodendrocyte Precursor Cells. PLoS ONE 2009, 4. [Google Scholar] [CrossRef] [Green Version]
- Ghiani, C.; Gallo, V. Inhibition of Cyclin E-Cyclin-Dependent Kinase 2 Complex Formation and Activity Is Associated with Cell Cycle Arrest and Withdrawal in Oligodendrocyte Progenitor Cells. J. Neurosci. 2001, 21, 1274–1282. [Google Scholar] [CrossRef] [Green Version]
- Caillava, C.; Vandenbosch, R.; Jablonska, B.; Deboux, C.; Spigoni, G.; Gallo, V.; Malgrange, B.; Baron-Van Evercooren, A. Cdk2 Loss Accelerates Precursor Differentiation and Remyelination in the Adult Central Nervous System. J. Cell Biol. 2011, 193, 397–407. [Google Scholar] [CrossRef] [Green Version]
- Belachew, S.; Aguirre, A.A.; Wang, H.; Vautier, F.; Yuan, X.; Anderson, S.; Kirby, M.; Gallo, V. Cyclin-Dependent Kinase-2 Controls Oligodendrocyte Progenitor Cell Cycle Progression and Is Downregulated in Adult Oligodendrocyte Progenitors. J. Neurosci. 2002, 22, 8553–8562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caillava, C.; Baron-Van Evercooren, A. Differential Requirement of Cyclin-Dependent Kinase 2 for Oligodendrocyte Progenitor Cell Proliferation and Differentiation. Cell Div. 2012, 7, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Q.; He, C.; Liu, H.; Liao, X.; Dai, B.; Chen, Y.; Yang, Y.; Zhao, B.; Bihl, J.; Ma, X. Microvascular Endothelial Cells-Derived Microvesicles Imply in Ischemic Stroke by Modulating Astrocyte and Blood Brain Barrier Function and Cerebral Blood Flow. Mol. Brain 2016, 9, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Didier, N.; Romero, I.A.; Créminon, C.; Wijkhuisen, A.; Grassi, J.; Mabondzo, A. Secretion of Interleukin-1beta by Astrocytes Mediates Endothelin-1 and Tumour Necrosis Factor-Alpha Effects on Human Brain Microvascular Endothelial Cell Permeability. J. Neurochem. 2003, 86, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, G.; Granger, D.N. Cell Adhesion Molecules and Ischemic Stroke. Neurol. Res. 2008, 30, 783–793. [Google Scholar] [CrossRef]
- Clark, W.M.; Lauten, J.D.; Lessov, N.; Woodward, W.; Coull, B.M. The Influence of Antiadhesion Therapies on Leukocyte Subset Accumulation in Central Nervous System Ischemia in Rats. J. Mol. Neurosci. MN 1995, 6, 43–50. [Google Scholar] [CrossRef]
- Bowes, M.P.; Rothlein, R.; Fagan, S.C.; Zivin, J.A. Monoclonal Antibodies Preventing Leukocyte Activation Reduce Experimental Neurologic Injury and Enhance Efficacy of Thrombolytic Therapy. Neurology 1995, 45, 815–819. [Google Scholar] [CrossRef]
- Wong, R.; Lénárt, N.; Hill, L.; Toms, L.; Coutts, G.; Martinecz, B.; Császár, E.; Nyiri, G.; Papaemmanouil, A.; Waisman, A.; et al. Interleukin-1 Mediates Ischaemic Brain Injury via Distinct Actions on Endothelial Cells and Cholinergic Neurons. Brain. Behav. Immun. 2019, 76, 126–138. [Google Scholar] [CrossRef]
- Berberich, N.; Uhl, B.; Joore, J.; Schmerwitz, U.K.; Mayer, B.A.; Reichel, C.A.; Krombach, F.; Zahler, S.; Vollmar, A.M.; Fürst, R. Roscovitine Blocks Leukocyte Extravasation by Inhibition of Cyclin-Dependent Kinases 5 and 9. Br. J. Pharmacol. 2011, 163, 1086–1098. [Google Scholar] [CrossRef] [Green Version]
- Liebl, J.; Weitensteiner, S.B.; Vereb, G.; Takács, L.; Fürst, R.; Vollmar, A.M.; Zahler, S. Cyclin-Dependent Kinase 5 Regulates Endothelial Cell Migration and Angiogenesis. J. Biol. Chem. 2010, 285, 35932–35943. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Zhang, Y.; Zhang, R.; Zhao, Z.; Zhang, H.; Wu, J.; Shen, W.; Zhong, M. Cyclin-Dependent Kinase 1 Disruption Inhibits Angiogenesis by Inducing Cell Cycle Arrest and Apoptosis. Exp. Ther. Med. 2019, 18, 3062–3070. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Walsh, K.; Wang, J. Regulation of Cdk2 Activity in Endothelial Cells That Are Inhibited from Growth by Cell Contact. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 629–635. [Google Scholar] [CrossRef] [Green Version]
- Shi, C.-S.; Kuo, K.-L.; Chen, M.-S.; Chow, P.-M.; Liu, S.-H.; Chang, Y.-W.; Lin, W.-C.; Liao, S.-M.; Hsu, C.-H.; Hsu, F.-S.; et al. Suppression of Angiogenesis by Targeting Cyclin-Dependent Kinase 7 in Human Umbilical Vein Endothelial Cells and Renal Cell Carcinoma: An In Vitro and In Vivo Study. Cells 2019, 8, 1469. [Google Scholar] [CrossRef] [Green Version]
- Schmerwitz, U.K.; Sass, G.; Khandoga, A.G.; Joore, J.; Mayer, B.A.; Berberich, N.; Totzke, F.; Krombach, F.; Tiegs, G.; Zahler, S.; et al. Flavopiridol Protects against Inflammation by Attenuating Leukocyte-Endothelial Interaction via Inhibition of Cyclin-Dependent Kinase 9. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 280–288. [Google Scholar] [CrossRef] [Green Version]
- Fumagalli, S.; Perego, C.; Pischiutta, F.; Zanier, E.R.; De Simoni, M.-G. The Ischemic Environment Drives Microglia and Macrophage Function. Front. Neurol. 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Benakis, C.; Garcia-Bonilla, L.; Iadecola, C.; Anrather, J. The Role of Microglia and Myeloid Immune Cells in Acute Cerebral Ischemia. Front. Cell. Neurosci. 2014, 8, 461. [Google Scholar] [CrossRef] [PubMed]
- Schilling, M.; Strecker, J.-K.; Schäbitz, W.-R.; Ringelstein, E.B.; Kiefer, R. Effects of Monocyte Chemoattractant Protein 1 on Blood-Borne Cell Recruitment after Transient Focal Cerebral Ischemia in Mice. Neuroscience 2009, 161, 806–812. [Google Scholar] [CrossRef]
- Rajan, W.D.; Wojtas, B.; Gielniewski, B.; Gieryng, A.; Zawadzka, M.; Kaminska, B. Dissecting Functional Phenotypes of Microglia and Macrophages in the Rat Brain after Transient Cerebral Ischemia. Glia 2019, 67, 232–245. [Google Scholar] [CrossRef]
- Jian, Z.; Liu, R.; Zhu, X.; Smerin, D.; Zhong, Y.; Gu, L.; Fang, W.; Xiong, X. The Involvement and Therapy Target of Immune Cells After Ischemic Stroke. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Han, D.; Liu, H.; Gao, Y. The Role of Peripheral Monocytes and Macrophages in Ischemic Stroke. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 2020. [Google Scholar] [CrossRef]
- Du, J.; Wei, N.; Guan, T.; Xu, H.; An, J.; Pritchard, K.A.; Shi, Y. Inhibition of CDKS by Roscovitine Suppressed LPS-Induced ·NO Production through Inhibiting NFκB Activation and BH4 Biosynthesis in Macrophages. Am. J. Physiol.—Cell Physiol. 2009, 297, C742–C749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfänder, P.; Fidan, M.; Burret, U.; Lipinski, L.; Vettorazzi, S. Cdk5 Deletion Enhances the Anti-Inflammatory Potential of GC-Mediated GR Activation During Inflammation. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Jhou, R.-S.; Sun, K.-H.; Sun, G.-H.; Wang, H.-H.; Chang, C.-I.; Huang, H.-C.; Lu, S.-Y.; Tang, S.-J. Inhibition of Cyclin-Dependent Kinases by Olomoucine and Roscovitine Reduces Lipopolysaccharide-Induced Inflammatory Responses via down-Regulation of Nuclear Factor ΚB. Cell Prolif. 2009, 42, 141–149. [Google Scholar] [CrossRef]
- Xu, J.; Xue, Z.; Zhang, C.; Liu, Y.; Busuttil, R.W.; Zhang, J.; Kupiec-Weglinski, J.W.; Ji, H. Inhibition of Cyclin-Dependent Kinase 2 Signaling Prevents Liver Ischemia and Reperfusion Injury. Transplantation 2019, 103, 724–732. [Google Scholar] [CrossRef]
- Na, Y.R.; Jung, D.; Gu, G.J.; Jang, A.R.; Suh, Y.-H.; Seok, S.H. The Early Synthesis of P35 and Activation of CDK5 in LPS-Stimulated Macrophages Suppresses Interleukin-10 Production. Sci. Signal. 2015, 8, ra121. [Google Scholar] [CrossRef]
- Rajan, W.D.; Wojtas, B.; Gielniewski, B.; Miró-Mur, F.; Pedragosa, J.; Zawadzka, M.; Pilanc, P.; Planas, A.M.; Kaminska, B. Defining Molecular Identity and Fates of CNS-Border Associated Macrophages after Ischemic Stroke in Rodents and Humans. Neurobiol. Dis. 2020, 137, 104722. [Google Scholar] [CrossRef] [PubMed]
- Jickling, G.C.; Liu, D.; Ander, B.P.; Stamova, B.; Zhan, X.; Sharp, F.R. Targeting Neutrophils in Ischemic Stroke: Translational Insights from Experimental Studies. J. Cereb. Blood Flow Metab. 2015, 35, 888–901. [Google Scholar] [CrossRef] [Green Version]
- Weston, R.M.; Jones, N.M.; Jarrott, B.; Callaway, J.K. Inflammatory Cell Infiltration after Endothelin-1-Induced Cerebral Ischemia: Histochemical and Myeloperoxidase Correlation with Temporal Changes in Brain Injury. J. Cereb. Blood Flow Metab. 2007, 27, 100–114. [Google Scholar] [CrossRef] [Green Version]
- Strecker, J.-K.; Schmidt, A.; Schäbitz, W.-R.; Minnerup, J. Neutrophil Granulocytes in Cerebral Ischemia—Evolution from Killers to Key Players. Neurochem. Int. 2017, 107, 117–126. [Google Scholar] [CrossRef] [Green Version]
- del Zoppo, G.J.; Schmid-Schönbein, G.W.; Mori, E.; Copeland, B.R.; Chang, C.M. Polymorphonuclear Leukocytes Occlude Capillaries Following Middle Cerebral Artery Occlusion and Reperfusion in Baboons. Stroke 1991, 22, 1276–1283. [Google Scholar] [CrossRef] [Green Version]
- Ludewig, P.; Sedlacik, J.; Gelderblom, M.; Bernreuther, C.; Korkusuz, Y.; Wagener, C.; Gerloff, C.; Fiehler, J.; Magnus, T.; Horst, A.K. Carcinoembryonic Antigen-Related Cell Adhesion Molecule 1 Inhibits MMP-9-Mediated Blood-Brain-Barrier Breakdown in a Mouse Model for Ischemic Stroke. Circ. Res. 2013, 113, 1013–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Bonilla, L.; Moore, J.M.; Racchumi, G.; Zhou, P.; Butler, J.M.; Iadecola, C.; Anrather, J. Inducible Nitric Oxide Synthase in Neutrophils and Endothelium Contributes to Ischemic Brain Injury in Mice. J. Immunol. 2014, 193, 2531–2537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otxoa-de-Amezaga, A.; Gallizioli, M.; Pedragosa, J.; Justicia, C.; Miró-Mur, F.; Salas-Perdomo, A.; Díaz-Marugan, L.; Gunzer, M.; Planas, A.M. Location of Neutrophils in Different Compartments of the Damaged Mouse Brain After Severe Ischemia/Reperfusion. Stroke 2019, 50, 1548–1557. [Google Scholar] [CrossRef]
- Cartwright, J.A.; Lucas, C.D.; Rossi, A.G. Inflammation Resolution and the Induction of Granulocyte Apoptosis by Cyclin-Dependent Kinase Inhibitor Drugs. Front. Pharmacol. 2019, 10. [Google Scholar] [CrossRef]
- Leitch, A.E.; Lucas, C.D.; Marwick, J.A.; Duffin, R.; Haslett, C.; Rossi, A.G. Cyclin-Dependent Kinases 7 and 9 Specifically Regulate Neutrophil Transcription and Their Inhibition Drives Apoptosis to Promote Resolution of Inflammation. Cell Death Differ. 2012, 19, 1950–1961. [Google Scholar] [CrossRef] [Green Version]
- Rossi, A.G.; Sawatzky, D.A.; Walker, A.; Ward, C.; Sheldrake, T.A.; Riley, N.A.; Caldicott, A.; Martinez-Losa, M.; Walker, T.R.; Duffin, R.; et al. Cyclin-Dependent Kinase Inhibitors Enhance the Resolution of Inflammation by Promoting Inflammatory Cell Apoptosis. Nat. Med. 2006, 12, 1056–1064. [Google Scholar] [CrossRef]
- Leitch, A.E.; Riley, N.A.; Sheldrake, T.A.; Festa, M.; Fox, S.; Duffin, R.; Haslett, C.; Rossi, A.G. The Cyclin-Dependent Kinase Inhibitor R-Roscovitine down-Regulates Mcl-1 to Override pro-Inflammatory Signalling and Drive Neutrophil Apoptosis. Eur. J. Immunol. 2010, 40, 1127–1138. [Google Scholar] [CrossRef]
- Farahi, N.; Uller, L.; Juss, J.K.; Langton, A.J.; Cowburn, A.S.; Gibson, A.; Foster, M.R.; Farrow, S.N.; Marco-Casanova, P.; Sobolewski, A.; et al. Effects of the Cyclin-Dependent Kinase Inhibitor R-Roscovitine on Eosinophil Survival and Clearance. Clin. Exp. Allergy J. Br. Soc. Allergy Clin. Immunol. 2011, 41, 673–687. [Google Scholar] [CrossRef] [PubMed]
- Gautam, S.; Kirschnek, S.; Wiesmeier, M.; Vier, J.; Häcker, G. Roscovitine-Induced Apoptosis in Neutrophils and Neutrophil Progenitors Is Regulated by the Bcl-2-Family Members Bim, Puma, Noxa and Mcl-1. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [Green Version]
- Rosales, J.L.; Ernst, J.D.; Hallows, J.; Lee, K.-Y. GTP-Dependent Secretion from Neutrophils Is Regulated by Cdk5. J. Biol. Chem. 2004, 279, 53932–53936. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.-Y.; Liu, L.; Jin, Y.; Fu, S.; Rosales, J.L. Cdk5 Mediates Vimentin Ser56 Phosphorylation during GTP-Induced Secretion by Neutrophils. J. Cell. Physiol. 2012, 227, 739–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Z.-X.; Qiu, S.; Lou, B.-S.; Yang, Y.; Wang, W.-C.; Lin, X.-F. Roscovitine Ameliorates Endotoxin-Induced Uveitis through Neutrophil Apoptosis. Mol. Med. Rep. 2016, 14, 1083–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Hampson, P.; Hazeldine, J.; Krystof, V.; Strnad, M.; Pechan, P.M.J. Cyclin-Dependent Kinase 9 Activity Regulates Neutrophil Spontaneous Apoptosis. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [Green Version]
- Klausen, P.; Bjerregaard, M.D.; Borregaard, N.; Cowland, J.B. End-Stage Differentiation of Neutrophil Granulocytes in Vivo Is Accompanied by up-Regulation of P27kip1 and down-Regulation of CDK2, CDK4, and CDK6. J. Leukoc. Biol. 2004, 75, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Pareek, T.K.; Lam, E.; Zheng, X.; Askew, D.; Kulkarni, A.B.; Chance, M.R.; Huang, A.Y.; Cooke, K.R.; Letterio, J.J. Cyclin-Dependent Kinase 5 Activity Is Required for T Cell Activation and Induction of Experimental Autoimmune Encephalomyelitis. J. Exp. Med. 2010, 207, 2507–2519. [Google Scholar] [CrossRef]
- Hoodless, L.J.; Lucas, C.D.; Duffin, R.; Denvir, M.A.; Haslett, C.; Tucker, C.S.; Rossi, A.G. Genetic and Pharmacological Inhibition of CDK9 Drives Neutrophil Apoptosis to Resolve Inflammation in Zebrafish in Vivo. Sci. Rep. 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Ma, L.; Lin, T.; Li, S.-J.; Chen, L.-L.; Wang, D.-Z. The Significance of Eosinophils in Predicting the Severity of Acute Ischemic Stroke. Oncotarget 2017, 8, 104238–104246. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.-B.; Liu, S.; Zhang, F.; Mao, G.-S.; Sun, L.-Z.; Liu, Y. The Role of Eosinophils in Stroke: A Pilot Study. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 3643–3648. [Google Scholar]
- Jucevičiūtė, N.; Mikužis, P.; Balnytė, R. Absolute Blood Eosinophil Count Could Be a Potential Biomarker for Predicting Haemorrhagic Transformation after Intravenous Thrombolysis for Acute Ischaemic Stroke. BMC Neurol. 2019, 19, 127. [Google Scholar] [CrossRef]
- Zhao, H.-M.; Qin, W.-Q.; Wang, P.-J.; Wen, Z.-M. Eosinopenia Is a Predictive Factor for the Severity of Acute Ischemic Stroke. Neural Regen. Res. 2019, 14, 1772–1779. [Google Scholar] [CrossRef]
- Duffin, R.; Leitch, A.E.; Sheldrake, T.A.; Hallett, J.M.; Meyer, C.; Fox, S.; Alessandri, A.L.; Martin, M.C.; Brady, H.J.; Teixeira, M.M.; et al. The CDK Inhibitor, R-Roscovitine, Promotes Eosinophil Apoptosis by down-Regulation of Mcl-1. FEBS Lett. 2009, 583, 2540–2546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odemuyiwa, S.O.; Ilarraza, R.; Davoine, F.; Logan, M.R.; Shayeganpour, A.; Wu, Y.; Majaesic, C.; Adamko, D.J.; Moqbel, R.; Lacy, P. Cyclin-Dependent Kinase 5 Regulates Degranulation in Human Eosinophils. Immunology 2015, 144, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Gelderblom, M.; Leypoldt, F.; Steinbach, K.; Behrens, D.; Choe, C.U.; Siler, D.A.; Arumugam, T.V.; Orthey, E.; Gerloff, C.; Tolosa, E.; et al. Temporal and Spatial Dynamics of Cerebral Immune Cell Accumulation in Stroke. Stroke 2009, 40, 1849–1857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jander, S.; Kraemer, M.; Schroeter, M.; Witte, O.W.; Stoll, G. Lymphocytic Infiltration and Expression of Intercellular Adhesion Molecule-1 in Photochemically Induced Ischemia of the Rat Cortex. J. Cereb. Blood Flow Metab. 1995, 15, 42–51. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Liao, S.; Wei, C.; Jia, D.; Wood, K.; Liu, Q.; Wang, X.; Shi, F.-D.; Jin, W.-N. Infiltration and Persistence of Lymphocytes during Late-Stage Cerebral Ischemia in Middle Cerebral Artery Occlusion and Photothrombotic Stroke Models. J. Neuroinflammation 2017, 14, 248. [Google Scholar] [CrossRef] [Green Version]
- Jones, K.A.; Maltby, S.; Plank, M.W.; Kluge, M.; Nilsson, M.; Foster, P.S.; Walker, F.R. Peripheral Immune Cells Infiltrate into Sites of Secondary Neurodegeneration after Ischemic Stroke. Brain. Behav. Immun. 2018, 67, 299–307. [Google Scholar] [CrossRef]
- Selvaraj, U.M.; Stowe, A.M. Long-Term T Cell Responses in the Brain After an Ischemic Stroke. Discov. Med. 2017, 24, 323–333. [Google Scholar]
- Yilmaz, G.; Arumugam, T.V.; Stokes, K.Y.; Granger, D.N. Role of T Lymphocytes and Interferon-Gamma in Ischemic Stroke. Circulation 2006, 113, 2105–2112. [Google Scholar] [CrossRef] [Green Version]
- Gelderblom, M.; Weymar, A.; Bernreuther, C.; Velden, J.; Arunachalam, P.; Steinbach, K.; Orthey, E.; Arumugam, T.V.; Leypoldt, F.; Simova, O.; et al. Neutralization of the IL-17 Axis Diminishes Neutrophil Invasion and Protects from Ischemic Stroke. Blood 2012, 120, 3793–3802. [Google Scholar] [CrossRef] [Green Version]
- Kleinschnitz, C.; Schwab, N.; Kraft, P.; Hagedorn, I.; Dreykluft, A.; Schwarz, T.; Austinat, M.; Nieswandt, B.; Wiendl, H.; Stoll, G. Early Detrimental T-Cell Effects in Experimental Cerebral Ischemia Are Neither Related to Adaptive Immunity nor Thrombus Formation. Blood 2010, 115, 3835–3842. [Google Scholar] [CrossRef]
- Planas, A.M.; Chamorro, A. Regulatory T Cells Protect the Brain after Stroke. Nat. Med. 2009, 15, 138–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liesz, A.; Suri-Payer, E.; Veltkamp, C.; Doerr, H.; Sommer, C.; Rivest, S.; Giese, T.; Veltkamp, R. Regulatory T Cells Are Key Cerebroprotective Immunomodulators in Acute Experimental Stroke. Nat. Med. 2009, 15, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Wang, L.; Zhou, Y.; Gan, Y.; Zhu, W.; Xia, Y.; Jiang, X.; Watkins, S.; Vazquez, A.; Thomson, A.W.; et al. C-C Chemokine Receptor Type 5 (CCR5)-Mediated Docking of Transferred Tregs Protects Against Early Blood-Brain Barrier Disruption After Stroke. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Kleinschnitz, C.; Kraft, P.; Dreykluft, A.; Hagedorn, I.; Göbel, K.; Schuhmann, M.K.; Langhauser, F.; Helluy, X.; Schwarz, T.; Bittner, S.; et al. Regulatory T Cells Are Strong Promoters of Acute Ischemic Stroke in Mice by Inducing Dysfunction of the Cerebral Microvasculature. Blood 2013, 121, 679–691. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, Q.; Leskov, K.S.; Wu, X.; Duan, J.; Zhang, G.L.; Hall, M.; Rosenbaum, J.T. Roscovitine Suppresses CD4+ T Cells and T Cell-Mediated Experimental Uveitis. PLoS ONE 2013, 8, e81154. [Google Scholar] [CrossRef] [Green Version]
- Lam, E.; Pareek, T.K.; Letterio, J.J. Cdk5 Controls IL-2 Gene Expression via Repression of the MSin3a-HDAC Complex. Cell Cycle Georget. Tex 2015, 14, 1327–1336. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Wang, H.; Kim, J.S.; Pihan, G.; Boussiotis, V.A. The Cyclin Dependent Kinase Inhibitor (R)-Roscovitine Prevents Alloreactive T Cell Clonal Expansion and Protects against Acute GvHD. Cell Cycle Georget. Tex 2009, 8, 1794–1802. [Google Scholar] [CrossRef]
- Pezzotta, A.; Mister, M.; Monteferrante, G.; Cassis, L.; Azzollini, N.; Aiello, S.; Satta, M.; Benigni, A.; Remuzzi, G.; Noris, M. Effect of Seliciclib (CYC202, R-Roscovitine) on Lymphocyte Alloreactivity and Acute Kidney Allograft Rejection in Rat. Transplantation 2008, 85, 1476–1482. [Google Scholar] [CrossRef]
- Zoja, C.; Casiraghi, F.; Conti, S.; Corna, D.; Rottoli, D.; Cavinato, R.A.; Remuzzi, G.; Benigni, A. Cyclin-Dependent Kinase Inhibition Limits Glomerulonephritis and Extends Lifespan of Mice with Systemic Lupus. Arthritis Rheum. 2007, 56, 1629–1637. [Google Scholar] [CrossRef]
- Malyshkina, A.; Littwitz-Salomon, E.; Sutter, K.; Zelinskyy, G.; Windmann, S.; Schimmer, S.; Paschen, A.; Streeck, H.; Hasenkrug, K.J.; Dittmer, U. Fas Ligand-Mediated Cytotoxicity of CD4+ T Cells during Chronic Retrovirus Infection. Sci. Rep. 2017, 7, 7785. [Google Scholar] [CrossRef]
- Hassin, D.; Garber, O.G.; Meiraz, A.; Schiffenbauer, Y.S.; Berke, G. Cytotoxic T Lymphocyte Perforin and Fas Ligand Working in Concert Even When Fas Ligand Lytic Action Is Still Not Detectable. Immunology 2011, 133, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Sairanen, T.; Karjalainen-Lindsberg, M.-L.; Paetau, A.; Ijäs, P.; Lindsberg, P.J. Apoptosis Dominant in the Periinfarct Area of Human Ischaemic Stroke—A Possible Target of Antiapoptotic Treatments. Brain 2006, 129, 189–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullah, I.; Chung, K.; Oh, J.; Beloor, J.; Bae, S.; Lee, S.C.; Lee, M.; Kumar, P.; Lee, S.-K. Intranasal Delivery of a Fas-Blocking Peptide Attenuates Fas-Mediated Apoptosis in Brain Ischemia. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Torgler, R.; Jakob, S.; Ontsouka, E.; Nachbur, U.; Mueller, C.; Green, D.R.; Brunner, T. Regulation of Activation-Induced Fas (CD95/Apo-1) Ligand Expression in T Cells by the Cyclin B1/Cdk1 Complex. J. Biol. Chem. 2004, 279, 37334–37342. [Google Scholar] [CrossRef] [Green Version]
- Chunder, N.; Wang, L.; Chen, C.; Hancock, W.W.; Wells, A.D. Cyclin-Dependent Kinase 2 Controls Peripheral Immune Tolerance. J. Immunol. 2012, 189, 5659–5666. [Google Scholar] [CrossRef] [Green Version]
- Leucci, E.; De Falco, G.; Onnis, A.; Cerino, G.; Cocco, M.; Luzzi, A.; Crupi, D.; Tigli, C.; Bellan, C.; Tosi, P.; et al. The Role of the Cdk9/Cyclin T1 Complex in T Cell Differentiation. J. Cell. Physiol. 2007, 212, 411–415. [Google Scholar] [CrossRef]
- Dolati, S.; Ahmadi, M.; Khalili, M.; Taheraghdam, A.A.; Siahmansouri, H.; Babaloo, Z.; Aghebati-Maleki, L.; Jadidi-Niaragh, F.; Younesi, V.; Yousefi, M. Peripheral Th17/Treg Imbalance in Elderly Patients with Ischemic Stroke. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 2018, 39, 647–654. [Google Scholar] [CrossRef]
- Cipollini, V.; Anrather, J.; Orzi, F.; Iadecola, C. Th17 and Cognitive Impairment: Possible Mechanisms of Action. Front. Neuroanat. 2019, 13. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, H.; Kotani, H.; Kondo, T.; Tani, I.; Wei, X.; Tsuruta, S.; Kimura, A.; Asakawa, M.; Ito, M.; Nagai, S.; et al. CDK Inhibitors Suppress Th17 and Promote ITreg Differentiation, and Ameliorate Experimental Autoimmune Encephalomyelitis in Mice. Biochem. Biophys. Res. Commun. 2013, 435, 378–384. [Google Scholar] [CrossRef]
- Khor, B.; Gagnon, J.D.; Goel, G.; Roche, M.I.; Conway, K.L.; Tran, K.; Aldrich, L.N.; Sundberg, T.B.; Paterson, A.M.; Mordecai, S.; et al. The Kinase DYRK1A Reciprocally Regulates the Differentiation of Th17 and Regulatory T Cells. eLife 2015, 4, e05920. [Google Scholar] [CrossRef]
- Meijer, L.; Nelson, D.J.; Riazanski, V.; Gabdoulkhakova, A.G.; Hery-Arnaud, G.; Le Berre, R.; Loaëc, N.; Oumata, N.; Galons, H.; Nowak, E.; et al. Modulating Innate and Adaptive Immunity by (R)-Roscovitine: Potential Therapeutic Opportunity in Cystic Fibrosis. J. Innate Immun. 2016, 8, 330–349. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Akiyoshi, K.; Dziennis, S.; Vandenbark, A.A.; Herson, P.S.; Hurn, P.D.; Offner, H. Regulatory B Cells Limit CNS Inflammation and Neurologic Deficits in Murine Experimental Stroke. J. Neurosci. 2011, 31, 8556–8563. [Google Scholar] [CrossRef] [PubMed]
- Bodhankar, S.; Chen, Y.; Lapato, A.; Vandenbark, A.A.; Murphy, S.J.; Saugstad, J.A.; Offner, H. Regulatory CD8(+)CD122 (+) T-Cells Predominate in CNS after Treatment of Experimental Stroke in Male Mice with IL-10-Secreting B-Cells. Metab. Brain Dis. 2015, 30, 911–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortega, S.B.; Torres, V.O.; Latchney, S.E.; Whoolery, C.W.; Noorbhai, I.Z.; Poinsatte, K.; Selvaraj, U.M.; Benson, M.A.; Meeuwissen, A.J.M.; Plautz, E.J.; et al. B Cells Migrate into Remote Brain Areas and Support Neurogenesis and Functional Recovery after Focal Stroke in Mice. Proc. Natl. Acad. Sci. USA 2020, 117, 4983–4993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuhmann, M.K.; Langhauser, F.; Kraft, P.; Kleinschnitz, C. B Cells Do Not Have a Major Pathophysiologic Role in Acute Ischemic Stroke in Mice. J. Neuroinflammation 2017, 14. [Google Scholar] [CrossRef] [Green Version]
- Doyle, K.P.; Quach, L.N.; Solé, M.; Axtell, R.C.; Nguyen, T.-V.V.; Soler-Llavina, G.J.; Jurado, S.; Han, J.; Steinman, L.; Longo, F.M.; et al. B-Lymphocyte-Mediated Delayed Cognitive Impairment Following Stroke. J. Neurosci. 2015, 35, 2133–2145. [Google Scholar] [CrossRef] [PubMed]
- Hahntow, I.N.; Schneller, F.; Oelsner, M.; Weick, K.; Ringshausen, I.; Fend, F.; Peschel, C.; Decker, T. Cyclin-Dependent Kinase Inhibitor Roscovitine Induces Apoptosis in Chronic Lymphocytic Leukemia Cells. Leukemia 2004, 18, 747–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zolnierczyk, J.D.; Błoński, J.Z.; Robak, T.; Kiliańska, Z.M.; Wesierska-Gadek, J. Roscovitine Triggers Apoptosis in B-Cell Chronic Lymphocytic Leukemia Cells with Similar Efficiency as Combinations of Conventional Purine Analogs with Cyclophosphamide. Ann. N. Y. Acad. Sci. 2009, 1171, 124–131. [Google Scholar] [CrossRef]
- Tanguay, D.A.; Chiles, T.C. Cell Cycle-Specific Induction of Cdk2 Expression in B Lymphocytes Following Antigen Receptor Cross-Linking. Mol. Immunol. 1994, 31, 643–649. [Google Scholar] [CrossRef]
- Wołowiec, D.; Deviller, P.; Simonin, D.; Souchier, C.; Rimokh, R.; Benchaib, M.; Bryon, P.A.; Ffrench, M. Cdk1 Is a Marker of Proliferation in Human Lymphoid Cells. Int. J. Cancer 1995, 61, 381–388. [Google Scholar] [CrossRef]
- Faber, A.C.; Chiles, T.C. Inhibition of Cyclin-Dependent Kinase-2 Induces Apoptosis in Human Diffuse Large B-Cell Lymphomas. Cell Cycle Georget. Tex 2007, 6, 2982–2989. [Google Scholar] [CrossRef] [PubMed]
- Farina, F.M.; Inguscio, A.; Kunderfranco, P.; Cortesi, A.; Elia, L.; Quintavalle, M. MicroRNA-26a/Cyclin-Dependent Kinase 5 Axis Controls Proliferation, Apoptosis and in Vivo Tumor Growth of Diffuse Large B-Cell Lymphoma Cell Lines. Cell Death Dis. 2017, 8, e2890. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Kim, D.H.; Yoon, D.H.; Suh, C.; Choi, C.-M.; Lee, J.C.; Hong, J.Y.; Rho, J.K. Efficacy of the Novel CDK7 Inhibitor QS1189 in Mantle Cell Lymphoma. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- De Falco, G.; Leucci, E.; Onnis, A.; Bellan, C.; Tigli, C.; Wirths, S.; Cerino, G.; Cocco, M.; Crupi, D.; De Luca, A.; et al. Cdk9/Cyclin T1 Complex: A Key Player during the Activation/Differentiation Process of Normal Lymphoid B Cells. J. Cell. Physiol. 2008, 215, 276–282. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le Roy, L.; Letondor, A.; Le Roux, C.; Amara, A.; Timsit, S. Cellular and Molecular Mechanisms of R/S-Roscovitine and CDKs Related Inhibition under Both Focal and Global Cerebral Ischemia: A Focus on Neurovascular Unit and Immune Cells. Cells 2021, 10, 104. https://doi.org/10.3390/cells10010104
Le Roy L, Letondor A, Le Roux C, Amara A, Timsit S. Cellular and Molecular Mechanisms of R/S-Roscovitine and CDKs Related Inhibition under Both Focal and Global Cerebral Ischemia: A Focus on Neurovascular Unit and Immune Cells. Cells. 2021; 10(1):104. https://doi.org/10.3390/cells10010104
Chicago/Turabian StyleLe Roy, Lucas, Anne Letondor, Cloé Le Roux, Ahmed Amara, and Serge Timsit. 2021. "Cellular and Molecular Mechanisms of R/S-Roscovitine and CDKs Related Inhibition under Both Focal and Global Cerebral Ischemia: A Focus on Neurovascular Unit and Immune Cells" Cells 10, no. 1: 104. https://doi.org/10.3390/cells10010104
APA StyleLe Roy, L., Letondor, A., Le Roux, C., Amara, A., & Timsit, S. (2021). Cellular and Molecular Mechanisms of R/S-Roscovitine and CDKs Related Inhibition under Both Focal and Global Cerebral Ischemia: A Focus on Neurovascular Unit and Immune Cells. Cells, 10(1), 104. https://doi.org/10.3390/cells10010104