Cytokine-Mediated Crosstalk between Immune Cells and Epithelial Cells in the Gut


Abstract
:1. Introduction
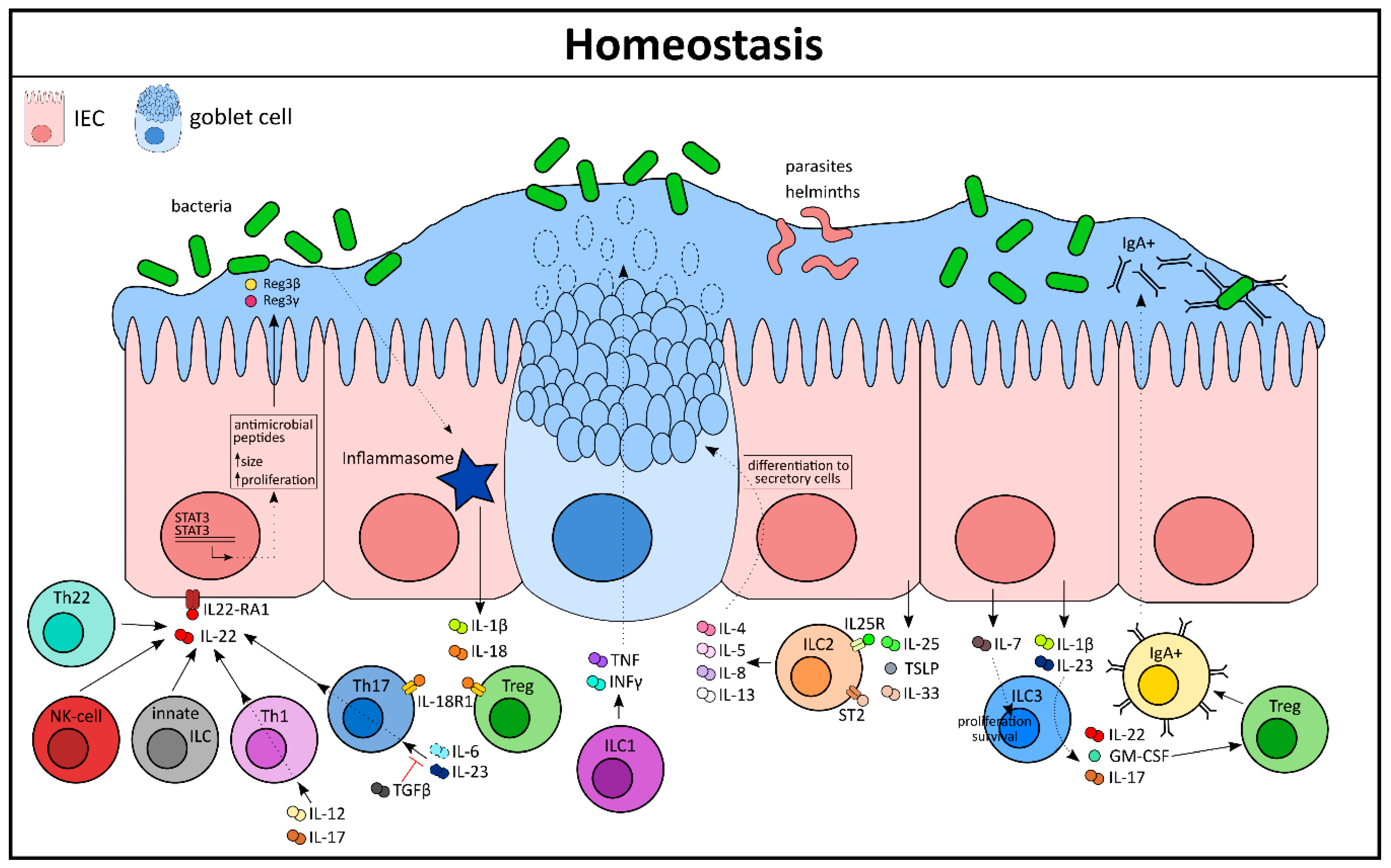
2. Tranquility in the Gut: Cytokines Maintaining Homeostasis
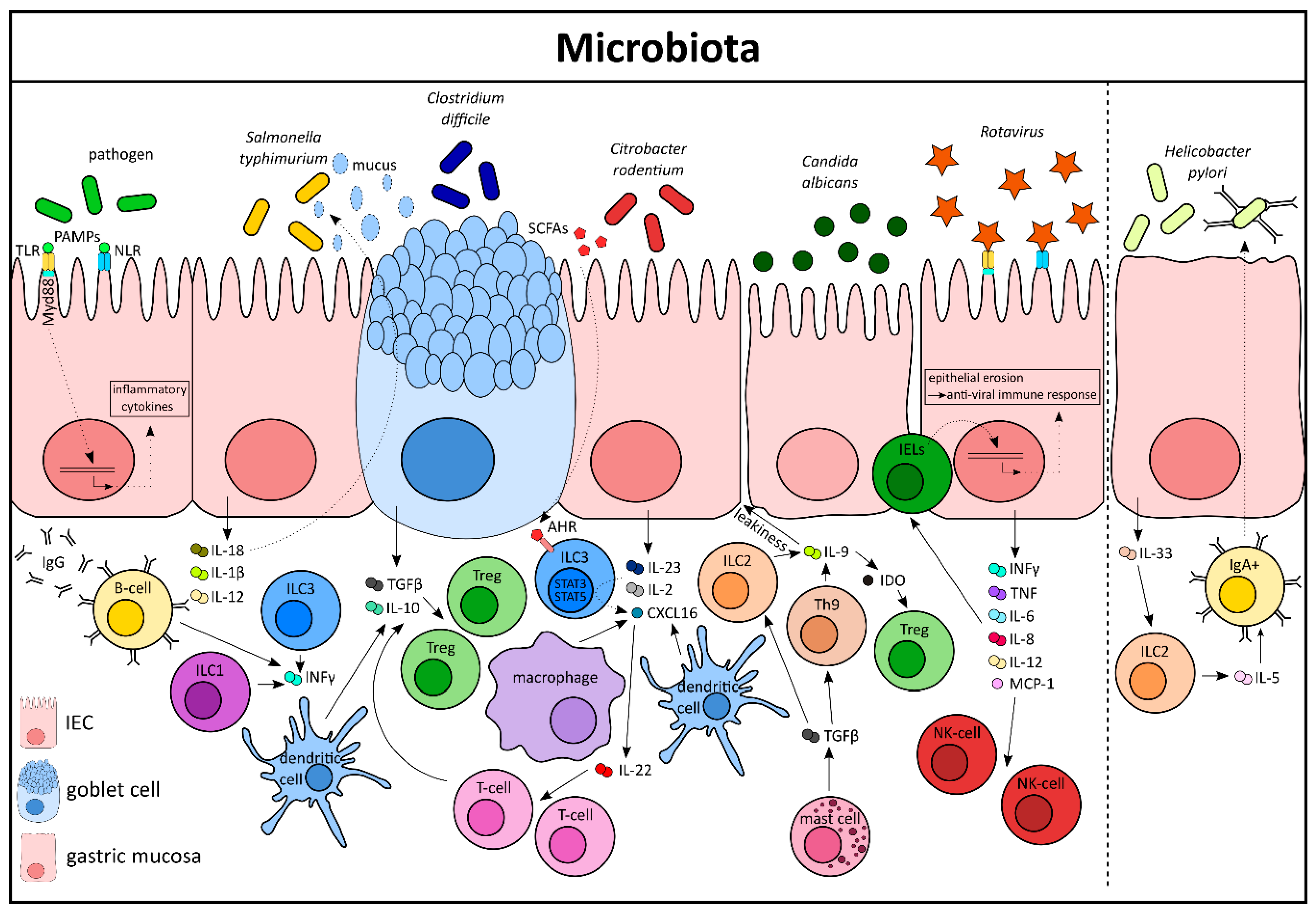
3. Intestinal Microbiota Influences Cytokine Production
4. Cytokine Regulation in Intestinal Pathology
4.1. Irritable Bowel Syndrome and Celiac Disease
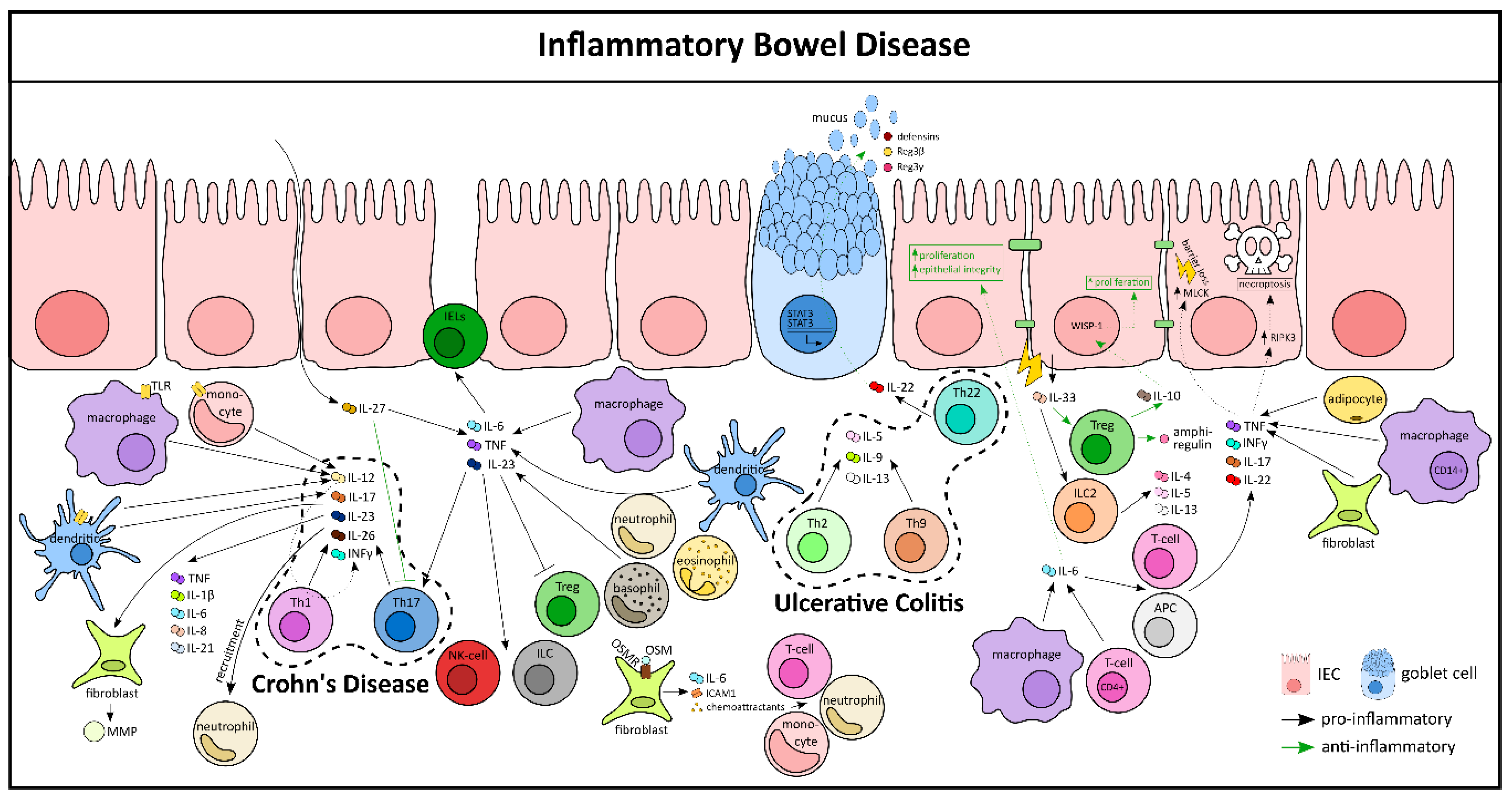
4.2. Inflammatory Bowel Diseases
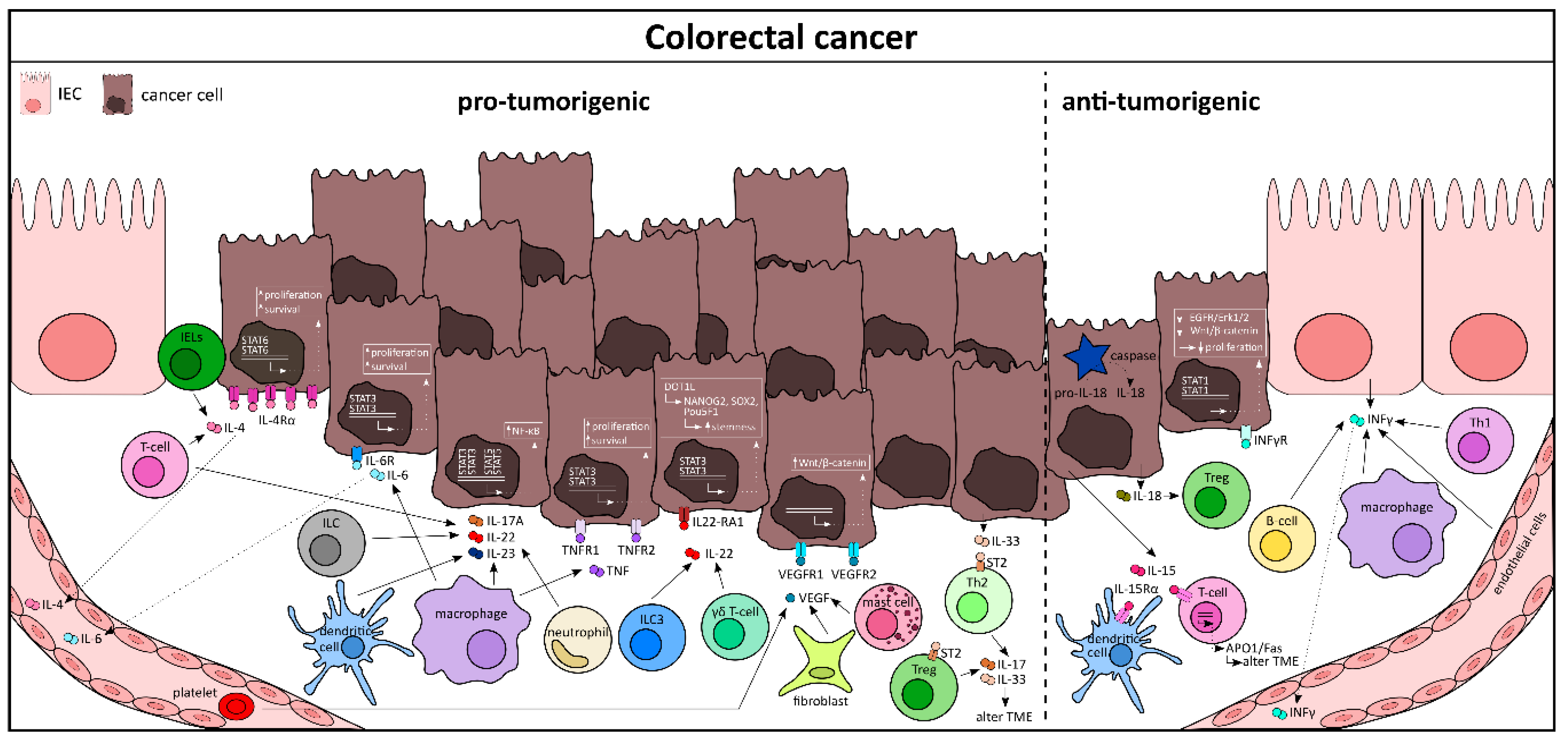
4.3. Cytokine Networks in Colorectal Cancer
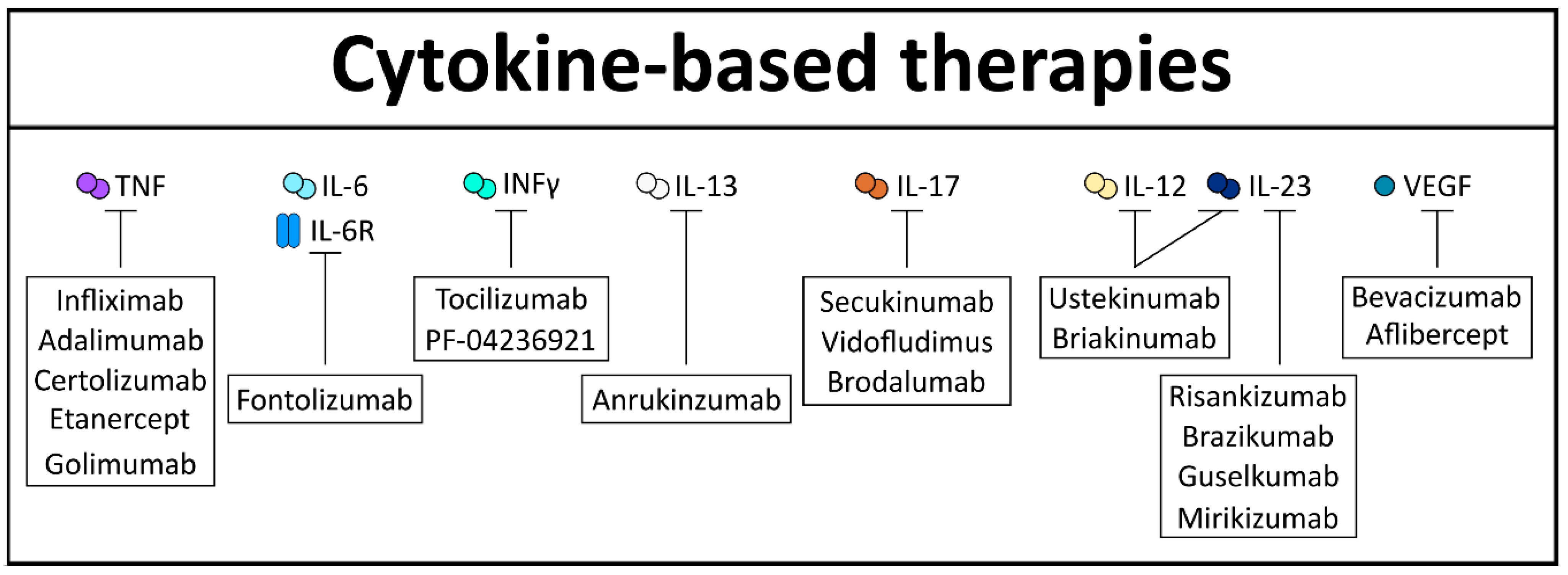
5. Cytokine-Mediated Therapy for Gut Diseases
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Peterson, L.W.; Artis, D. Intestinal epithelial cells: Regulators of barrier function and immune homeostasis. Nat. Rev. Immunol. 2014, 14, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Burgueño, J.F.; Abreu, M.T. Epithelial Toll-like receptors and their role in gut homeostasis and disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 263–278. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, M.; Pohin, M.; Powrie, F. Cytokine Networks in the Pathophysiology of Inflammatory Bowel Disease. Immunity 2019, 50, 992–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rathinam, V.A.; Chan, F.K.-M. Inflammasome, Inflammation, and Tissue Homeostasis. Trends Mol. Med. 2018, 24, 304–318. [Google Scholar] [CrossRef] [PubMed]
- Palomo, J.; Dietrich, D.; Martin, P.; Palmer, G.; Gabay, C. The interleukin (IL)-1 cytokine family—Balance between agonists and antagonists in inflammatory diseases. Cytokine 2015, 76, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Nowarski, R.; Jackson, R.; Gagliani, N.; De Zoete, M.R.; Palm, N.W.; Bailis, W.; Low, J.S.; Harman, C.C.D.; Graham, M.; Elinav, E.; et al. Epithelial IL-18 Equilibrium Controls Barrier Function in Colitis. Cell 2015, 163, 1444–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, O.J.; Srinivasan, N.; Pott, J.W.R.; Schiering, C.; Krausgruber, T.; Ilott, N.E.; Maloy, K.J. Epithelial-derived IL-18 regulates Th17 cell differentiation and Foxp3+ Treg cell function in the intestine. Mucosal Immunol. 2015, 8, 1226–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shohan, M.; Dehghani, R.; Khodadadi, A.; Dehnavi, S.; Ahmadi, R.; Joudaki, N.; Houshmandfar, S.; Shamshiri, M.; Shojapourian, S.; Bagheri, N. Interleukin-22 and intestinal homeostasis: Protective or destructive? IUBMB Life 2020, 72, 1585–1602. [Google Scholar] [CrossRef]
- Liang, S.C.; Tan, X.-Y.; Luxenberg, D.P.; Karim, R.; Dunussi-Joannopoulos, K.; Collins, M.; Fouser, L.A. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J. Exp. Med. 2006, 203, 2271–2279. [Google Scholar] [CrossRef]
- Zheng, Y.; Danilenko, D.M.; Valdez, P.A.; Kasman, I.; Eastham-Anderson, J.; Wu, J.; Ouyang, W. Interleukin-22, a TH17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature 2006, 445, 648–651. [Google Scholar] [CrossRef]
- Rutz, S.; Noubade, R.; Eidenschenk, C.; Ota, N.; Zeng, W.; Zheng, Y.; Hackney, J.; Ding, J.; Singh, H.; Ouyang, W. Transcription factor c-Maf mediates the TGF-beta-dependent suppression of IL-22 production in T(H)17 cells. Nat. Immunol. 2011, 12, 1238–1245. [Google Scholar] [CrossRef] [PubMed]
- Satoh-Takayama, N.; Vosshenrich, C.A.J.; Lesjean-Pottier, S.; Sawa, S.; Lochner, M.; Rattis, F.; Mention, J.-J.; Thiam, K.; Cerf-Bensussan, N.; Mandelboim, O.; et al. Microbial Flora Drives Interleukin 22 Production in Intestinal NKp46+ Cells that Provide Innate Mucosal Immune Defense. Immunity 2008, 29, 958–970. [Google Scholar] [CrossRef] [PubMed]
- Lindemans, C.A.; Calafiore, M.; Mertelsmann, A.M.; O’connor, M.H.; Dudakov, J.A.; Jenq, R.R.; Velardi, E.; Young, L.F.; Smith, O.M.; Lawrence, G.; et al. Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration. Nature 2015, 528, 560–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zwarycz, B.; Gracz, A.D.; Rivera, K.R.; Williamson, I.A.; Samsa, L.A.; Starmer, J.; Daniele, M.A.; Salter-Cid, L.; Zhao, Q.; Magness, S.T. IL22 Inhibits Epithelial Stem Cell Expansion in an Ileal Organoid Model. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Valdez, P.A.; Danilenko, D.M.; Hu, Y.; Sa, S.M.; Gong, Q.; Abbas, A.R.; Modrusan, Z.; Ghilardi, N.; De Sauvage, F.J.; et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat. Med. 2008, 14, 282–289. [Google Scholar] [CrossRef]
- Geremia, A.; Arancibia-Cárcamo, C.V. Innate Lymphoid Cells in Intestinal Inflammation. Front. Immunol. 2017, 8, 1296. [Google Scholar] [CrossRef]
- Spits, H.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate lymphoid cells—A proposal for uniform nomenclature. Nat. Rev. Immunol. 2013, 13, 145–149. [Google Scholar] [CrossRef]
- Krämer, B.; Goeser, F.; Lutz, P.; Glässner, A.; Boesecke, C.; Schwarze-Zander, C.; Kaczmarek, D.J.; Nischalke, H.D.; Branchi, V.; Manekeller, S.; et al. Compartment-specific distribution of human intestinal innate lymphoid cells is altered in HIV patients under effective therapy. PLoS Pathog. 2017, 13, e1006373. [Google Scholar] [CrossRef]
- Klose, C.S.; Kiss, E.A.; Schwierzeck, V.; Ebert, K.; Hoyler, T.; d’Hargues, Y.; Göppert, N.; Croxford, A.L.; Waisman, A.; Tanriver, Y.; et al. A T-bet gradient controls the fate and function of CCR6-RORgammat+ innate lymphoid cells. Nature 2013, 494, 261–265. [Google Scholar] [CrossRef]
- Klose, C.S.; Flach, M.; Möhle, L.; Rogell, L.; Hoyler, T.; Ebert, K.; Fabiunke, C.; Pfeifer, D.; Sexl, V.; Fonseca-Pereira, D.; et al. Differentiation of Type 1 ILCs from a Common Progenitor to All Helper-like Innate Lymphoid Cell Lineages. Cell 2014, 157, 340–356. [Google Scholar] [CrossRef] [Green Version]
- Mjösberg, J.; Spits, H. Human innate lymphoid cells. J. Allergy Clin. Immunol. 2016, 138, 1265–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Paul, W.E. Inflammatory group 2 innate lymphoid cells. Int. Immunol. 2015, 28, dxv044-8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Cornelissen, F.; Papazian, N.; Reijmers, R.M.; Llorian, M.; Cupedo, T.; Coles, M.; Seddon, B. IL-7–dependent maintenance of ILC3s is required for normal entry of lymphocytes into lymph nodes. J. Exp. Med. 2018, 215, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Forkel, M.; Mjösberg, J. Dysregulation of Group 3 Innate Lymphoid Cells in the Pathogenesis of Inflammatory Bowel Disease. Curr. Allergy Asthma Rep. 2016, 16, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, T.; Shitara, S.; Imai, K.; Miyachi, H.; Kitano, S.; Yao, H.; Tani-Ichi, S.; Ikuta, K. Identification of IL-7–Producing Cells in Primary and Secondary Lymphoid Organs Using IL-7–GFP Knock-In Mice. J. Immunol. 2012, 189, 1577–1584. [Google Scholar] [CrossRef] [Green Version]
- Maloy, K.J.; Kullberg, M.C. IL-23 and Th17 cytokines in intestinal homeostasis. Mucosal Immunol. 2008, 1, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Sonnenberg, G.F.; Monticelli, L.A.; Elloso, M.M.; Fouser, L.A.; Artis, D. CD4+ Lymphoid Tissue-Inducer Cells Promote Innate Immunity in the Gut. Immunity 2011, 34, 122–134. [Google Scholar] [CrossRef] [Green Version]
- Gladiator, A.; Wangler, N.; Trautwein-Weidner, K.; LeibundGut-Landmann, S. Cutting Edge: IL-17–Secreting Innate Lymphoid Cells Are Essential for Host Defense against Fungal Infection. J. Immunol. 2012, 190, 521–525. [Google Scholar] [CrossRef] [Green Version]
- Luu, M.; Steinhoff, U.; Visekruna, A. Functional heterogeneity of gut-resident regulatory T cells. Clin. Transl. Immunol. 2017, 6, e156. [Google Scholar] [CrossRef]
- Goto, Y.; Obata, T.; Kunisawa, J.; Sato, S.; Ivanov, I.I.; Lamichhane, A.; Takeyama, N.; Kamioka, M.; Sakamoto, M.; Matsuki, T.; et al. Innate lymphoid cells regulate intestinal epithelial cell glycosylation. Science 2014, 345, 1254009. [Google Scholar] [CrossRef] [Green Version]
- Sabihi, M.; Böttcher, M.; Pelczar, P.; Huber, S. Microbiota-Dependent Effects of IL-22. Cells 2020, 9, 2205. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Kolodziejczyk, A.A.; Thaiss, C.A.; Elinav, E. Dysbiosis and the immune system. Nat. Rev. Immunol. 2017, 17, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Thaiss, C.A.; Levy, M.; Suez, J.; Elinav, E. The interplay between the innate immune system and the microbiota. Curr. Opin. Immunol. 2014, 26, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Frantz, A.L.; Rogier, E.W.; Weber, C.R.; Shen, L.; Cohen, D.A.; Fenton, L.A.; Bruno, M.E.C.; Kaetzel, C.S. Targeted deletion of MyD88 in intestinal epithelial cells results in compromised antibacterial immunity associated with downregulation of polymeric immunoglobulin receptor, mucin-2, and antibacterial peptides. Mucosal Immunol. 2012, 5, 501–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouskra, D.; Brézillon, C.; Bérard, M.; Werts, C.; Varona, R.; Boneca, I.G.; Eberl, G. Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature 2008, 456, 507–510. [Google Scholar] [CrossRef]
- Henao-Mejia, J.; Elinav, E.; Thaiss, C.A.; Flavell, R.A. Inflammasomes and Metabolic Disease. Annu. Rev. Physiol. 2014, 76, 57–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elinav, E.; Strowig, T.; Kau, A.L.; Henao-Mejia, J.; Thaiss, C.A.; Booth, C.J.; Peaper, D.R.; Bertin, J.; Eisenbarth, S.C.; Gordon, J.I.; et al. NLRP6 Inflammasome Regulates Colonic Microbial Ecology and Risk for Colitis. Cell 2011, 145, 745–757. [Google Scholar] [CrossRef] [Green Version]
- Ghimire, L.; Paudel, S.; Jin, L.; Jeyaseelan, S. The NLRP6 inflammasome in health and disease. Mucosal Immunol. 2020, 13, 388–398. [Google Scholar] [CrossRef] [Green Version]
- Belkaid, Y.; Hand, T.W. Role of the Microbiota in Immunity and Inflammation. Cell 2014, 157, 121–141. [Google Scholar] [CrossRef] [Green Version]
- Raupach, B.; Peuschel, S.K.; Monack, D.M.; Zychlinsky, A. Caspase-1-mediated activation of interleukin-1beta (IL-1beta) and IL-18 contributes to innate immune defenses against Salmonella enterica serovar Typhimurium infection. Infect. Immun. 2006, 74, 4922–4926. [Google Scholar] [CrossRef] [Green Version]
- Jarret, A.; Jackson, R.; Duizer, C.; Healy, M.E.; Zhao, J.; Rone, J.M.; Bielecki, P.; Sefik, E.; Roulis, M.; Rice, T.; et al. Enteric Nervous System-Derived IL-18 Orchestrates Mucosal Barrier Immunity. Cell 2020, 180, 813–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, K.; Ohno, H.; Satoh-Takayama, N. Innate Lymphoid Cells: Important Regulators of Host–Bacteria Interaction for Border Defense. Microorganisms 2020, 8, 1342. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.; Yu, J.; Hu, B.; Lu, C.; Liu, X.; Gao, X.; Li, W.; Zhou, L.; Wang, J.; Wang, D.; et al. Runx3 Mediates Resistance to Intracellular Bacterial Infection by Promoting IL12 Signaling in Group 1 ILC and NCR+ILC3. Front. Immunol. 2018, 9, 2101. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.M.; Kenny, D.J.; Xavier, R.J. Gut Microbiota Regulation of T Cells During Inflammation and Autoimmunity. Annu. Rev. Immunol. 2019, 37, 599–624. [Google Scholar] [CrossRef] [PubMed]
- Reid-Yu, S.A.; Small, C.-L.N.; Coombes, B.K. CD3−NK1.1+cells aid in the early induction of a Th1 response to an attaching and effacing enteric pathogen. Eur. J. Immunol. 2013, 43, 2638–2649. [Google Scholar] [CrossRef]
- Qiu, J.; Heller, J.J.; Guo, X.; Chen, Z.-M.E.; Fish, K.; Fu, Y.-X.; Zhou, L. The Aryl Hydrocarbon Receptor Regulates Gut Immunity through Modulation of Innate Lymphoid Cells. Immunity 2012, 36, 92–104. [Google Scholar] [CrossRef] [Green Version]
- Kiss, E.A.; Vonarbourg, C.; Kopfmann, S.; Hobeika, E.; Finke, D.; Esser, C.; Diefenbach, A. Natural Aryl Hydrocarbon Receptor Ligands Control Organogenesis of Intestinal Lymphoid Follicles. Science 2011, 334, 1561–1565. [Google Scholar] [CrossRef]
- Chen, J.; Waddell, A.E.; Lin, Y.-D.; Cantorna, M.T. Dysbiosis caused by vitamin D receptor deficiency confers colonization resistance to Citrobacter rodentium through modulation of innate lymphoid cells. Mucosal Immunol. 2015, 8, 618–626. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Yu, T.; Huang, X.; Bilotta, A.J.; Xu, L.; Lu, Y.; Sun, J.; Pan, F.; Zhou, J.; Zhang, W.; et al. Intestinal microbiota-derived short-chain fatty acids regulation of immune cell IL-22 production and gut immunity. Nat. Commun. 2020, 11, 4457. [Google Scholar] [CrossRef]
- Manta, C.; Heupel, E.; Radulovic, K.; Rossini, V.; Garbi, N.; Riedel, C.U.; Niess, J.H. CX3CR1+ macrophages support IL-22 production by innate lymphoid cells during infection with Citrobacter rodentium. Mucosal Immunol. 2012, 6, 177–188. [Google Scholar] [CrossRef] [Green Version]
- Satoh-Takayama, N.; Serafini, N.; Verrier, T.; Rekiki, A.; Renauld, J.-C.; Frankel, G.; Di Santo, J.P. The Chemokine Receptor CXCR6 Controls the Functional Topography of Interleukin-22 Producing Intestinal Innate Lymphoid Cells. Immunity 2014, 41, 776–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longman, R.S.; Diehl, G.E.; Victorio, D.A.; Huh, J.R.; Galan, C.; Miraldi, E.R.; Swaminath, A.; Bonneau, R.; Scherl, E.J.; Littman, D.R. CX3CR1+ mononuclear phagocytes support colitis-associated innate lymphoid cell production of IL-22. J. Exp. Med. 2014, 211, 1571–1583. [Google Scholar] [CrossRef] [PubMed]
- Bauché, D.; Joyce-Shaikh, B.; Fong, J.; Villarino, A.V.; Ku, K.S.; Jain, R.; Lee, Y.-C.; Annamalai, L.; Yearley, J.H.; Cua, D.J. IL-23 and IL-2 activation of STAT5 is required for optimal IL-22 production in ILC3s during colitis. Sci. Immunol. 2020, 5, eaav1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buzzelli, J.N.; Chalinor, H.V.; Pavlic, D.I.; Sutton, P.; Menheniott, T.R.; Giraud, A.S.; Judd, L.M. IL33 Is a Stomach Alarmin That Initiates a Skewed Th2 Response to Injury and Infection. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 203–221.e3. [Google Scholar] [CrossRef] [Green Version]
- Satoh-Takayama, N.; Kato, T.; Motomura, Y.; Kageyama, T.; Taguchi-Atarashi, N.; Kinoshita-Daitoku, R.; Kuroda, E.; Di Santo, J.P.; Mimuro, H.; Moro, K.; et al. Bacteria-Induced Group 2 Innate Lymphoid Cells in the Stomach Provide Immune Protection through Induction of IgA. Immunity 2020, 52, 635–649.e4. [Google Scholar] [CrossRef]
- Li, R.; Jiang, X.-X.; Zhang, L.-F.; Liu, X.-M.; Hu, T.-Z.; Xia, X.-J.; Li, M.; Xu, C.-X. Group 2 Innate Lymphoid Cells Are Involved in Skewed Type 2 Immunity of Gastric Diseases Induced by Helicobacter pylori Infection. Mediat. Inflamm. 2017, 2017, 4927964. [Google Scholar] [CrossRef] [Green Version]
- Conteduca, V.; Sansonno, D.; Lauletta, G.; Russi, S.; Ingravallo, G.; Dammacco, F.H. pylori infection and gastric cancer: State of the art (review). Int. J. Oncol. 2013, 42, 5–18. [Google Scholar] [CrossRef] [Green Version]
- Sonoyama, K.; Miki, A.; Sugita, R.; Goto, H.; Nakata, M.; Yamaguchi, N. Gut colonization by Candida albicans aggravates inflammation in the gut and extra-gut tissues in mice. Med. Mycol. 2011, 49, 237–247. [Google Scholar] [CrossRef]
- Renga, G.; Moretti, S.; Oikonomou, V.; Borghi, M.; Zelante, T.; Paolicelli, G.; Costantini, C.; De Zuani, M.; Villella, V.R.; Raia, V.; et al. IL-9 and Mast Cells Are Key Players of Candida albicans Commensalism and Pathogenesis in the Gut. Cell Rep. 2018, 23, 1767–1778. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Wang, C.; Tang, C.; He, Q.; Li, N.; Li, J. Dysbiosis of Gut Fungal Microbiota is Associated With Mucosal Inflammation in Crohn’s Disease. J. Clin. Gastroenterol. 2014, 48, 513–523. [Google Scholar] [CrossRef] [Green Version]
- Perry, A.K.; Chen, G.; Zheng, D.; Tang, H.; Cheng, G. The host type I interferon response to viral and bacterial infections. Cell Res. 2005, 15, 407–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boshuizen, J.A.; Reimerink, J.; Male, A.M.K.-V.; Van Ham, V.J.J.; Koopmans, M.P.G.; Büller, H.A.; Dekker, J.; Einerhand, A.W.C. Changes in small intestinal homeostasis, morphology, and gene expression during rotavirus infection of infant mice. J. Virol. 2003, 77, 13005–13016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldridge, M.T.; Lee, S.; Brown, J.J.; McAllister, N.; Urbanek, K.; Dermody, T.S.; Nice, T.J.; Virgin, H.W. Expression of Ifnlr1 on Intestinal Epithelial Cells Is Critical to the Antiviral Effects of Interferon Lambda against Norovirus and Reovirus. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pott, J.; Mahlakõiv, T.; Mordstein, M.; Duerr, C.U.; Michiels, T.; Stockinger, S.; Staeheli, P.; Hornef, M.W. IFN-lambda determines the intestinal epithelial antiviral host defense. Proc. Natl. Acad. Sci. USA 2011, 108, 7944–7949. [Google Scholar] [CrossRef] [Green Version]
- Villena, J.; Vizoso-Pinto, M.G.; Kitazawa, H. Intestinal Innate Antiviral Immunity and Immunobiotics: Beneficial Effects against Rotavirus Infection. Front. Immunol. 2016, 7, 563. [Google Scholar] [CrossRef] [Green Version]
- Clayburgh, D.R.; Shen, L.; Turner, J.R. A porous defense: The leaky epithelial barrier in intestinal disease. Lab. Investig. 2004, 84, 282–291. [Google Scholar] [CrossRef] [Green Version]
- Chatila, R.; Merhi, M.; Hariri, E.; Sabbah, N.; Deeb, M.E. Irritable bowel syndrome: Prevalence, risk factors in an adult Lebanese population. BMC Gastroenterol. 2017, 17, 137. [Google Scholar] [CrossRef]
- Lazaridis, N.; Germanidis, G. Current insights into the innate immune system dysfunction in irritable bowel syndrome. Ann. Gastroenterol. 2018, 31, 171–187. [Google Scholar] [CrossRef]
- O’Mahony, L.; McCarthy, J.; Kelly, P.; Hurley, G.; Luo, F.; Chen, K.; O’Sullivan, G.C.; Kiely, B.; Collins, J.K.; Shanahan, F.; et al. Lactobacillus and bifidobacterium in irritable bowel syndrome: Symptom responses and relationship to cytokine profiles. Gastroenterology 2005, 128, 541–551. [Google Scholar] [CrossRef]
- Zhen, Y.; Chu, C.; Zhou, S.; Qi, M.; Shu, R. Imbalance of tumor necrosis factor-?, interleukin-8 and interleukin-10 production evokes barrier dysfunction, severe abdominal symptoms and psychological disorders in patients with irritable bowel syndrome-associated diarrhea. Mol. Med. Rep. 2015, 12, 5239–5245. [Google Scholar] [CrossRef] [Green Version]
- Liebregts, T.; Adam, B.; Bredack, C.; Röth, A.; Heinzel, S.; Lester, S.; Downie–Doyle, S.; Smith, E.; Drew, P.; Talley, N.J.; et al. Immune Activation in Patients With Irritable Bowel Syndrome. Gastroenterology 2007, 132, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Del Valle-Pinero, A.Y.; Martino, A.C.; Taylor, T.J.; Majors, B.L.; Patel, N.S.; Heitkemper, M.M.; Henderson, W.A. Pro-inflammatory chemokine C-C motif ligand 16 (CCL-16) dysregulation in irritable bowel syndrome (IBS): A pilot study. Neurogastroenterol. Motil. 2011, 23, 1092–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tülübaş, F.; Oran, M.; Mete, R.; Turan, F.; Yilmaz, A.; Yildiz, Z.D.; Gürel, A. Investigation of serum macrophage migration inhibitor factor and monocyte chemotactic protein-1 levels in irritable bowel syndrome. Turk. J. Med Sci. 2014, 44, 967–971. [Google Scholar] [CrossRef]
- Barkhordari, E.; Rezaei, N.; Ansaripour, B.; Larki, P.; Alighardashi, M.; Ahmadi-Ashtiani, H.R.; Mahmoudi, M.; Keramati, M.R.; Habibollahi, P.; Bashashati, M.; et al. Proinflammatory Cytokine Gene Polymorphisms in Irritable Bowel Syndrome. J. Clin. Immunol. 2009, 30, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Villani, A.; Lemire, M.; Thabane, M.; Belisle, A.; Geneau, G.; Garg, A.X.; Clark, W.F.; Moayyedi, P.; Collins, S.M.; Franchimont, D.; et al. Genetic Risk Factors for Post-Infectious Irritable Bowel Syndrome Following a Waterborne Outbreak of Gastroenteritis. Gastroenterology 2010, 138, 1502–1513. [Google Scholar] [CrossRef]
- Dunne, M.R.; Byrne, G.; Chirdo, F.G.; Feighery, C. Coeliac Disease Pathogenesis: The Uncertainties of a Well-Known Immune Mediated Disorder. Front. Immunol. 2020, 11, 1374. [Google Scholar] [CrossRef]
- Di Sabatino, A. Epithelium derived interleukin 15 regulates intraepithelial lymphocyte Th1 cytokine production, cytotoxicity, and survival in coeliac disease. Gut 2006, 55, 469–477. [Google Scholar] [CrossRef] [Green Version]
- Drago, S.; El Asmar, R.; Di Pierro, M.; Clemente, M.G.; Sapone, A.T.A.; Thakar, M.; Iacono, G.; Carroccio, A.; D’Agate, C.; Not, T.; et al. Gliadin, zonulin and gut permeability: Effects on celiac and non-celiac intestinal mucosa and intestinal cell lines. Scand. J. Gastroenterol. 2006, 41, 408–419. [Google Scholar] [CrossRef]
- Matysiak-Budnik, T.; Candalh, C.; Dugave, C.; Namane, A.; Cellier, C.; Cerf-Bensussan, N.; Heyman, M. Alterations of the intestinal transport and processing of gliadin peptides in celiac disease. Gastroenterology 2003, 125, 696–707. [Google Scholar] [CrossRef]
- Clemente, M.G.; De Virgiliis, S.; Kang, J.S.; Macatagney, R.; Musu, M.P.; Di Pierro, M.R.; Drago, S.; Congia, M.; Fasano, A. Early effects of gliadin on enterocyte intracellular signalling involved in intestinal barrier function. Gut 2003, 52, 218–223. [Google Scholar] [CrossRef]
- Meresse, B.; Chen, Z.; Ciszewski, C.; Tretiakova, M.; Bhagat, G.; Krausz, T.N.; Raulet, D.H.; Lanier, L.L.; Groh, V.; Spies, T.; et al. Coordinated Induction by IL15 of a TCR-Independent NKG2D Signaling Pathway Converts CTL into Lymphokine-Activated Killer Cells in Celiac Disease. Immunity 2004, 21, 357–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abadie, V.; Jabri, B. IL-15: A central regulator of celiac disease immunopathology. Immunol. Rev. 2014, 260, 221–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dieterich, W.; Ehnis, T.; Bauer, M.; Donner, P.; Volta, U.; Riecken, E.O.; Schuppan, D. Identification of tissue transglutaminase as the autoantigen of celiac disease. Nat. Med. 1997, 3, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Garrote, J.A.; Gómez-González, E.; Bernardo, D.; Arranz, E.; Chirdo, F. Celiac Disease Pathogenesis: The Proinflammatory Cytokine Network. J. Pediatr. Gastroenterol. Nutr. 2008, 47, S27–S32. [Google Scholar] [CrossRef] [PubMed]
- Pickert, G.; Wirtz, S.; Matzner, J.; Ashfaq-Khan, M.; Heck, R.; Rosigkeit, S.; Thies, D.; Surabattula, R.; Ehmann, D.; Wehkamp, J.; et al. Wheat Consumption Aggravates Colitis in Mice via Amylase Trypsin Inhibitor–mediated Dysbiosis. Gastroenterology 2020, 159, 257–272.e17. [Google Scholar] [CrossRef] [PubMed]
- Marotto, D.; Atzeni, F.; Ardizzone, S.; Monteleone, G.; Giorgi, V.; Sarzi-Puttini, P. Extra-intestinal manifestations of inflammatory bowel diseases. Pharmacol. Res. 2020, 161, 105206. [Google Scholar] [CrossRef]
- Marafini, I.; Sedda, S.; DiNallo, V.; Monteleone, G. Inflammatory cytokines: From discoveries to therapies in IBD. Expert Opin. Biol. Ther. 2019, 19, 1207–1217. [Google Scholar] [CrossRef]
- de Souza, H.S.; Fiocchi, C. Immunopathogenesis of IBD: Current state of the art. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 13–27. [Google Scholar] [CrossRef]
- Neurath, M.F. IL-23 in inflammatory bowel diseases and colon cancer. Cytokine Growth Factor Rev. 2019, 45, 1–8. [Google Scholar] [CrossRef]
- Gately, M.K.; Renzetti, L.M.; Magram, J.; Stern, A.S.; Adorini, L.; Gubler, U.; Presky, D.H. The interleukin-12/interleukin-12-receptor system: Role in Normal and Pathologic Immune Responses. Annu. Rev. Immunol. 1998, 16, 495–521. [Google Scholar] [CrossRef]
- Langrish, C.L.; McKenzie, B.S.; Wilson, N.J.; Malefyt, R.D.W.; Kastelein, R.A.; Cua, D.J. IL-12 and IL-23: Master regulators of innate and adaptive immunity. Immunol. Rev. 2004, 202, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Andrews, C.; McLean, M.H.; Durum, S.K. Interleukin-27 as a Novel Therapy for Inflammatory Bowel Disease: A Critical Review of the Literature. Inflamm. Bowel Dis. 2016, 22, 2255–2264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaoka, T.; Ito, M.; Yamashita, J.; Nakajima, K.; Tanaka, I.; Narita, M.; Hara, Y.; Hada, K.; Takahashi, M.; Ohno, Y.; et al. Treatment with IL-27 attenuates experimental colitis through the suppression of the development of IL-17-producing T helper cells. Am. J. Physiol. Liver Physiol. 2011, 300, G568–G576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanson, M.L.; Hixon, J.A.; Li, W.; Felber, B.K.; Anver, M.R.; Stewart, C.A.; Janelsins, B.M.; Datta, S.K.; Shen, W.; McLean, M.H.; et al. Oral Delivery of IL-27 Recombinant Bacteria Attenuates Immune Colitis in Mice. Gastroenterology 2014, 146, 210–221.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, J.H.; Kljavin, N.M.; Ramamoorthi, N.; Diehl, L.; Batten, M.; Ghilardi, N. IL-27 promotes T cell–dependent colitis through multiple mechanisms. J. Exp. Med. 2010, 208, 115–123. [Google Scholar] [CrossRef]
- Neurath, M.F. Cytokines in inflammatory bowel disease. Nat. Rev. Immunol. 2014, 14, 329–342. [Google Scholar] [CrossRef]
- Atreya, R.; Zimmer, M.; Bartsch, B.; Waldner, M.; Atreya, I.; Neumann, H.; Hildner, K.; Hoffman, A.; Kiesslich, R.; Rink, A.D.; et al. Antibodies Against Tumor Necrosis Factor (TNF) Induce T-Cell Apoptosis in Patients With Inflammatory Bowel Diseases via TNF Receptor 2 and Intestinal CD14+ Macrophages. Gastroenterology 2011, 141, 2026–2038. [Google Scholar] [CrossRef]
- Kamada, N.; Hisamatsu, T.; Okamoto, S.; Chinen, H.; Kobayashi, T.; Sato, T.; Sakuraba, A.; Kitazume, M.T.; Sugita, A.; Koganei, K.; et al. Unique CD14+ intestinal macrophages contribute to the pathogenesis of Crohn disease via IL-23/IFN-γ axis. J. Clin. Investig. 2008, 118, 2269–2280. [Google Scholar] [CrossRef] [Green Version]
- Günther, C.; Martini, E.; Wittkopf, N.; Amann, K.; Weigmann, B.; Neumann, H.; Waldner, M.J.; Hedrick, S.M.; Tenzer, S.; Neurath, M.F.; et al. Caspase-8 regulates TNF-α-induced epithelial necroptosis and terminal ileitis. Nature 2011, 477, 335–339. [Google Scholar] [CrossRef] [Green Version]
- Su, L.; Nalle, S.C.; Shen, L.; Turner, E.S.; Singh, G.; Breskin, L.A.; Khramtsova, E.A.; Khramtsova, G.; Tsai, P.; Fu, Y.; et al. TNFR2 Activates MLCK-Dependent Tight Junction Dysregulation to Cause Apoptosis-Mediated Barrier Loss and Experimental Colitis. Gastroenterology 2013, 145, 407–415. [Google Scholar] [CrossRef] [Green Version]
- Monteleone, G.; Caruso, R.; Fina, D.; Peluso, I.; Gioia, V.; Stolfi, C.; Fantini, M.C.; Caprioli, F.; Tersigni, R.; Alessandroni, L.; et al. Control of matrix metalloproteinase production in human intestinal fibroblasts by interleukin 21. Gut 2006, 55, 1774–1780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteleone, G.; Monteleone, I.; Fina, D.; Vavassori, P.; Blanco, G.D.V.; Caruso, R.; Tersigni, R.; Alessandroni, L.; Biancone, L.; Naccari, G.C.; et al. Interleukin-21 enhances T-helper cell type I signaling and interferon-gamma production in Crohn’s disease. Gastroenterology 2005, 128, 687–694. [Google Scholar] [CrossRef] [PubMed]
- Siakavellas, S.I.; Bamias, G. Role of the IL-23/IL-17 axis in Crohn’s disease. Discov. Med. 2012, 14, 253–262. [Google Scholar] [PubMed]
- Pickert, G.; Neufert, C.; Leppkes, M.; Zheng, Y.; Wittkopf, N.; Warntjen, M.; Lehr, H.-A.; Hirth, S.; Weigmann, B.; Wirtz, S.; et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J. Exp. Med. 2009, 206, 1465–1472. [Google Scholar] [CrossRef] [Green Version]
- Zenewicz, L.A.; Yancopoulos, G.D.; Valenzuela, D.M.; Murphy, A.J.; Stevens, S.; Flavell, R.A. Innate and Adaptive Interleukin-22 Protects Mice from Inflammatory Bowel Disease. Immunity 2008, 29, 947–957. [Google Scholar] [CrossRef] [Green Version]
- Latiano, A.; Palmieri, O.; Pastorelli, L.; Vecchi, M.; Pizarro, T.T.; Bossa, F.; Merla, G.; Augello, B.; Latiano, T.; Corritore, G.; et al. Associations between Genetic Polymorphisms in IL-33, IL1R1 and Risk for Inflammatory Bowel Disease. PLoS ONE 2013, 8, e62144. [Google Scholar] [CrossRef] [Green Version]
- Pastorelli, L.; De Salvo, C.; Vecchi, M.; Pizarro, T.T. The Role of IL-33 in Gut Mucosal Inflammation. Mediat. Inflamm. 2013, 2013, 608187. [Google Scholar] [CrossRef]
- Guan, Q.; Zhang, J. Recent Advances: The Imbalance of Cytokines in the Pathogenesis of Inflammatory Bowel Disease. Mediat. Inflamm. 2017, 2017, 4810258. [Google Scholar] [CrossRef] [Green Version]
- Sedhom, M.A.K.; Pichery, M.; Murdoch, J.R.; Foligné, B.; Ortega, N.; Normand, S.; Mertz, K.D.; Sanmugalingam, D.; Brault, L.; Grandjean, T.; et al. Neutralisation of the interleukin-33/ST2 pathway ameliorates experimental colitis through enhancement of mucosal healing in mice. Gut 2012, 62, 1714–1723. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Yang, F.; Sang, L.; Zhai, J.; Zhang, X.; Yue, D.; Li, S.; Li, Y.; Lu, C.; Sun, X. IL-33 Aggravates DSS-Induced Acute Colitis in Mouse Colon Lamina Propria by Enhancing Th2 Cell Responses. Mediat. Inflamm. 2015, 2015, 913041. [Google Scholar] [CrossRef] [Green Version]
- Pushparaj, P.N.; Li, D.; Komai-Koma, M.; Guabiraba, R.; Alexander, J.; McSharry, C.; Xu, D. Interleukin-33 exacerbates acute colitis via interleukin-4 in mice. Immunology 2013, 140, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Wang, Y.; Yang, F.; Sang, L.; Zhai, J.; Li, S.; Li, Y.; Wang, D.; Lu, C.-L.; Sun, X. IL-33 alleviates DSS-induced chronic colitis in C57BL/6 mice colon lamina propria by suppressing Th17 cell response as well as Th1 cell response. Int. Immunopharmacol. 2015, 29, 846–853. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Chen, J.; Zhang, H.; Yang, H.; Zhu, P.; Xiong, A.; Xia, Q.; Zheng, F.; Tan, Z.; Gong, F.; et al. Interleukin-33 Ameliorates Experimental Colitis through Promoting Th2/Foxp3+ Regulatory T-Cell Responses in Mice. Mol. Med. 2012, 18, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Monticelli, L.A.; Osborne, L.C.; Noti, M.; Tran, S.V.; Zaiss, D.M.; Artis, D. IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin-EGFR interactions. Proc. Natl. Acad. Sci. USA 2015, 112, 10762–10767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, N.R.; Oxford IBD Cohort Investigators; Hegazy, A.N.; Owens, B.M.J.; Bullers, S.J.; Linggi, B.; Buonocore, S.; Coccia, M.; Görtz, D.; This, S.; et al. Oncostatin M drives intestinal inflammation and predicts response to tumor necrosis factor–neutralizing therapy in patients with inflammatory bowel disease. Nat. Med. 2017, 23, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.M.; Kaser, A.; Blumberg, R.S. A role for oncostatin M in inflammatory bowel disease. Nat. Med. 2017, 23, 535–536. [Google Scholar] [CrossRef] [Green Version]
- Beigel, F.; Friedrich, M.; Probst, C.; Sotlar, K.; Göke, B.; Diegelmann, J.; Brand, S. Oncostatin M Mediates STAT3-Dependent Intestinal Epithelial Restitution via Increased Cell Proliferation, Decreased Apoptosis and Upregulation of SERPIN Family Members. PLoS ONE 2014, 9, e93498. [Google Scholar] [CrossRef]
- Quirós, M.; Nishio, H.; Neumann, P.A.; Siuda, D.; Brazil, J.C.; Azcutia, V.; Hilgarth, R.; O’Leary, M.N.; Garcia-Hernandez, V.; Leoni, G.; et al. Macrophage-derived IL-10 mediates mucosal repair by epithelial WISP-1 signaling. J. Clin. Investig. 2017, 127, 3510–3520. [Google Scholar] [CrossRef]
- Wei, H.-X.; Wang, B.; Li, B. IL-10 and IL-22 in Mucosal Immunity: Driving Protection and Pathology. Front. Immunol. 2020, 11, 1315. [Google Scholar] [CrossRef]
- Leman, J.K.H.; Munoz-Erazo, L.; Kemp, R.A. The Intestinal Tumour Microenvironment. Cannabinoids Neuropsychiatr. Disord. 2020, 1226, 1–22. [Google Scholar] [CrossRef]
- Li, J.; Huang, L.; Zhao, H.; Yan, Y.; Lu, J. The Role of Interleukins in Colorectal Cancer. Int. J. Biol. Sci. 2020, 16, 2323–2339. [Google Scholar] [CrossRef] [PubMed]
- Mager, L.F.; Wasmer, M.H.; Rau, T.T.; Krebs, P. Cytokine-Induced Modulation of Colorectal Cancer. Front. Oncol. 2016, 6, 96. [Google Scholar] [CrossRef] [PubMed]
- Koller, F.L.; Hwang, D.G.; Dozier, E.; Fingleton, B. Epithelial interleukin-4 receptor expression promotes colon tumor growth. Carcinogenesis 2010, 31, 1010–1017. [Google Scholar] [CrossRef] [PubMed]
- Tosolini, M.; Kirilovsky, A.; Mlecnik, B.; Fredriksen, T.; Mauger, S.; Bindea, G.; Berger, A.; Bruneval, P.; Fridman, W.-H.; Pagès, F.; et al. Clinical Impact of Different Classes of Infiltrating T Cytotoxic and Helper Cells (Th1, Th2, Treg, Th17) in Patients with Colorectal Cancer. Cancer Res. 2011, 71, 1263–1271. [Google Scholar] [CrossRef] [Green Version]
- Sarrabayrouse, G.; Corvaisier, M.; Ouisse, L.-H.; Bossard, C.; Le Mével, B.; Potiron, L.; Meurette, G.; Gervois, N.; Jotereau, F. Tumor-reactive CD4+ CD8αβ+ CD103+ αβT cells: A prevalent tumor-reactive T-cell subset in metastatic colorectal cancers. Int. J. Cancer 2011, 128, 2923–2932. [Google Scholar] [CrossRef]
- Ingram, N.; Northwood, E.L.; Perry, S.L.; Marston, G.; Snowden, H.; Taylor, J.C.; Scott, N.; Bishop, D.T.; Coletta, P.L.; Hull, M.A. Reduced type II interleukin-4 receptor signalling drives initiation, but not progression, of colorectal carcinogenesis: Evidence from transgenic mouse models and human case–control epidemiological observations. Carcinogenesis 2013, 34, 2341–2349. [Google Scholar] [CrossRef]
- Nelms, K.; Keegan, A.D.; Zamorano, J.; Ryan, J.J.; Paul, W.E. THE IL-4 RECEPTOR: Signaling Mechanisms and Biologic Functions. Annu. Rev. Immunol. 1999, 17, 701–738. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.-G.; Ye, Y.-J.; Yuan, J.; Liu, F.-F.; Zhang, H.; Wang, S. EZH2 and STAT6 expression profiles are correlated with colorectal cancer stage and prognosis. World J. Gastroenterol. 2010, 16, 2421–2427. [Google Scholar] [CrossRef]
- Kantola, T.; Klintrup, K.; Väyrynen, J.P.; Vornanen, J.; Bloigu, R.; Karhu, T.; Herzig, K.-H.; Näpänkangas, J.; Mäkelä, J.; Karttunen, T.J.; et al. Stage-dependent alterations of the serum cytokine pattern in colorectal carcinoma. Br. J. Cancer 2012, 107, 1729–1736. [Google Scholar] [CrossRef] [Green Version]
- Becker, C.; Fantini, M.; Schramm, C.; Lehr, H.; Wirtz, S.; Burg, J.; Strand, S.; Kiesslich, R.; Huber, S.; Galle, P.; et al. TGF-beta suppresses tumor progression in colon cancer by inhibition of IL–6 trans-signaling. Zeitschrift für Gastroenterologie 2004, 42, 491–501. [Google Scholar] [CrossRef]
- Atreya, R.; Mudter, J.; Finotto, S.; Müllberg, J.; Jostock, T.; Wirtz, S.; Schütz, M.; Bartsch, B.; Holtmann, M.; Becker, C.; et al. Blockade of interleukin 6 trans signaling suppresses T-cell resistance against apoptosis in chronic intestinal inflammation: Evidence in Crohn disease and experimental colitis in vivo. Nat. Med. 2000, 6, 583–588. [Google Scholar] [CrossRef]
- Kusaba, T.; Nakayama, T.; Yamazumi, K.; Yakata, Y.; Yoshizaki, A.; Inoue, K.; Nagayasu, T.; Sekine, I. Activation of STAT3 is a marker of poor prognosis in human colorectal cancer. Oncol. Rep. 2006, 15, 1445–1451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ljujic, B.; Radosavljevic, G.; Jovanovic, I.P.; Pavlovic, S.; Zdravkovic, N.; Milovanovic, M.; Acimovic, L.; Knežević, M.; Banković, D.; Zdravkovic, D.; et al. Elevated Serum Level of IL-23 Correlates with Expression of VEGF in Human Colorectal Carcinoma. Arch. Med Res. 2010, 41, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Langowski, J.L.; Zhang, X.; Wu, L.; Mattson, J.D.; Chen, T.; Smith, K.; Basham, B.; McClanahan, T.; Kastelein, R.A.; Oft, M. IL-23 promotes tumour incidence and growth. Nature 2006, 442, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Hue, S.; Ahern, P.; Buonocore, S.; Kullberg, M.C.; Cua, D.J.; McKenzie, B.S.; Powrie, F.; Maloy, K.J. Interleukin-23 drives innate and T cell–mediated intestinal inflammation. J. Exp. Med. 2006, 203, 2473–2483. [Google Scholar] [CrossRef] [Green Version]
- Yen, D.; Cheung, J.; Scheerens, H.; Poulet, F.; McClanahan, T.; McKenzie, B.; Kleinschek, M.A.; Owyang, A.; Mattson, J.; Blumenschein, W.; et al. IL-23 is essential for T cell–mediated colitis and promotes inflammation via IL-17 and IL-6. J. Clin. Investig. 2006, 116, 1310–1316. [Google Scholar] [CrossRef] [PubMed]
- Khare, V.; Paul, G.; Movadat, O.; Frick, A.; Jambrich, M.; Krnjic, A.; Marian, B.; Wrba, F.; Gasche, C. IL10R2 Overexpression Promotes IL22/STAT3 Signaling in Colorectal Carcinogenesis. Cancer Immunol. Res. 2015, 3, 1227–1235. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Li, J.; Li, L.; Zhang, J.; Wang, X.; Yang, C.; Li, Y.; Lan, F.; Lin, P. IL-23 selectively promotes the metastasis of colorectal carcinoma cells with impaired Socs3 expression via the STAT5 pathway. Carcinogenesis 2014, 35, 1330–1340. [Google Scholar] [CrossRef]
- Gronke, K.; Hernández, P.P.; Zimmermann, J.; Klose, C.S.N.; Kofoed-Branzk, M.; Guendel, F.; Witkowski, M.; Tizian, C.; Amann, L.; Schumacher, F.; et al. Interleukin-22 protects intestinal stem cells against genotoxic stress. Nature 2019, 566, 249–253. [Google Scholar] [CrossRef]
- Kryczek, I.; Lin, Y.; Nagarsheth, N.; Peng, D.; Zhao, L.; Zhao, E.; Vatan, L.; Szeliga, W.; Dou, Y.; Owens, S.; et al. IL-22+CD4+ T Cells Promote Colorectal Cancer Stemness via STAT3 Transcription Factor Activation and Induction of the Methyltransferase DOT1L. Immunity 2014, 40, 772–784. [Google Scholar] [CrossRef] [Green Version]
- Waldner, M.J.; Wirtz, S.; Jefremow, A.; Warntjen, M.; Neufert, C.; Atreya, R.; Becker, C.; Weigmann, B.; Vieth, M.; Rose-John, S.; et al. VEGF receptor signaling links inflammation and tumorigenesis in colitis-associated cancer. J. Exp. Med. 2010, 207, 2855–2868. [Google Scholar] [CrossRef] [PubMed]
- Goodlad, R.A.; Ryan, A.; Wedge, S.; Pyrah, I.; Alferez, D.; Poulsom, R.; Smith, N.; Mandir, N.; Watkins, A.; Wilkinson, R.W. Inhibiting vascular endothelial growth factor receptor-2 signaling reduces tumor burden in the ApcMin/+ mouse model of early intestinal cancer. Carcinogenesis 2006, 27, 2133–2139. [Google Scholar] [CrossRef] [PubMed]
- Easwaran, V.; Lee, S.H.; Inge, L.; Guo, L.; Goldbeck, C.; Garrett, E.; Wiesmann, M.; Garcia, P.D.; Fuller, J.H.; Chan, V.; et al. beta-Catenin regulates vascular endothelial growth factor expression in colon cancer. Cancer Res. 2003, 63, 3145–3153. [Google Scholar] [PubMed]
- Lakatos, P.L.; Lakatos, L. Risk for colorectal cancer in ulcerative colitis: Changes, causes and management strategies. World J. Gastroenterol. 2008, 14, 3937–3947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobelt, D.; Zhang, C.; Clayton-Lucey, I.A.; Glauben, R.; Voss, C.; Siegmund, B.; Stein, U. Pro-inflammatory TNF-alpha and IFN-gamma Promote Tumor Growth and Metastasis via Induction of MACC1. Front. Immunol. 2020, 11, 980. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.R. Tumour necrosis factor and cancer. Nat. Rev. Cancer 2009, 9, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Cox, G.W.; Melillo, G.; Chattopadhyay, U.; Mullet, D.; Fertel, R.H.; Varesio, L. Tumor necrosis factor-alpha-dependent production of reactive nitrogen intermediates mediates IFN-gamma plus IL-2-induced murine macrophage tumoricidal activity. J. Immunol. 1992, 149, 3290–3296. [Google Scholar]
- Zins, K.; Abraham, D.; Sioud, M.; Aharinejad, S. Colon Cancer Cell–Derived Tumor Necrosis Factor-α Mediates the Tumor Growth–Promoting Response in Macrophages by Up-regulating the Colony-Stimulating Factor-1 Pathway. Cancer Res. 2007, 67, 1038–1045. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, K.E.; Simmons, J.G.; Ding, S.; Van Landeghem, L.; Lund, P.K. Cytokine Induction of Tumor Necrosis Factor Receptor 2 Is Mediated by STAT3 in Colon Cancer Cells. Mol. Cancer Res. 2011, 9, 1718–1731. [Google Scholar] [CrossRef] [Green Version]
- Popivanova, B.K.; Kitamura, K.; Wu, Y.; Kondo, T.; Kagaya, T.; Kaneko, S.; Oshima, M.; Fujii, C.; Mukaida, N. Blocking TNF-α in mice reduces colorectal carcinogenesis associated with chronic colitis. J. Clin. Investig. 2008, 118, 560–570. [Google Scholar] [CrossRef]
- Maywald, R.L.; Doerner, S.K.; Pastorelli, L.; De Salvo, C.; Benton, S.M.; Dawson, E.P.; Lanza, D.G.; Berger, N.A.; Markowitz, S.D.; Lenz, H.J.; et al. IL-33 activates tumor stroma to promote intestinal polyposis. Proc. Natl. Acad. Sci. USA 2015, 112, E2487–E2496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Z.; Chen, L.; Souto, F.O.; Canasto-Chibuque, C.; Bongers, G.; Deshpande, M.; Harpaz, N.; Ko, H.M.; Kelley, K.; Furtado, G.C.; et al. Epithelial-derived IL-33 promotes intestinal tumorigenesis in Apc Min/+ mice. Sci. Rep. 2017, 7, 5520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, K.M.; Minaya, M.K.; Vaish, V.; Peña, M.M.O. The Role of IL-33/ST2 Pathway in Tumorigenesis. Int. J. Mol. Sci. 2018, 19, 2676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastille, E.; Wasmer, M.-H.; Adamczyk, A.; Vu, V.P.; Mager, L.F.; Phuong, N.N.T.; Palmieri, V.; Simillion, C.; Hansen, W.; Kasper, S.; et al. The IL-33/ST2 pathway shapes the regulatory T cell phenotype to promote intestinal cancer. Mucosal Immunol. 2019, 12, 990–1003. [Google Scholar] [CrossRef] [Green Version]
- Mahapatro, M.; Foersch, S.; Hefele, M.; He, G.-W.; Giner-Ventura, E.; Mchedlidze, T.; Kindermann, M.; Vetrano, S.; Danese, S.; Günther, C.; et al. Programming of Intestinal Epithelial Differentiation by IL-33 Derived from Pericryptal Fibroblasts in Response to Systemic Infection. Cell Rep. 2016, 15, 1743–1756. [Google Scholar] [CrossRef] [Green Version]
- Waugh, D.J.; Wilson, C. The Interleukin-8 Pathway in Cancer. Clin. Cancer Res. 2008, 14, 6735–6741. [Google Scholar] [CrossRef] [Green Version]
- Yoshizaki, A.; Nakayama, T.; Yamazumi, K.; Yakata, Y.; Taba, M.; Sekine, I. Expression of interleukin (IL)-11 and IL-11 receptor in human colorectal adenocarcinoma: IL-11 up-regulation of the invasive and proliferative activity of human colorectal carcinoma cells. Int. J. Oncol. 2006, 29, 869–876. [Google Scholar] [CrossRef] [Green Version]
- Putoczki, T.L.; Thiem, S.; Loving, A.; Busuttil, R.A.; Wilson, N.J.; Ziegler, P.K.; Nguyen, P.M.; Preaudet, A.; Farid, R.; Edwards, K.M.; et al. Interleukin-11 Is the Dominant IL-6 Family Cytokine during Gastrointestinal Tumorigenesis and Can Be Targeted Therapeutically. Cancer Cell 2013, 24, 257–271. [Google Scholar] [CrossRef] [Green Version]
- Evans, C.P.; Morrison, I.; Heriot, A.G.; Bartlett, J.B.; Finlayson, C.; Dalgleish, A.G.; Kumar, D. The correlation between colorectal cancer rates of proliferation and apoptosis and systemic cytokine levels; plus their influence upon survival. Br. J. Cancer 2006, 94, 1412–1419. [Google Scholar] [CrossRef]
- Osawa, E.; Nakajima, A.; Fujisawa, T.; Kawamura, Y.I.; Toyama-Sorimachi, N.; Nakagama, H.; Dohi, T. Predominant T helper type 2-inflammatory responses promote murine colon cancers. Int. J. Cancer 2005, 118, 2232–2236. [Google Scholar] [CrossRef]
- Wang, L.; Wang, Y.; Song, Z.; Chu, J.; Qu, X. Deficiency of Interferon-Gamma or Its Receptor Promotes Colorectal Cancer Development. J. Interf. Cytokine Res. 2015, 35, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Sullivan, L.; Caligiuri, M.A. Molecular Pathways: Interleukin-15 Signaling in Health and in Cancer. Clin. Cancer Res. 2014, 20, 2044–2050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahri, R.; Pateras, I.S.; D’Orlando, O.; Goyeneche-Patino, D.A.; Campbell, M.; Polansky, J.K.; Sandig, H.; Papaioannou, M.; Evangelou, K.; Foukas, P.G.; et al. IL-15 suppresses colitis-associated colon carcinogenesis by inducing antitumor immunity. OncoImmunology 2015, 4, e1002721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jabri, B.; Abadie, V. IL-15 functions as a danger signal to regulate tissue-resident T cells and tissue destruction. Nat. Rev. Immunol. 2015, 15, 771–783. [Google Scholar] [CrossRef]
- Fung, K.Y.; Nguyen, P.M.; Putoczki, T.L. Emerging Roles for Interleukin-18 in the Gastrointestinal Tumor Microenvironment. Optogenetics 2020, 1240, 59–72. [Google Scholar] [CrossRef]
- Salcedo, R.; Worschech, A.; Cardone, M.; Jones, Y.; Gyulai, Z.; Dai, R.-M.; Wang, E.; Ma, W.; Haines, D.; O’Huigin, C.; et al. MyD88-mediated signaling prevents development of adenocarcinomas of the colon: Role of interleukin 18. J. Exp. Med. 2010, 207, 1625–1636. [Google Scholar] [CrossRef]
- Kunze, F.A.; Bauer, M.; Komuczki, J.; Lanzinger, M.; Gunasekera, K.; Hopp, A.K.; Lehmann, M.; Becher, B.; Müller, A.; Hottiger, M.O. ARTD1 in Myeloid Cells Controls the IL-12/18-IFN-gamma Axis in a Model of Sterile Sepsis, Chronic Bacterial Infection, and Cancer. J. Immunol. 2019, 202, 1406–1416. [Google Scholar] [CrossRef]
- Tong, Z.; Yang, X.O.; Yan, H.; Liu, W.; Niu, X.; Shi, Y.; Fang, W.; Xiong, B.; Wan, Y.; Dong, C. A Protective Role by Interleukin-17F in Colon Tumorigenesis. PLoS ONE 2012, 7, e34959. [Google Scholar] [CrossRef] [Green Version]
- Razi, S.; Noveiry, B.B.; Keshavarz-Fathi, M.; Rezaei, N. IL-17 and colorectal cancer: From carcinogenesis to treatment. Cytokine 2019, 116, 7–12. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, Z.; Wu, D.; Min, Z.; Quan, Y. Upregulation of interleukin-17F in colorectal cancer promotes tumor invasion by inducing epithelial-mesenchymal transition. Oncol. Rep. 2019, 42, 1141–1148. [Google Scholar] [CrossRef]
- Girondel, C.; Lévesque, K.; Langlois, M.-J.; Pasquin, S.; Saba-El-Leil, M.K.; Rivard, N.; Friesel, R.; Servant, M.J.; Gauchat, J.-F.; Lesage, S.; et al. Loss of interleukin-17 receptor D promotes chronic inflammation-associated tumorigenesis. Oncogene 2020, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-C.; Liao, T.-T.; Yang, M. Immune Adaptation of Colorectal Cancer Stem Cells and Their Interaction With the Tumor Microenvironment. Front. Oncol. 2020, 10, 588542. [Google Scholar] [CrossRef] [PubMed]
- Czajka-Francuz, P.; Francuz, T.; Cisoń-Jurek, S.; Czajka, A.; Fajkis, M.; Szymczak, B.; Kozaczka, M.; Malinowski, K.P.; Zasada, W.; Wojnar, J.; et al. Serum cytokine profile as a potential prognostic tool in colorectal cancer patients—One center study. Rep. Pr. Oncol. Radiother. 2020, 25, 867–875. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, H.I.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus Irinotecan, Fluorouracil, and Leucovorin for Metastatic Colorectal Cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef] [Green Version]
- Saltz, L.; Clarke, S.; Díaz-Rubio, E.; Scheithauer, W.; Figer, A.; Wong, R.; Koski, S.; Lichinitser, M.; Yang, T.-S.; Rivera, F.; et al. Bevacizumab in Combination With Oxaliplatin-Based Chemotherapy As First-Line Therapy in Metastatic Colorectal Cancer: A Randomized Phase III Study. J. Clin. Oncol. 2008, 26, 2013–2019. [Google Scholar] [CrossRef] [Green Version]
- Tabernero, J.; Van Cutsem, E.; Lakomý, R.; Prausová, J.; Ruff, P.; Van Hazel, G.A.; Moiseyenko, V.M.; Ferry, D.R.; McKendrick, J.J.; Soussan-Lazard, K.; et al. Aflibercept versus placebo in combination with fluorouracil, leucovorin and irinotecan in the treatment of previously treated metastatic colorectal cancer: Prespecified subgroup analyses from the VELOUR trial. Eur. J. Cancer 2014, 50, 320–331. [Google Scholar] [CrossRef] [Green Version]
- Neurath, M.F.; Fuss, I.; Pasparakis, M.; Alexopoulou, L.; Haralambous, S.; Strober, W.; Kollias, G.; Büschenfelde, K.-H.M.Z. Predominant pathogenic role of tumor necrosis factor in experimental colitis in mice. Eur. J. Immunol. 1997, 27, 1743–1750. [Google Scholar] [CrossRef]
- Present, D.H.; Rutgeerts, P.; Targan, S.; Hanauer, S.B.; Mayer, L.; Van Hogezand, R.A.; Podolsky, D.K.; Sands, B.E.; Braakman, T.; DeWoody, K.L.; et al. Infliximab for the treatment of fistulas in patients with Crohn’s disease. N. Engl. J. Med. 1999, 340, 1398–1405. [Google Scholar] [CrossRef] [Green Version]
- Rutgeerts, P.; Sandborn, W.J.; Feagan, B.G.; Reinisch, W.; Olson, A.; Johanns, J.; Travers, S.; Rachmilewitz, D.; Hanauer, S.B.; Lichtenstein, G.R.; et al. Infliximab for Induction and Maintenance Therapy for Ulcerative Colitis. N. Engl. J. Med. 2005, 353, 2462–2476. [Google Scholar] [CrossRef] [Green Version]
- Melsheimer, R.; Geldhof, A.; Apaolaza, I.; Schaible, T. Remicade((R)) (infliximab): 20 years of contributions to science and medicine. Biologics 2019, 13, 139–178. [Google Scholar]
- Baert, F.; Noman, M.; Vermeire, S.; Van Assche, G.; D’Haens, G.; Carbonez, A.; Rutgeerts, P. Influence of immunogenicity on the long-term efficacy of infliximab in Crohn’s disease. N. Engl. J. Med. 2003, 348, 601–608. [Google Scholar] [CrossRef]
- Keane, J.; Gershon, S.; Wise, R.P.; Mirabile-Levens, E.; Kasznica, J.; Schwieterman, W.D.; Siegel, J.N.; Braun, M.M. Tuberculosis Associated with Infliximab, a Tumor Necrosis Factor α–Neutralizing Agent. N. Engl. J. Med. 2001, 345, 1098–1104. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.; D’Amico, F.; Bonovas, S.; Danese, S.; Peyrin-Biroulet, L. TNF Inhibitors and Risk of Malignancy in Patients with Inflammatory Bowel Diseases: A Systematic Review. J. Crohn’s Coliti 2020. [Google Scholar] [CrossRef]
- Grivennikov, S.; Karin, E.; Terzic, J.; Mucida, D.; Yu, G.Y.; Vallabhapurapu, S.; Scheller, J.; Rose-John, S.; Cheroutre, H.; Eckmann, L.; et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 2009, 15, 103–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, H.; Takazoe, M.; Fukuda, Y.; Hibi, T.; Kusugami, K.; Andoh, A.; Matsumoto, T.; Yamamura, T.; Azuma, J.; Nishimoto, N. A pilot randomized trial of a human anti-interleukin-6 receptor monoclonal antibody in active Crohn’s disease☆. Gastroenterology 2004, 126, 989–996. [Google Scholar] [CrossRef] [PubMed]
- Danese, S.; Vermeire, S.; Hellstern, P.; Panaccione, R.; Rogler, G.; Fraser, G.; Kohn, A.; Desreumaux, P.; Leong, R.W.; Comer, G.M.; et al. Randomised trial and open-label extension study of an anti-interleukin-6 antibody in Crohn’s disease (ANDANTE I and II). Gut 2019, 68, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Siegmund, B.; Fantuzzi, G.; Rieder, F.; Gamboni-Robertson, F.; Lehr, H.A.; Hartmann, G.; Dinarello, C.A.; Endres, S.; Eigler, A. Neutralization of interleukin-18 reduces severity in murine colitis and intestinal IFN-gamma and TNF-alpha production. Am. J. Physiol. Integr. Comp. Physiol. 2001, 281, 1264–1273. [Google Scholar] [CrossRef]
- Wirtz, S.; Becker, C.; Blumberg, R.; Galle, P.R.; Neurath, M.F. Treatment of T cell-dependent experimental colitis in SCID mice by local administration of an adenovirus expressing IL-18 antisense mRNA. J. Immunol. 2002, 168, 411–420. [Google Scholar] [CrossRef] [Green Version]
- Sandborn, W.J.; Gasink, C.; Gao, L.L.; Blank, M.A.; Johanns, J.; Guzzo, C.; Sands, B.E.; Hanauer, S.B.; Targan, S.; Rutgeerts, P.; et al. Ustekinumab induction and maintenance therapy in refractory Crohn’s disease. N. Engl. J. Med. 2012, 367, 1519–1528. [Google Scholar] [CrossRef]
- Feagan, B.G.; Sandborn, W.J.; Gasink, C.; Jacobstein, D.; Lang, Y.; Friedman, J.R.; Blank, M.A.; Johanns, J.; Gao, L.-L.; Miao, Y.; et al. Ustekinumab as Induction and Maintenance Therapy for Crohn’s Disease. N. Engl. J. Med. 2016, 375, 1946–1960. [Google Scholar] [CrossRef]
- Rutgeerts, P.; Gasink, C.; Chan, D.; Lang, Y.; Pollack, P.; Colombel, J.-F.; Wolf, D.C.; Jacobstein, D.; Johanns, J.; Szapary, P.; et al. Efficacy of Ustekinumab for Inducing Endoscopic Healing in Patients With Crohn’s Disease. Gastroenterology 2018, 155, 1045–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandborn, W.J.; Rutgeerts, P.; Gasink, C.; Jacobstein, D.; Zou, B.; Johanns, J.; Sands, B.E.; Hanauer, S.B.; Targan, S.; Ghosh, S.; et al. Long-term efficacy and safety of ustekinumab for Crohn’s disease through the second year of therapy. Aliment. Pharmacol. Ther. 2018, 48, 65–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panaccione, R.; Sandborn, W.J.; Gordon, G.L.; Lee, S.D.; Safdi, A.; Sedghi, S.; Feagan, B.G.; Hanauer, S.; Reinisch, W.; Valentine, J.F.; et al. Briakinumab for treatment of Crohn’s disease: Results of a randomized trial. Inflamm. Bowel. Dis. 2015, 21, 1329–1340. [Google Scholar]
- Elson, C.O.; Cong, Y.; Weaver, C.T.; Schoeb, T.R.; McClanahan, T.K.; Fick, R.B.; Kastelein, R.A. Monoclonal Anti–Interleukin 23 Reverses Active Colitis in a T Cell–Mediated Model in Mice. Gastroenterology 2007, 132, 2359–2370. [Google Scholar] [CrossRef] [PubMed]
- Uhlig, H.H.; McKenzie, B.S.; Hue, S.; Thompson, C.; Joyce-Shaikh, B.; Stepankova, R.; Robinson, N.; Buonocore, S.; Tlaskalova-Hogenova, H.; Cua, D.J.; et al. Differential Activity of IL-12 and IL-23 in Mucosal and Systemic Innate Immune Pathology. Immunity 2006, 25, 309–318. [Google Scholar] [CrossRef] [Green Version]
- Kullberg, M.C.; Jankovic, D.; Feng, C.G.; Hue, S.; Gorelick, P.L.; McKenzie, B.S.; Cua, D.J.; Powrie, F.; Cheever, A.W.; Maloy, K.J.; et al. IL-23 plays a key role in Helicobacter hepaticus–induced T cell–dependent colitis. J. Exp. Med. 2006, 203, 2485–2494. [Google Scholar] [CrossRef] [Green Version]
- Almradi, A.; Hanzel, J.; Sedano, R.; Parker, C.E.; Feagan, B.G.; Ma, C.; Jairath, V. Clinical Trials of IL-12/IL-23 Inhibitors in Inflammatory Bowel Disease. BioDrugs 2020, 34, 713–721. [Google Scholar] [CrossRef]
- Sands, B.E.; Chen, J.; Feagan, B.G.; Penney, M.; Rees, W.A.; Danese, S.; Higgins, P.D.; Newbold, P.; Faggioni, R.; Patra, K.; et al. Efficacy and Safety of MEDI2070, an Antibody Against Interleukin 23, in Patients With Moderate to Severe Crohn’s Disease: A Phase 2a Study. Gastroenterology 2017, 153, 77–86.e6. [Google Scholar] [CrossRef] [Green Version]
- Sandborn, W.J.; Ferrante, M.; Bhandari, B.R.; Berliba, E.; Feagan, B.G.; Hibi, T.; Tuttle, J.L.; Klekotka, P.; Friedrich, S.; Durante, M.; et al. Efficacy and Safety of Mirikizumab in a Randomized Phase 2 Study of Patients With Ulcerative Colitis. Gastroenterology 2020, 158, 537–549.e10. [Google Scholar] [CrossRef] [Green Version]
- Hommes, D.W.; Mikhajlova, T.L.; Stoinov, S.; Štimac, D.; Vucelic, B.; Lonovics, J.; Zákuciová, M.; D’Haens, G.; Van Assche, G.; Ba, S.; et al. Fontolizumab, a humanised anti-interferon gamma antibody, demonstrates safety and clinical activity in patients with moderate to severe Crohn’s disease. Gut 2006, 55, 1131–1137. [Google Scholar] [CrossRef]
- Reinisch, W.; Panés, J.; Khurana, S.; Toth, G.; Hua, F.; Comer, G.M.; Hinz, M.; Page, K.; O’Toole, M.; Moorehead, T.M.; et al. Anrukinzumab, an anti-interleukin 13 monoclonal antibody, in active UC: Efficacy and safety from a phase IIa randomised multicentre study. Gut 2015, 64, 894–900. [Google Scholar] [CrossRef] [PubMed]
- Hueber, W.; Sands, B.E.; Lewitzky, S.; Vandemeulebroecke, M.; Reinisch, W.; Higgins, P.D.; Wehkamp, J.; Feagan, B.G.; Yao, M.D.; Karczewski, M.; et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: Unexpected results of a randomised, double-blind placebo-controlled trial. Gut 2012, 61, 1693–1700. [Google Scholar] [CrossRef] [PubMed]
- Herrlinger, K.; Diculescu, M.; Fellermann, K.; Hartmann, H.; Howaldt, S.; Nikolov, R.; Petrov, A.; Reindl, W.; Otte, J.; Stoynov, S.; et al. Efficacy, safety and tolerability of vidofludimus in patients with inflammatory bowel disease: The ENTRANCE study. J. Crohn’s Coliti 2013, 7, 636–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Targan, S.R.; Feagan, B.; Vermeire, S.; Panaccione, R.; Melmed, G.Y.; Landers, C.; Li, D.; Russell, C.; Newmark, R.; Zhang, N.; et al. A Randomized, Double-Blind, Placebo-Controlled Phase 2 Study of Brodalumab in Patients With Moderate-to-Severe Crohn’s Disease. Am. J. Gastroenterol. 2016, 111, 1599–1607. [Google Scholar] [CrossRef]
- Lee, J.S.; Tato, C.M.; Joyce-Shaikh, B.; Gulen, M.F.; Cayatte, C.; Chen, Y.; Blumenschein, W.M.; Judo, M.; Ayanoglu, G.; McClanahan, T.K.; et al. Interleukin-23-Independent IL-17 Production Regulates Intestinal Epithelial Permeability. Immunity 2015, 43, 727–738. [Google Scholar] [CrossRef] [Green Version]
- Maxwell, J.R.; Zhang, Y.; Brown, W.A.; Smith, C.L.; Byrne, F.R.; Fiorino, M.; Stevens, E.; Bigler, J.; Davis, J.A.; Rottman, J.B.; et al. Differential Roles for Interleukin-23 and Interleukin-17 in Intestinal Immunoregulation. Immunity 2015, 43, 739–750. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cytokine | Cellular Source(s) | Target Cell(s) | Function |
|---|---|---|---|
| IL-22 | ILC, Th22, NK cell, Th1, Th17 | IECs | Activation of STAT3 signaling and release of AMPs |
| IL-1β | IECs | Mϕ, Endothelial cells | Activation of T-cells and ILCs |
| IL-18 | IECs | Th17, Tregs | IEC proliferation, tissue regeneration, production of proinflammatory cytokines |
| IL-6 | IECs, fibroblasts, Mϕ | Th17, IELs, IECs | IEC proliferation and repair, activation of STAT3, crypt homeostasis |
| IL-23 | IECs, Mϕ, DC | IELs, ILC3, NK cell, T-cells | Proinflammatory cytokine secretion contributes to chronic inflammation |
| IL-12 | Monocyte, Mϕ, DC | Th1 | T-cell survival and differentiation, proliferation of NK cell |
| IL-17A | Th1, ILC3 | IECs | Antimicrobial response, maintenance of homeostasis |
| TNF | ILC1, Mϕ | IECs | Epithelial cell death, epithelial cell migration during wound healing, mucosal repair during inflammation |
| INF-γ | ILC1, Mϕ | IECs, DC, Tregs | Confers protection against pathogens, activation of STAT1 signaling, disruption of epithelial barrier |
| TGF-β | IECs, DC, Tregs, Mast cells | B-cells, Th9 cells, Mϕ | Expansion of Tregs, IgA secretion, IEL development, tight junction maintenance |
| IL-4 | ILC2, Th2, | IECs, Mast cells | Differentiation of IECs to secretory cells, confers protection against intestinal parasite infection, survival of malignant cells, activation of STAT6 signaling |
| IL-5 | ILC2, Th2, B-cells | IECs, B-cells, Eosinophills | Differentiation of IECs to secretory cells, confers protection against intestinal parasite infection |
| IL-9 | ILC2, Th9 | IECs | Differentiation of IECs to secretory cells, leakiness in gut barrier |
| IL-13 | ILC2, Th2 | IECs | Differentiation of IECs to secretory cells, mucin production, confers protection against intestinal parasite infection, activation of STAT6 signaling |
| IL-25 | IECs | ILC2, T-cells | Host protection against intestinal helminthes, type 2 immune response |
| TSLP | IECs, Mast cells, DC | Th2 cells, ILC2 | Type 2 immune response, T- and B-cell activation |
| IL-33 | IECs, intestinal myofiboblasts | ILC2, Tregs, Th2 cells IECs | Type 2 immune response, IEC differentiation, intestinal inflammation |
| IL-7 | IECs | ILC3, Tregs, T effector cells | Proinflammatory cytokine secretion, IEC homeostasis |
| GM-CSF | T cells, ILC3 | Monocyte, Mϕ, Tregs | Mϕ differentiation, IgA secretion from B-cells, bacterial clearance, epithelial repair during wound healing |
| IL-2 | T cells | Th1, Tregs | Activation of STAT3/5 signaling, differentiation of T-cells, intestinal homeostasis |
| IL-10 | Mϕ, Tregs | IECs | Intestinal homeostasis, IEC proliferation |
| IL-11 | Mϕ | Malignant IECs | Activation of JAK/STAT signaling, tumor cell survival |
| IL-15 | IECs | T-cells, IELs | Epithelial barrier disruption, antitumorigenic functions |
| OSM | T cells, DC | Stromal cells | Proinflammatory cytokine secretion, activation of JAK/STAT signaling |
| Areg | Tregs, ILC2 | IECs | Tissue repair after damage, fibrosis |
| VEGF | Stromal cells, Mast cells, Platelets | IECs | Malignant cell survival, angiogenesis, intestinal stem cell proliferation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahapatro, M.; Erkert, L.; Becker, C. Cytokine-Mediated Crosstalk between Immune Cells and Epithelial Cells in the Gut. Cells 2021, 10, 111. https://doi.org/10.3390/cells10010111
Mahapatro M, Erkert L, Becker C. Cytokine-Mediated Crosstalk between Immune Cells and Epithelial Cells in the Gut. Cells. 2021; 10(1):111. https://doi.org/10.3390/cells10010111
Chicago/Turabian StyleMahapatro, Mousumi, Lena Erkert, and Christoph Becker. 2021. "Cytokine-Mediated Crosstalk between Immune Cells and Epithelial Cells in the Gut" Cells 10, no. 1: 111. https://doi.org/10.3390/cells10010111
APA StyleMahapatro, M., Erkert, L., & Becker, C. (2021). Cytokine-Mediated Crosstalk between Immune Cells and Epithelial Cells in the Gut. Cells, 10(1), 111. https://doi.org/10.3390/cells10010111