Chronic Myeloid Leukemia: A Model Disease of the Past, Present and Future


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. The History of Chronic Myeloid Leukemia
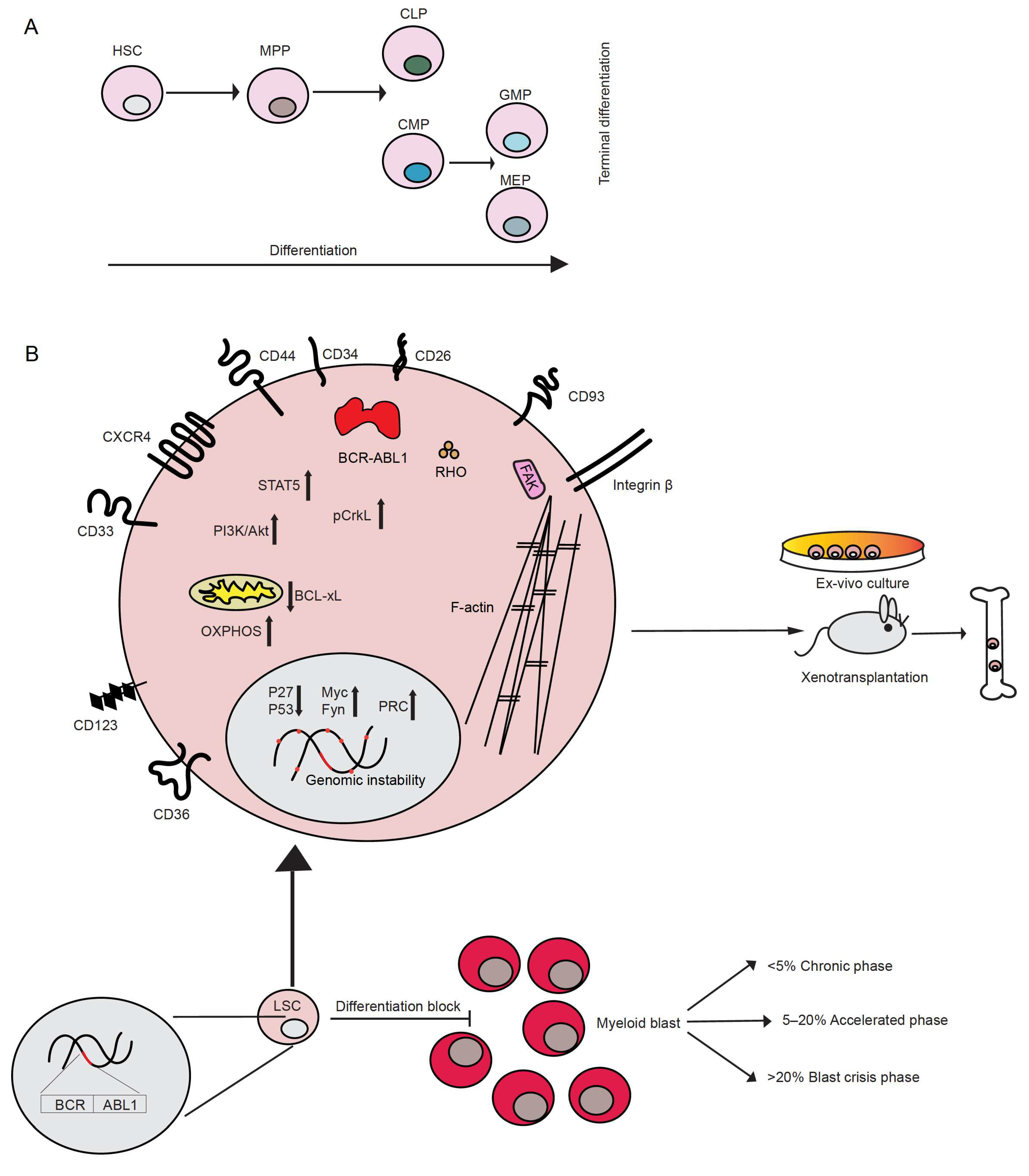
2. Characteristics of CML
3. CML Stem Cells
- (a) the sialic acid receptor CD33, which was expressed in CP LSC but only variably in BP LSC [23],
- (d) the lectin transmembrane receptor CD93, which labels a population with increased stem cell characteristics, robust engraftment in xenotransplantation models and correlation with relapse upon TKI withdrawal [28],
- (f) the interleukin-2 receptor α CD25 [30] and
- (g) the interleukin 3 receptor subunit (CD123), a known marker for acute myeloid leukemia stem cells, but also CP and BP LSC [31].
4. Signaling in CML
5. Additional Molecular Alterations beyond BCR-ABL1
6. Diagnosis of CML
7. Treatment of CML
Targeted Therapy of CML and Resistance
8. Treatment Free Remission
9. Metabolic Targeting in CML
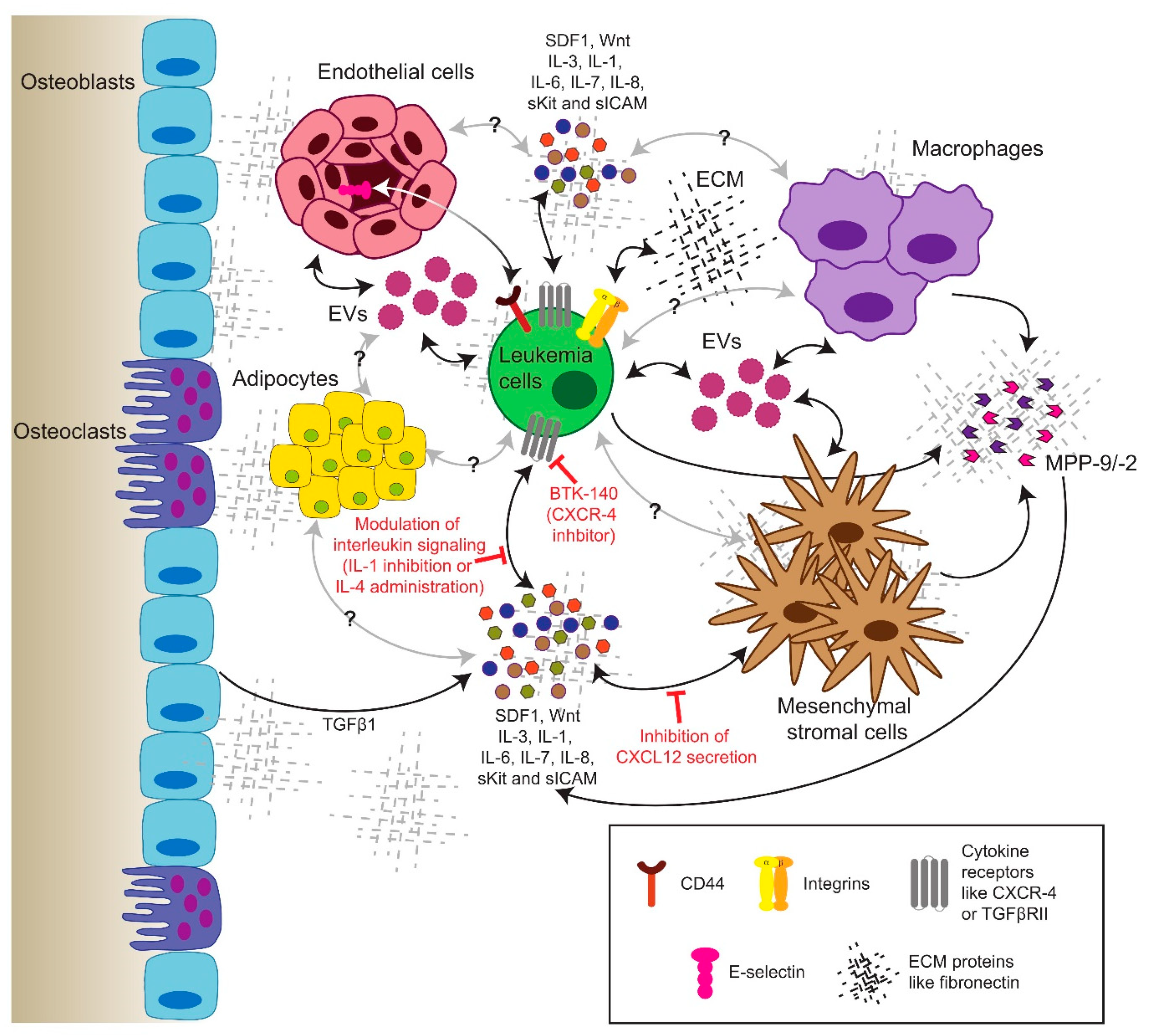
10. The Role of the Bone Marrow Microenvironment in CML
10.1. Soluble Factors in the Extracellular Milieu
10.2. Extracellular Vesicles
11. Molecular Targets beyond TKI and Combination Treatments
11.1. Targeting of Alternative Signaling Pathways
11.2. Targeting Epigenetic Modification
11.3. Autophagy
12. Conclusions
Funding
Conflicts of Interest
References
- Rudolf Virchow (1821–1902). CA Cancer J. Clin. 1975, 25, 91–92. [CrossRef] [PubMed]
- Bennett, J.H. Case of Hypertrophy of the Spleen and Liver, which Death Took Place from Suppuration of the Blood. Edinb. Med. Surg. J. 1845, 413–423. [Google Scholar]
- Neumann, E. Ein Fall von Leukämie mit Erkrankung des Knochenmarks Anhang: Salkowski: Chemische Untersuchungen des leukämischen Markes. Archiv der Heilkunde 1871, 1–15. [Google Scholar]
- Peter, C.; Nowell, D.A.H. A minute chromosome in human chronic granulocytic leukemia. Science 1960, 132, 1497–1499. [Google Scholar]
- Rowley, J.D. Letter: A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature 1973, 243, 290–293. [Google Scholar] [CrossRef]
- Heisterkamp, N.; Stephenson, J.R.; Groffen, J.; Hansen, P.F.; de Klein, A.; Bartram, C.R.; Grosveld, G. Localization of the c-abl oncogene adjacent to a translocation break point in chronic myelocytic leukaemia. Nature 1983, 306, 239–242. [Google Scholar] [CrossRef]
- Bartram, C.R.; De Klein, A.; Hagemeijer, A.; Grosveld, G.; Heisterkamp, N.; Groffen, J. Localization of the human c-sis oncogene in Ph1-positive and Ph1-negative chronic myelocytic leukemia by in situ hybridization. Blood 1984, 63, 223–225. [Google Scholar] [CrossRef]
- Bartram, C.R.; de Klein, A.; Hagemeijer, A.; van Agthoven, T.; van Kessel, A.G.; Bootsma, D.; Groffen, J. Translocation of c-abl oncogene correlates with the presence of a Philadelphia chromosome in chronic myelocytic leukaemia. Nature 1983, 306, 277–280. [Google Scholar] [CrossRef]
- Groffen, J.; Stephenson, J.R.; Heisterkamp, N.; De Klein, A.; Bartram, C.R.; Grosveld, G. Philadelphia chromosomal breakpoints are clustered within a limited region, bcr, on chromosome. Cell 1984, 36, 93–99. [Google Scholar] [CrossRef]
- Jain, P.; Kantarjian, H.; Patel, K.P.; Gonzalez, G.N.; Luthra, R.; Shamanna, R.K.; Sasaki, K.; Jabbour, E.; Romo, C.G.; Kadia, T.M.; et al. Impact of BCR-ABL transcript type on outcome in patients with chronic-phase CML treated with tyrosine kinase inhibitors. Blood 2016, 127, 1269–1275. [Google Scholar] [CrossRef]
- Shtivelman, E.; Lifshitz, B.; Gale, R.P.; Canaani, E. Fused transcript of abl and bcr genes in chronic myelogenous leukaemia. Nat. Cell Biol. 1985, 315, 550–554. [Google Scholar] [CrossRef] [PubMed]
- Stam, K.; Heisterkamp, N.; Grosveld, G.; de Klein, A.; Verma, R.S.; Coleman, M.; Dosik, H.; Groffen, J. Evidence of a new chimeric bcr/c-abl mRNA in patients with chronic myelocytic leukemia and the Philadelphia chromosome. N. Engl. J. Med. 1985, 313, 1429–1433. [Google Scholar] [CrossRef] [PubMed]
- Konopka, J.B.; Watanabe, S.M.; Witte, O.N. An alteration of the human c-abl protein in K562 leukemia cells unmasks associated tyrosine kinase activity. Cell 1984, 37, 1035–1042. [Google Scholar] [CrossRef]
- Ben-Neriah, Y.; Daley, G.Q.; Mes-Masson, A.M.; Witte, O.N.; Baltimore, D. The chronic myelogenous leukemia-specific P210 protein is the product of the bcr/abl hybrid gene. Science 1986, 233, 212–214. [Google Scholar] [CrossRef] [PubMed]
- Lugo, T.G.; Pendergast, A.M.; Muller, A.J.; Witte, O.N. Tyrosine kinase activity and transformation potency of bcr-abl oncogene products. Science 1990, 247, 1079–1082. [Google Scholar] [CrossRef]
- Daley, G.Q.; Baltimore, D. Transformation of an interleukin 3-dependent hematopoietic cell line by the chronic myelogenous leukemia-specific P210bcr/abl protein. Proc. Natl. Acad. Sci. USA 1988, 85, 9312–9316. [Google Scholar] [CrossRef] [Green Version]
- Daley, G.Q.; Van Etten, R.A.; Baltimore, D. Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science 1990, 247, 824–830. [Google Scholar] [CrossRef] [Green Version]
- Jamieson, C.H.; Ailles, L.E.; Dylla, S.J.; Muijtjens, M.; Jones, C.; Zehnder, J.L.; Gotlib, J.; Li, K.; Manz, M.G.; Keating, A.; et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis, C.M.L. N. Engl. J. Med. 2004, 351, 657–667. [Google Scholar] [CrossRef] [Green Version]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- Vetrie, D.; Helgason, G.V.; Copland, M. The leukaemia stem cell: Similarities, differences and clinical prospects in CML and AML. Nat. Rev. Cancer 2020, 20, 158–173. [Google Scholar] [CrossRef] [PubMed]
- Giustacchini, A.; Thongjuea, S.; Barkas, N.; Woll, P.S.; Povinelli, B.J.; Booth, C.A.G.; Sopp, P.; Norfo, R.; Rodriguez-Meira, A.; Ashley, N.; et al. Single-cell transcriptomics uncovers distinct molecular signatures of stem cells in chronic myeloid leukemia. Nat. Med. 2017, 23, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, H.; Cerny-Reiterer, S.; Gleixner, K.V.; Blatt, K.; Herndlhofer, S.; Rabitsch, W.; Jääger, E.; Mitterbauer-Hohendanner, G.; Streubel, B.; Selzer, E.; et al. CD34(+)/CD38(-) stem cells in chronic myeloid leukemia express Siglec-3 (CD33) and are responsive to the CD33-targeting drug gemtuzumab/ozogamicin. Haematologica 2012, 97, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Landberg, N.; Von Palffy, S.; Askmyr, M.; Lilljebjörn, H.; Sandén, C.; Rissler, M.; Mustjoki, S.; Hjorth-Hansen, H.; Richter, J.; Ågerstam, H.; et al. CD36 defines primitive chronic myeloid leukemia cells less responsive to imatinib but vulnerable to antibody-based therapeutic targeting. Haematologica 2017, 103, 447–455. [Google Scholar] [CrossRef] [Green Version]
- Ye, H.; Adane, B.; Khan, N.; Sullivan, T.; Minhajuddin, M.; Gasparetto, M.; Stevens, B.; Pei, S.; Balys, M.; Ashton, J.M.; et al. Leukemic Stem Cells Evade Chemotherapy by Metabolic Adaptation to an Adipose Tissue Niche. Cell Stem Cell 2016, 19, 23–37. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, H.; Sadovnik, I.; Cerny-Reiterer, S.; Rülicke, T.; Stefanzl, G.; Willmann, M.; Hörmann, G.; Bilban, M.; Blatt, K.; Herndlhofer, S.; et al. Dipeptidylpeptidase IV (CD26) defines leukemic stem cells (LSC) in chronic myeloid leukemia. Blood 2014, 123, 3951–3962. [Google Scholar] [CrossRef] [Green Version]
- Warfvinge, R.; Geironson, L.; Sommarin, M.N.E.; Lang, S.; Karlsson, C.; Roschupkina, T.; Stenke, L.; Stentoft, J.; Olsson-Strömberg, U.; Hjorth-Hansen, H.; et al. Single-cell molecular analysis defines therapy response and immunophenotype of stem cell subpopulations in CML. Blood 2017, 129, 2384–2394. [Google Scholar] [CrossRef]
- Kinstrie, R.; Horne, G.A.; Morrison, H.; Irvine, D.; Munje, C.; Castañeda, E.G.; Copland, M. CD93 is expressed on chronic myeloid leukemia stem cells and identifies a quiescent population which persists after tyrosine kinase inhibitor therapy. Leukemia 2020, 34, 1613–1625. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Chu, S.; Agarwal, P.; Campbell, V.L.; Hopcroft, L.; Jørgensen, H.G.; Lin, A.; Gaal, K.; Holyoake, T.L.; Bhatia, R. Inhibition of interleukin-1 signaling enhances elimination of tyrosine kinase inhibitor–treated CML stem cells. Blood 2016, 128, 2671–2682. [Google Scholar] [CrossRef] [Green Version]
- Sadovnik, I.; Herrmann, H.; Eisenwort, G.; Blatt, K.; Hoermann, G.; Mueller, N.; Sperr, W.R.; Valent, P. Expression of CD25 on leukemic stem cells in BCR-ABL1 + CML: Potential diagnostic value and functional implications. Exp. Hematol. 2017, 51, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Nievergall, E.; Ramshaw, H.; Yong, A.S.; Biondo, M.; Busfield, S.J.; Vairo, G.; Lopez, A.F.; Hughes, T.P.; White, D.L.; Hiwase, D.K. Monoclonal antibody targeting of IL-3 receptor α with CSL362 effectively depletes CML progenitor and stem cells. Blood 2014, 123, 1218–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Chen, Y.; Douglas, L.; Li, S. beta-Catenin is essential for survival of leukemic stem cells insensitive to kinase inhibition in mice with BCR-ABL-induced chronic myeloid leukemia. Leukemia 2009, 23, 109–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neviani, P.; Harb, J.G.; Oaks, J.J.; Santhanam, R.; Walker, C.J.; Ellis, J.J.; Ferenchak, G.; Dorrance, A.M.; Paisie, C.A.; Eiring, A.M.; et al. PP2A-activating drugs selectively eradicate TKI-resistant chronic myeloid leukemic stem cells. J. Clin. Investig. 2013, 123, 4144–4157. [Google Scholar] [CrossRef] [PubMed]
- Abraham, S.A.; Hopcroft, L.E.M.; Carrick, E.; Drotar, M.E.; Dunn, K.; Williamson, A.J.K.; Korfi, K.; Baquero, P.; Park, L.E.; Scott, M.T.; et al. Dual targeting of p53 and c-MYC selectively eliminates leukaemic stem cells. Nat. Cell Biol. 2016, 534, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Park, C.S.; Lewis, A.H.; Chen, T.J.; Bridges, C.S.; Shen, Y.; Suppipat, K.; Puppi, M.; Tomolonis, J.A.; Pang, P.D.; Mistretta, T.-A.; et al. A KLF4-DYRK2–mediated pathway regulating self-renewal in CML stem cells. Blood 2019, 134, 1960–1972. [Google Scholar] [CrossRef] [PubMed]
- Kuntz, E.M.; Baquero, P.; Michie, A.M.; Dunn, K.; Tardito, S.; Holyoake, T.L.; Helgason, G.V.; Gottlieb, E. Targeting mitochondrial oxidative phosphorylation eradicates therapy-resistant chronic myeloid leukemia stem cells. Nat. Med. 2017, 23, 1234–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellodi, C.; Lidonnici, M.R.; Hamilton, A.; Helgason, G.V.; Soliera, A.R.; Ronchetti, M.; Calabretta, B. Targeting autophagy potentiates tyrosine kinase inhibitor–induced cell death in Philadelphia chromosome–positive cells, including primary CML stem cells. J. Clin. Investig. 2009, 119, 1109–1123. [Google Scholar] [CrossRef]
- Karvela, M.; Baquero, P.; Kuntz, E.M.; Mukhopadhyay, A.; Mitchell, R.; Allan, E.K.; Chan, E.; Kranc, K.R.; Calabretta, B.; Salomoni, P.; et al. ATG7 regulates energy metabolism, differentiation and survival of Philadelphia-chromosome-positive cells. Autophagy 2016, 12, 936–948. [Google Scholar] [CrossRef] [Green Version]
- Corbin, A.S.; Agarwal, A.; Loriaux, M.; Cortes, J.; Deininger, M.W.; Druker, B.J. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J. Clin. Invest. 2011, 121, 396–409. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, A.; Helgason, G.V.; Schemionek, M.; Zhang, B.; Myssina, S.; Allan, E.K.; Nicolini, F.E.; Mueller-Tidow, C.; Bhatia, R.; Brunton, V.G.; et al. Chronic myeloid leukemia stem cells are not dependent on Bcr-Abl kinase activity for their survival. Blood 2012, 119, 1501–1510. [Google Scholar] [CrossRef] [Green Version]
- Houshmand, M.; Simonetti, G.; Circosta, P.; Gaidano, V.; Cignetti, A.; Martinelli, G.; Saglio, G.; Gale, R.P. Chronic myeloid leukemia stem cells. Leukemia 2019, 33, 1543–1556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, A.J.; Young, J.C.; Pendergast, A.M.; Pondel, M.; Landau, N.R.; Littman, D.R.; Witte, O.N. BCR first exon sequences specifically activate the BCR/ABL tyrosine kinase oncogene of Philadelphia chromosome-positive human leukemias. Mol. Cell. Biol. 1991, 11, 1785–1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, M.Y.; Dowding, C.R.; Riley, G.; Goldman, J.M.; Greaves, M.F. Altered adhesive interactions with marrow stroma of haematopoietic progenitor cells in chronic myeloid leukaemia. Nat. Cell Biol. 1987, 328, 342–344. [Google Scholar] [CrossRef] [PubMed]
- Verfaillie, C.M.; McCarthy, J.B.; McGlave, P.B. Mechanisms underlying abnormal trafficking of malignant progenitors in chronic myelogenous leukemia. Decreased adhesion to stroma and fibronectin but increased adhesion to the basement membrane components laminin and collagen type IV. J. Clin. Investig. 1992, 90, 1232–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uemura, N.; Griffin, J.D. The adapter protein Crkl links Cbl to C3G after integrin ligation and enhances cell migration. J. Biol. Chem. 1999, 274, 37525–37532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salgia, R.; Brunkhorst, B.; Pisick, E.; Li, J.L.; Lo, S.H.; Chen, L.B.; Griffin, J.D. Increased tyrosine phosphorylation of focal adhesion proteins in myeloid cell lines expressing, p.2.1.0.B.C.R./.A.B.L. Oncogene 1995, 11, 1149–1155. [Google Scholar] [PubMed]
- Van Etten, R.A.; Jackson, P.K.; Baltimore, D.; Sanders, M.C.; Matsudaira, P.T.; Janmey, P.A. The COOH terminus of the c-Abl tyrosine kinase contains distinct F- and G-actin binding domains with bundling activity. J. Cell Biol. 1994, 124, 325–340. [Google Scholar] [CrossRef] [Green Version]
- Salgia, R.; Li, J.L.; Ewaniuk, D.S.; Pear, W.; Pisick, E.; Burky, S.A.; Ernst, T.; Sattler, M.; Chen, L.B.; Griffin, J.D. BCR/ABL induces multiple abnormalities of cytoskeletal function. J. Clin. Investig. 1997, 100, 46–57. [Google Scholar] [CrossRef]
- Wertheim, J.A.; Perera, S.A.; Hammer, A.D.; Ren, R.; Boettiger, D.; Pear, W.S. Localization of BCR-ABL to F-actin regulates cell adhesion but does not attenuate CML development. Blood 2003, 102, 2220–2228. [Google Scholar] [CrossRef]
- Thomas, E.K.; Cancelas, A.J.; Zheng, Y.; Williams, D.A. Rac GTPases as key regulators of p210-BCR-ABL-dependent leukemogenesis. Leukemia 2008, 22, 898–904. [Google Scholar] [CrossRef] [Green Version]
- Krause, D.S.; Lazarides, K.; Von Andrian, U.H.; Van Etten, A.R. Requirement for CD44 in homing and engraftment of BCR-ABL–expressing leukemic stem cells. Nat. Med. 2006, 12, 1175–1180. [Google Scholar] [CrossRef] [PubMed]
- Godavarthy, P.S.; Kumar, R.; Herkt, S.C.; Pereira, R.S.; Hayduk, N.; Weissenberger, E.S.; Aggoune, D.; Manavski, Y.; Lucas, T.; Pan, K.-T.; et al. The vascular bone marrow niche influences outcome in chronic myeloid leukemia via the E-selectin - SCL/TAL1 - CD44 axis. Haematologica 2019, 105, 136–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deininger, M.W.; Vieira, S.; Mendiola, R.; Schultheis, B.; Goldman, J.M.; Melo, J.V. BCR-ABL tyrosine kinase activity regulates the expression of multiple genes implicated in the pathogenesis of chronic myeloid leukemia. Cancer Res. 2000, 60. [Google Scholar]
- Bazzoni, G.; Carlesso, N.; Griffin, J.D.; Hemler, E.M. Bcr/Abl expression stimulates integrin function in hematopoietic cell lines. J. Clin. Investig. 1996, 98, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Krause, D.S.; Lazarides, K.; Lewis, J.B.; Von Andrian, U.H.; Van Etten, R.A. Selectins and their ligands are required for homing and engraftment of BCR-ABL1+ leukemic stem cells in the bone marrow niche. Blood 2014, 123, 1361–1371. [Google Scholar] [CrossRef] [Green Version]
- Steelman, L.S.; Abrams, S.L.; Whelan, J.; Bertrand, E.F.; Ludwig, E.D.; Bäsecke, J.; Libra, M.; Stivala, F.; Milella, M.; Tafuri, A.; et al. Contributions of the Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways to leukemia. Leukemia 2008, 22, 686–707. [Google Scholar] [CrossRef] [Green Version]
- Ilaria, R.L.; Van Etten, A.R. P210 and P190(BCR/ABL) induce the tyrosine phosphorylation and DNA binding activity of multiple specific STAT family members. J. Biol. Chem. 1996, 271. [Google Scholar] [CrossRef] [Green Version]
- Moriggl, R.H.; Sexl, V.; Kenner, L.; Duntsch, C.; Stangl, K.; Gingras, S.; Hoffmeyer, A.; Bauer, A.; Piekorz, R.; Wang, D.; et al. Stat5 tetramer formation is associated with leukemogenesis. Cancer Cell 2005, 7, 87–99. [Google Scholar] [CrossRef] [Green Version]
- Ye, D.; Wolff, N.; Li, L.; Zhang, S.; Ilaria, R.L. STAT5 signaling is required for the efficient induction and maintenance of CML in mice. Blood 2006, 107, 4917–4925. [Google Scholar] [CrossRef]
- José-Enériz, E.S.; Román-Gómez, J.; Cordeu, L.; Ballestar, E.; Garate, L.; Andreu, E.J.; Isidro, I.; Guruceaga, E.; Jiménez-Velasco, A.; Heiniger, A.; et al. BCR-ABL1-induced expression of HSPA8 promotes cell survival in chronic myeloid leukaemia. Br. J. Haematol. 2008, 142, 571–582. [Google Scholar] [CrossRef]
- Hoelbl, A.; Schuster, C.; Kovacic, B.; Zhu, B.; Wickre, M.; Hoelzl, M.A.; Fajmann, S.; Grebien, F.; Warsch, W.; Stengl, G.; et al. Stat5 is indispensable for the maintenance of bcr/abl -positive leukaemia. EMBO Mol. Med. 2010, 2, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Gesbert, F.; Griffin, J.D. Bcr/Abl activates transcription of the Bcl-X gene through STAT. Blood J. Am. Soc. Hematol. 2000, 96, 2269–2276. [Google Scholar]
- Skorski, T.; Kanakaraj, P.; Nieborowska-Skorska, M.; Ratajczak, M.; Wen, S.; Zon, G.; Gewirtz, A.; Perussia, B.; Calabretta, B. Phosphatidylinositol-3 kinase activity is regulated by BCR/ABL and is required for the growth of Philadelphia chromosome-positive cells. Blood 1995, 86, 726–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franke, T.F.; Kaplan, D.R.; Cantley, L.C. PI3K: Downstream AKTion Blocks Apoptosis. Cell 1997, 88, 435–437. [Google Scholar] [CrossRef] [Green Version]
- Sawyers, C.L.; McLaughlin, J.; Witte, O.N. Genetic requirement for Ras in the transformation of fibroblasts and hematopoietic cells by the Bcr-Abl oncogene. J. Exp. Med. 1995, 181, 307–313. [Google Scholar] [CrossRef] [Green Version]
- Jonuleit, T.; van der Kuip, H.; Miething, C.; Michels, H.; Hallek, M.; Duyster, J.; Aulitzky, W.E. Bcr-Abl kinase down-regulates cyclin-dependent kinase inhibitor p27 in human and murine cell lines. Blood J. Am. Soc. Hematol. 2000, 96, 1933–1939. [Google Scholar]
- Agarwal, A.; MacKenzie, R.J.; Besson, A.; Jeng, S.; Carey, A.; Latocha, D.H.; Fleischman, A.G.; Duquesnes, N.; Eide, C.A.; Vasudevan, K.B.; et al. BCR-ABL1 promotes leukemia by converting p27 into a cytoplasmic oncoprotein. Blood 2014, 124, 3260–3273. [Google Scholar] [CrossRef] [Green Version]
- Dasgupta, Y.; Koptyra, M.; Hoser, G.; Kantekure, K.; Roy, D.; Górnicka, B.; Nieborowska-Skorska, M.; Bolton-Gillespie, E.; Cerny-Reiterer, S.; Müschen, M.; et al. Normal ABL1 is a tumor suppressor and therapeutic target in human and mouse leukemias expressing oncogenic ABL1 kinases. Blood 2016, 127, 2131–2143. [Google Scholar] [CrossRef] [Green Version]
- Skorski, T. Genomic instability: The cause and effect of BCR/ABL tyrosine kinase. Curr. Hematol. Malign. Rep. 2007, 2, 69–74. [Google Scholar] [CrossRef]
- O’Dwyer, M.; Mauro, M.J.; Blasdel, C.; Farnsworth, M.; Kurilik, G.; Hsieh, Y.-C.; Mori, M.; Druker, B.J. Clonal evolution and lack of cytogenetic response are adverse prognostic factors for hematologic relapse of chronic phase CML patients treated with imatinib mesylate. Blood 2004, 103, 451–455. [Google Scholar] [CrossRef] [Green Version]
- Cortes, J.; Talpaz, M.; Giles, F.; O’Brien, S.; Rios, M.B.; Shan, J.; Garcia-Manero, G.; Faderl, S.; Thomas, D.; Wierda, W.; et al. Prognostic significance of cytogenetic clonal evolution in patients with chronic myelogenous leukemia on imatinib mesylate therapy. Blood 2003, 101, 3794–3800. [Google Scholar] [CrossRef] [PubMed]
- Lahaye, T.; Riehm, B.; Berger, U.; Paschka, P.; Müller, M.C.; Kreil, S.; Merx, K.; Schwindel, U.; Schoch, C.; Hehlmann, R.; et al. Response and resistance in 300 patients with BCR-ABL-positive leukemias treated with imatinib in a single center. Cancer 2005, 103, 1659–1669. [Google Scholar] [CrossRef] [PubMed]
- Johansson, B.; Fioretos, T.; Mitelman, F. Cytogenetic and Molecular Genetic Evolution of Chronic Myeloid Leukemia. Acta Haematol. 2002, 107, 76–94. [Google Scholar] [CrossRef] [PubMed]
- Verma, D.; Kantarjian, H.; Shan, J.; O’Brien, S.; Estrov, Z.; Garcia-Manero, G.; Koller, C.; Borthakur, G.; Cortes, J. Survival outcomes for clonal evolution in chronic myeloid leukemia patients on second generation tyrosine kinase inhibitor therapy. Cancer 2010, 116, 2673–2681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilaria, R.L. Pathobiology of Lymphoid and Myeloid Blast Crisis and Management Issues. Hematology 2005, 2005, 188–194. [Google Scholar] [CrossRef]
- Honda, H.; Ushijima, T.; Wakazono, K.; Oda, H.; Tanaka, Y.; Aizawa, S.-I.; Ishikawa, T.; Yazaki, Y.; Hirai, H. Acquired loss of p53 induces blastic transformation in p210bcr/abl-expressing hematopoietic cells: A transgenic study for blast crisis of human CML. Blood 2000, 95, 1144–1150. [Google Scholar] [CrossRef]
- Peterson, L.F.; Mitrikeska, E.; Giannola, D.; Lui, Y.; Sun, H.; Bixby, D.; Malek, S.N.; Donato, N.J.; Wang, S.; Talpaz, M. p53 stabilization induces apoptosis in chronic myeloid leukemia blast crisis cells. Leukemia 2011, 25, 761–769. [Google Scholar] [CrossRef] [Green Version]
- Ko, T.K.; Javed, A.; Lee, K.L.; Pathiraja, T.N.; Liu, X.; Malik, S.; Soh, S.X.; Heng, X.T.; Takahashi, N.; Tan, J.H.J.; et al. An integrative model of pathway convergence in genetically heterogeneous blast crisis chronic myeloid leukemia. Blood 2020, 135, 2337–2353. [Google Scholar] [CrossRef]
- Schmidt, M.; Rinke, J.; Schafer, V.; Schnittger, S.; Kohlmann, A.; Obstfelder, E.; Kunert, C.; Ziermann, J.; Winkelmann, N.; Eigendorff, E.; et al. Molecular-defined clonal evolution in patients with chronic myeloid leukemia independent of the BCR-ABL status. Leukemia 2014, 28, 2292–2299. [Google Scholar] [CrossRef]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [Green Version]
- Ban, K.; Gao, Y.; Amin, H.M.; Howard, A.; Miller, C.; Lin, Q.; Leng, X.; Munsell, M.; Bar-Eli, M.; Arlinghaus, R.B.; et al. BCR-ABL1 mediates up-regulation of Fyn in chronic myelogenous leukemia. Blood 2008, 111, 2904–2908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, M.M.; Howard, A.; Irwin, M.E.; Gao, Y.; Lu, X.; Multani, A.; Chandra, J. Expression and Activity of Fyn Mediate Proliferation and Blastic Features of Chronic Myelogenous Leukemia. PLoS ONE 2012, 7, e51611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Handa, H.; Hegde, U.P.; Kotelnikov, V.M.; Mundle, S.D.; Dong, L.-M.; Burke, P.; Rose, S.; Gaskin, F.; Raza, A.; Preisler, H.D. bcl-2 and c-myc Expression, cell cycle kinetics and apoptosis during the progression of chronic myelogenous leukemia from diagnosis to blastic phase. Leuk. Res. 1997, 21, 479–489. [Google Scholar] [CrossRef]
- Beverly, L.J.; Varmus, H.E. MYC-induced myeloid leukemogenesis is accelerated by all six members of the antiapoptotic BCL family. Oncogene 2009, 28, 1274–1279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Casares, M.T.; Garcia-Alegria, E.; Lopez-Jorge, C.E.; Ferrandiz, N.; Blanco, R.; Alvarez, S.; Vaqué, J.P.; Bretones, G.; Caraballo, J.M.; Sánchez-Bailón, P.; et al. MYC antagonizes the differ-entiation induced by imatinib in chronic myeloid leukemia cells through downregulation of p27(KIP1.). Oncogene 2013, 32, 2239–2246. [Google Scholar] [CrossRef]
- Srutova, K.; Curik, N.; Burda, P.; Savvulidi, F.; Silvestri, G.; Trotta, R.; Klamova, H.; Pecherkova, P.; Sovova, Z.; Koblihova, J.; et al. BCR-ABL1 mediated miR-150 downregulation through MYC contributed to myeloid differentiation block and drug resistance in chronic myeloid leukemia. Haematologica 2018, 103, 2016–2025. [Google Scholar] [CrossRef]
- Albajar, M.; Gómez-Casares, M.T.; Llorca, J.; Mauleon, I.; Vaqué, J.P.; Acosta, J.A.T.; Bermúdez, A.; Donato, N.J.; Delgado, M.D.; León, J. MYC in Chronic Myeloid Leukemia: Induction of Aberrant DNA Synthesis and Association with Poor Response to Imatinib. Mol. Cancer Res. 2011, 9, 564–576. [Google Scholar] [CrossRef] [Green Version]
- Hochhaus, A.; Baccarani, M.; Silver, R.T.; Schiffer, C.; Apperley, J.F.; Cervantes, F.; Hehlmann, R. European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia 2020, 34, 966–984. [Google Scholar] [CrossRef] [Green Version]
- Hukku, S.; Baboo, H.A.; Venkataratnam, S.; Vidyasagar, M.S.; Patel, N.L. Splenic Irradiation in Chronic Myeloid Leukemia. Acta Radiol. Oncol. 1983, 22, 9–12. [Google Scholar] [CrossRef]
- Morstyn, G.; Sullivan, J.; Fairhead, S.; Cowling, D.; Hurley, T. Effect of High Dose Busulphan on Leukaemic Progenitor Cells in Chronic Myeloid Leukaemia. Aust. N. Z. J. Med. 1981, 11, 609–614. [Google Scholar] [CrossRef]
- Kennedy, B.J. Hydroxyurea therapy in chronic myelogenous leukemia. Cancer 1972, 29, 1052–1056. [Google Scholar] [CrossRef]
- Talpaz, M.; McCredie, K.; Jian, H.K.; Trujillo, J.; Keating, M.; Gutterman, J. Chronic myelogenous leukaemia: Haematological remissions with alpha interferon. Br. J. Haematol. 1986, 64, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Yoffe, G.; Blick, M.; Kantarjian, H.; Spitzer, G.; Gutterman, J.; Talpaz, M. Molecular analysis of interferon-induced suppression of Philadelphia chromosome in patients with chronic myeloid leukemia. Blood 1987, 69, 961–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonifazi, F.; De Vivo, A.; Rosti, G.; Guilhot, F.; Guilhot, J.; Trabacchi, E.; Hehlmann, R.; Hochhaus, A.; Shepherd, P.C.A.; Steegmann, J.L.; et al. Chronic myeloid leukemia and interferon-α: A study of complete cytogenetic responders. Blood 2001, 98, 3074–3081. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.M.; O’Brien, S.; Cortes, J.E.; Shan, J.; Giles, F.J.; Rios, M.B.; Talpaz, M. Complete cytogenetic and molecular responses to interferon-α-based therapy for chronic myelogenous leukemia are associated with excellent long-term prognosis. Cancer Interdiscip. Int. J. Am. Cancer Soc. 2003, 97, 1033–1041. [Google Scholar] [CrossRef]
- Fefer, A.; Cheever, M.A.; Thomas, E.D.; Boyd, C.; Ramberg, R.; Glucksberg, H.; Buckner, C.D.; Storb, R. Disappearance of pH1-Positive Cells in Four Patients with Chronic Granulocytic Leukemia after Chemotherapy, Irradiation and Marrow Transplantation from an Identical Twin. N. Engl. J. Med. 1979, 300, 333–337. [Google Scholar] [CrossRef]
- Goldman, J.M.; McCarthy, D.M.; Hows, J.M.; Catovsky, D.; Goolden AW, G.; Baughan AS, J.; Galton, D.A.G. Marrow transplantation for patients in the chronic phase of chronic granulocytic leukaemia. Lancet 1982, 320, 623–625. [Google Scholar] [CrossRef]
- McGlave, P.B.; Arthur, D.C.; Kim, T.H.; Ramsay, N.K.; Hurd, D.D.; Kersey, J. Successful allogeneic bone-marrow transplan-tation for patients in the accelerated phase of chronic granulocytic leukaemia. Lancet 1982, 2, 625–627. [Google Scholar] [CrossRef]
- Horowitz, M.M.; Gale, R.P.; Sondel, P.M.; Goldman, J.M.; Kersey, J.; Kolb, H.J.; Speck, B. Graft-versus-leukemia reactions after bone marrow transplantation. Blood 1990, 75, 555–562. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.H.; Coleman, S. Chronic myeloid leukemia as an immunological target. Am. J. Hematol. 1997, 54, 61–67. [Google Scholar] [CrossRef]
- El-Shami, K.; Smith, B.D. Immunotherapy for myeloid leukemias: Current status and future directions. Leukemia 2008, 22, 1658–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchdunger, E.; Zimmermann, J.; Mett, H.; Meyer, T.; Müller, M.; Druker, B.J.; Lydon, N.B. Inhibition of the Abl protein-tyrosine kinase in vitro and in vivo by a 2-phenylaminopyrimidine derivative. Cancer Res. 1996, 56. [Google Scholar]
- Druker, B.J.; Tamura, S.; Buchdunger, E.; Ohno, S.; Segal, G.M.; Fanning, S.; Zimmermann, J.; Lydon, N.B. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr–Abl positive cells. Nat. Med. 1996, 2, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Deininger, M.W.; Goldman, J.M.; Lydon, N.; Melo, J.V. The Tyrosine Kinase Inhibitor CGP57148B Selectively Inhibits the Growth of BCR-ABL–Positive Cells. Blood 1997, 90, 3691–3698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hochhaus, A.; Larson, R.A.; Guilhot, F.; Radich, J.P.; Branford, S.; Hughes, T.P.; Baccarani, M.; Deininger, M.W.; Cervantes, F.; Fujihara, S.; et al. Long-Term Outcomes of Imatinib Treatment for Chronic Myeloid Leukemia. N. Engl. J. Med. 2017, 376, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Braun, T.P.; Eide, C.A.; Druker, B.J. Response and Resistance to BCR-ABL1-Targeted Therapies. Cancer Cell 2020, 37, 530–542. [Google Scholar] [CrossRef]
- Weisberg, E.; Manley, P.W.; Breitenstein, W.; Bruggen, J.; Cowan-Jacob, S.W.; Ray, A.; Huntly, B.; Fabbro, D.; Fendrich, G.; Hall-Meyers, E.; et al. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell. 2005, 7, 129–141. [Google Scholar] [CrossRef] [Green Version]
- Shah, N.P.; Tran, C.; Lee, F.Y.; Chen, P.; Norris, D.; Sawyers, C.L. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science 2004, 305, 399–401. [Google Scholar] [CrossRef] [Green Version]
- Golas, J.M.; Arndt, K.; Etienne, C.; Lucas, J.; Nardin, D.; Gibbons, J.; Frost, P.; Ye, F.; Boschelli, D.H.; Boschelli, F. SKI-606, a 4-anilino-3-quinolinecarbonitrile dual inhibitor of Src and Abl kinases, is a potent antiproliferative agent against chronic myelogenous leukemia cells in culture and causes re-gression of K562 xenografts in nude mice. Cancer Res. 2003, 63, 375–381. [Google Scholar]
- O’Hare, T.; Shakespeare, W.C.; Zhu, X.; Eide, C.A.; Rivera, V.M.; Wang, F.; Adrian, L.T.; Zhou, T.; Huang, W.-S.; Xu, Q.; et al. AP24534, a Pan-BCR-ABL Inhibitor for Chronic Myeloid Leukemia, Potently Inhibits the T315I Mutant and Overcomes Mutation-Based Resistance. Cancer Cell 2009, 16, 401–412. [Google Scholar] [CrossRef] [Green Version]
- Wylie, A.A.; Schoepfer, J.; Jahnke, W.; Cowan-Jacob, S.W.; Loo, A.; Furet, P.; Marzinzik, A.L.; Pelle, X.; Donovan, J.; Zhu, W.; et al. The allosteric inhibitor ABL001 enables dual targeting of BCR–ABL1. Nat. Cell Biol. 2017, 543, 733–737. [Google Scholar] [CrossRef] [PubMed]
- Bower, H.; Björkholm, M.; Dickman, P.W.; Höglund, M.; Lambert, P.C.; Andersson, T.M.-L. Life Expectancy of Patients With Chronic Myeloid Leukemia Approaches the Life Expectancy of the General Population. J. Clin. Oncol. 2016, 34, 2851–2857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Efficace, F.; Baccarani, M.; Breccia, M.; Cottone, F.; Alimena, G.; Deliliers, G.L.; Baratè, C.; Specchia, G.; Di Lorenzo, R.; Luciano, L.; et al. Chronic fatigue is the most important factor limiting health-related quality of life of chronic myeloid leukemia patients treated with imatinib. Leukemia 2013, 27, 1511–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Efficace, F.; Baccarani, M.; Breccia, M.; Alimena, G.; Rosti, G.; Cottone, F.; Deliliers, G.L.; Baratè, C.; Rossi, A.R.; Fioritoni, G.; et al. Health-related quality of life in chronic myeloid leukemia patients receiving long-term therapy with imatinib compared with the general population. Blood 2011, 118, 4554–4560. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Ferrer, S.; Bonnet, D.; Steensma, D.P.; Hasserjian, R.P.; Ghobrial, I.M.; Gribben, J.G.; Andreeff, M.; Krause, D.S. Bone marrow niches in haematological malignancies. Nat. Rev. Cancer 2020, 20, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Bocchia, M.; Sicuranza, A.; Abruzzese, E.; Iurlo, A.; Sirianni, S.; Gozzini, A.; Galimberti, S.; Aprile, L.; Martino, B.; Pregno, P.; et al. Residual Peripheral Blood CD26+ Leukemic Stem Cells in Chronic Myeloid Leukemia Patients During TKI Therapy and During Treatment-Free Remission. Front. Oncol. 2018, 8, 194. [Google Scholar] [CrossRef] [Green Version]
- Briscoe, J.; Therond, P.P. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 2013, 14, 416–429. [Google Scholar] [CrossRef]
- Gao, J.; Graves, S.; Koch, U.; Liu, S.; Jankovic, V.; Buonamici, S.; El Andaloussi, A.; Nimer, S.D.; Kee, B.L.; Taichman, R.S.; et al. Hedgehog Signaling Is Dispensable for Adult Hematopoietic Stem Cell Function. Cell Stem Cell 2009, 4, 548–558. [Google Scholar] [CrossRef] [Green Version]
- Dierks, C.; Beigi, R.; Guo, G.R.; Zirlik, K.; Stegert, M.R.; Manley, P.; Trussel, C.; Schmitt-Graeff, A.; Landwerlin, K.; Veelken, H.; et al. Expansion of Bcr-Abl-positive leukemic stem cells is dependent on Hedgehog pathway activation. Cancer Cell. 2008, 14, 238–249. [Google Scholar] [CrossRef] [Green Version]
- Irvine, D.A.; Zhang, B.; Kinstrie, R.; Tarafdar, A.; Morrison, H.; Campbell, V.L.; Moka, H.A.; Ho, Y.; Nixon, C.; Manley, P.W.; et al. Deregulated hedgehog pathway signaling is inhibited by the smoothened antagonist LDE225 (Sonidegib) in chronic phase chronic myeloid leukaemia. Sci. Rep. 2016, 6, 25476. [Google Scholar] [CrossRef] [Green Version]
- Zeng, X.; Zhao, H.; Li, Y.; Fan, J.; Sun, Y.; Wang, S.; Wang, Z.; Song, P.; Ju, D. Targeting Hedgehog signaling pathway and autophagy overcomes drug resistance of BCR-ABL-positive chronic myeloid leukemia. Autophagy 2015, 11, 355–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoxhaj, G.; Manning, B.D. The PI3K–AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2019, 20, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Airiau, K.; Mahon, F.-X.; Josselin, M.; Jeanneteau, M.; Belloc, F. PI3K/mTOR pathway inhibitors sensitize chronic myeloid leukemia stem cells to nilotinib and restore the response of progenitors to nilotinib in the presence of stem cell factor. Cell Death Dis. 2013, 4, e827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alves, R.; Goncalves, A.C.; Jorge, J.; Marques, G.; Luis, D.; Ribeiro, A.B.; Sarmento-Ribeiro, A.B. MicroRNA signature refine response prediction in, C.M.L. Sci Rep. 2019, 9, 9666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.-Z.; Pu, Q.-H.; Lin, X.-H.; Liu, M.-Y.; Wu, L.-R.; Wu, Q.-Q.; Chen, Y.-H.; Liao, F.-F.; Zhu, J.; Jin, X.-B. Silencing of miR-21 sensitizes CML CD34+ stem/progenitor cells to imatinib-induced apoptosis by blocking PI3K/AKT pathway. Leuk. Res. 2015, 39, 1117–1124. [Google Scholar] [CrossRef]
- Trojani, A.; Pungolino, E.; Molin, A.D.; Lodola, M.; Rossi, G.; D’Adda, M.; Perego, A.; Elena, C.; Turrini, M.; Borin, L.; et al. Nilotinib interferes with cell cycle, ABC transporters and JAK-STAT signaling pathway in CD34+/lin- cells of patients with chronic phase chronic myeloid leukemia after 12 months of treatment. PLoS ONE 2019, 14, e0218444. [Google Scholar] [CrossRef] [Green Version]
- Gallipoli, P.; Cook, A.; Rhodes, S.; Hopcroft, L.; Wheadon, H.; Whetton, A.D.; Jørgensen, H.G.; Bhatia, R.; Holyoake, T.L. JAK2/STAT5 inhibition by nilotinib with ruxolitinib contributes to the elimination of CML CD34+ cells in vitro and in vivo. Blood 2014, 124, 1492–1501. [Google Scholar] [CrossRef]
- Chen, M.; Gallipoli, P.; DeGeer, D.; Sloma, I.; Forrest, D.L.; Chan, M.; Lai, D.; Jorgensen, H.; Ringrose, A.; Wang, H.M.; et al. Targeting Primitive Chronic Myeloid Leukemia Cells by Effective Inhibition of a New AHI-1–BCR-ABL–JAK2 Complex. J. Natl. Cancer Inst. 2013, 105, 405–423. [Google Scholar] [CrossRef]
- Kuepper, M.K.; Bütow, M.; Herrmann, O.; Ziemons, J.; Chatain, N.; Maurer, A.; Kirschner, M.; Maié, T.; Costa, I.G.; Eschweiler, J.; et al. Stem cell persistence in CML is mediated by extrinsically activated JAK1-STAT3 signaling. Leukemia 2019, 33, 1964–1977. [Google Scholar] [CrossRef]
- Zhou, H.; Mak, P.Y.; Mu, H.; Mak, D.H.; Zeng, Z.; Cortes, J.; Carter, B.Z. Combined inhibition of beta-catenin and Bcr-Abl synergistically targets tyrosine kinase inhibitor-resistant blast crisis chronic myeloid leukemia blasts and progenitors in vitro and in vivo. Leukemia 2017, 31, 2065–2074. [Google Scholar] [CrossRef]
- Carter, B.Z.; Mak, P.Y.; Mu, H.; Wang, X.; Tao, W.; Mak, D.H.; Dettman, E.J.; Cardone, M.; Zernovak, O.; Seki, T.; et al. Combined inhibition of MDM2 and BCR-ABL1 tyrosine kinase targets chronic myeloid leukemia stem/progenitor cells in a murine model. Haematologica 2020, 105, 1274–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, B.Z.; Mak, P.Y.; Mu, H.; Zhou, H.; Mak, D.H.; Schober, W.; Leverson, J.D.; Zhang, B.; Bhatia, R.; Huang, X.; et al. Combined targeting of BCL-2 and BCR-ABL tyrosine kinase eradicates chronic myeloid leukemia stem cells. Sci. Transl. Med. 2016, 8, 355ra117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burchert, A.; Neubauer, A. Interferon a and T-cell responses in chronic myeloid leukemia. Leuk. Lymphoma 2005, 46, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Palandri, F.; Castagnetti, F.; Iacobucci, I.; Martinelli, G.; Amabile, M.; Gugliotta, G.; Poerio, A.; Testoni, N.; Breccia, M.; Bocchia, M.; et al. The response to imatinib and interfer-on-alpha is more rapid than the response to imatinib alone: A retrospective analysis of 495 Philadelphia-positive chronic myeloid leukemia patients in early chronic phase. Haematologica 2010, 95, 1415–1419. [Google Scholar] [CrossRef]
- Burchert, A.; Müller, M.C.; Kostrewa, P.; Erben, P.; Bostel, T.; Liebler, S.; Hehlmann, R.; Neubauer, A.; Hochhaus, A. Sustained Molecular Response With Interferon Alfa Maintenance After Induction Therapy With Imatinib Plus Interferon Alfa in Patients With Chronic Myeloid Leukemia. J. Clin. Oncol. 2010, 28, 1429–1435. [Google Scholar] [CrossRef] [PubMed]
- Preudhomme, C.; Guilhot, J.; Nicolini, F.E.; Guerci-Bresler, A.; Rigal-Huguet, F.; Maloisel, F.; Coiteux, V.; Gardembas, M.; Berthou, C.; Vekhoff, A.; et al. Imatinib plus Peginterferon Alfa-2a in Chronic Myeloid Leukemia. N. Engl. J. Med. 2010, 363, 2511–2521. [Google Scholar] [CrossRef]
- Hanfstein, B.; for the SAKK and the German CML Study Group; Müller, M.C.; Hehlmann, R.; Erben, P.; Lauseker, M.; Fabarius, A.; Schnittger, S.; Haferlach, C.; Göhring, G.; et al. Early molecular and cytogenetic response is predictive for long-term progression-free and overall survival in chronic myeloid leukemia (CML). Leukemia 2012, 26, 2096–2102. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, C.; Weinert, B.T.; Nishida, Y.; Verdin, E.; Mann, M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 536–550. [Google Scholar] [CrossRef]
- Zhang, B.; Strauss, A.C.; Chu, S.; Li, M.; Ho, Y.; Shiang, K.-D.; Snyder, D.S.; Huettner, C.S.; Shultz, L.; Holyoake, T.; et al. Effective Targeting of Quiescent Chronic Myelogenous Leukemia Stem Cells by Histone Deacetylase Inhibitors in Combination with Imatinib Mesylate. Cancer Cell 2010, 17, 427–442. [Google Scholar] [CrossRef] [Green Version]
- Wei, D.; Lu, T.; Ma, D.; Yu, K.; Zhang, T.; Xiong, J.; Wang, W.; Zhang, Z.; Fang, Q.; Wang, J. Synergistic activity of imatinib and AR-42 against chronic myeloid leukemia cells mainly through HDAC1 inhibition. Life Sci. 2018, 211, 224–237. [Google Scholar] [CrossRef]
- Matsuda, Y.; Yamauchi, T.; Hosono, N.; Uzui, K.; Negoro, E.; Morinaga, K.; Nishi, R.; Yoshida, A.; Kimura, S.; Maekawa, T.; et al. Combination of panobinostat with ponatinib synergistically overcomes imatinib-resistant CML cells. Cancer Sci. 2016, 107, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Byun, H.-M.; Eshaghian, S.; Douer, D.; Trent, J.; Garcia-Manero, G.; Bhatia, R.; Siegmund, K.; Hyang-Min, B. Impact of chromosomal rearrangement upon DNA methylation patterns in leukemia. Open Med. 2017, 12, 76–85. [Google Scholar] [CrossRef] [PubMed]
- San Jose-Eneriz, E.; Agirre, X.; Jimenez-Velasco, A.; Cordeu, L.; Martin, V.; Arqueros, V.; Gárate, L.; Fresquet, V.; Cervantes, F.; Martínez-Climent, J.A.; et al. Epigenetic down-regulation of BIM expression is associated with reduced optimal responses to imatinib treatment in chronic myeloid leukaemia. Eur. J. Cancer. 2009, 45, 1877–1889. [Google Scholar] [CrossRef] [PubMed]
- Scott, M.T.; Korfi, K.; Saffrey, P.; Hopcroft, L.E.; Kinstrie, R.; Pellicano, F.; Guenther, C.; Gallipoli, P.; Cruz, M.; Dunn, K.; et al. Epigenetic Reprogramming Sensitizes CML Stem Cells to Combined EZH2 and Tyrosine Kinase Inhibition. Cancer Discov. 2016, 6, 1248–1257. [Google Scholar] [CrossRef] [Green Version]
- Ghez, D.; Micol, J.-B.; Pasquier, F.; Auger, N.; Saada, V.; Spentchian, M.; Ianotto, J.-C.; Bourhis, J.-H.; Bennaceur-Griscelli, A.; Terre, C.; et al. Clinical efficacy of second generation tyrosine kinase inhibitor and 5-azacytidine combination in chronic myelogenous leukaemia in myeloid blast crisis. Eur. J. Cancer 2013, 49, 3666–3670. [Google Scholar] [CrossRef]
- Ruggiu, M.; Oberkampf, F.; Ghez, D.; Cony-Makhoul, P.; Beckeriche, F.; Cano, I.; Taksin, A.L.; Benbrahim, O.; Ghez, S.; Farhat, H.; et al. Azacytidine in combination with tyrosine kinase inhibitors induced durable responses in patients with advanced phase chronic myelogenous leukemia. Leuk. Lymphoma 2018, 59, 1659–1665. [Google Scholar] [CrossRef]
- Shao, S.; Li, S.; Qin, Y.; Wang, X.; Yang, Y.; Bai, H.; Zhou, L.; Zhao, C.; Wang, C. Spautin-1, a novel autophagy inhibitor, enhances imatinib-induced apoptosis in chronic myeloid leukemia. Int. J. Oncol. 2014, 44, 1661–1668. [Google Scholar] [CrossRef] [Green Version]
- Rothe, K.; Lin, H.; Lin, K.B.; Leung, A.; Wang, H.M.; Malekesmaeili, M.; Brinkman, R.R.; Forrest, L.; Gorski, M.; Jiang, X. The core autophagy protein ATG4B is a potential bi-omarker and therapeutic target in CML stem/progenitor cells. Blood 2014, 123, 3622–3634. [Google Scholar] [CrossRef]
- Baquero, P.; Dawson, A.; Mukhopadhyay, A.; Kuntz, E.M.; Mitchell, R.; Olivares, O.; Ianniciello, A.; Scott, M.T.; Dunn, K.; Nicastri, M.C.; et al. Targeting quiescent leukemic stem cells using second generation autophagy inhibitors. Leukemia 2019, 33, 981–994. [Google Scholar] [CrossRef] [Green Version]
- Horne, G.A.; Stobo, J.; Kelly, C.; Mukhopadhyay, A.; Latif, A.L.; Dixon-Hughes, J.; McMahon, L.; Cony-Makhoul, P.; Byrne, J.; Smith, G.; et al. A randomised phase II trial of hy-droxychloroquine and imatinib versus imatinib alone for patients with chronic myeloid leukaemia in major cytogenetic response with residual disease. Leukemia 2020, 34, 1775–1786. [Google Scholar] [CrossRef]
- Chomel, J.C.; Bonnet, M.L.; Sorel, N.; Sloma, I.; Bennaceur-Griscelli, A.; Rea, D.; Legros, L.; Marfaing-Koka, A.; Bourhis, J.H.; Ame, S. Leukemic stem cell persistence in chronic myeloid leukemia patients in deep molecular response induced by tyrosine kinase inhibitors and the impact of therapy dis-continuation. Oncotarget 2016, 7, 35293–35301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahon, F.-X.; Réa, D.; Guilhot, J.; Guilhot, F.; Huguet, F.; Nicolini, F.; Legros, L.; Charbonnier, A.; Guerci, A.; Varet, B.; et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: The prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol. 2010, 11, 1029–1035. [Google Scholar] [CrossRef]
- Ross, D.M.; Branford, S.; Seymour, J.F.; Schwarer, A.P.; Arthur, C.; Yeung, D.T.; Dang, P.; Goyne, J.M.; Slader, C.; Filshie, R.J.; et al. Safety and efficacy of imatinib cessation for CML patients with stable undetectable minimal residual disease: Results from the TWISTER study. Blood 2013, 122, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, S.L.; Savona, M.; Mauro, M.J. Considerations for Successful Treatment-free Remission in Chronic Myeloid Leukemia. Clin. Lymphoma Myeloma Leuk. 2018, 18, 98–105. [Google Scholar] [CrossRef] [Green Version]
- Rousselot, P.; Charbonnier, A.; Cony-Makhoul, P.; Agape, P.; Nicolini, F.E.; Varet, B.; Gardembas, M.; Etienne, G.; Réa, D.; Roy, L.; et al. Loss of Major Molecular Response As a Trigger for Restarting Tyrosine Kinase Inhibitor Therapy in Patients With Chronic-Phase Chronic Myelogenous Leukemia Who Have Stopped Imatinib After Durable Undetectable Disease. J. Clin. Oncol. 2014, 32, 424–430. [Google Scholar] [CrossRef] [PubMed]
- D’Adda, M.; Farina, M.; Schieppati, F.; Borlenghi, E.; Bottelli, C.; Cerqui, E.; Ferrari, S.; Gramegna, D.; Pagani, C.; Passi, A.; et al. The e13a2 BCR-ABL transcript negatively affects sustained deep molecular response and the achievement of treatment-free remission in patients with chronic myeloid leukemia who receive tyrosine kinase inhibitors. Cancer 2019, 125, 1674–1682. [Google Scholar] [CrossRef]
- Burchert, A.; Saussele, S.; Eigendorff, E.; Müller, M.C.; Sohlbach, K.; Inselmann, S.; Schütz, C.; Metzelder, S.K.; Ziermann, J.; Kostrewa, P.; et al. Interferon alpha 2 maintenance therapy may enable high rates of treatment discontinuation in chronic myeloid leukemia. Leukemia 2015, 29, 1331–1335. [Google Scholar] [CrossRef]
- Ohyashiki, K.; Katagiri, S.; Tauchi, T.; Ohyashiki, J.H.; Maeda, Y.; Matsumura, I.; Kyo, T.-I. Increased natural killer cells and decreased CD3+CD8+CD62L+ T cells in CML patients who sustained complete molecular remission after discontinuation of imatinib. Br. J. Haematol. 2012, 157, 254–256. [Google Scholar] [CrossRef]
- Ilander, M.; Olsson-Strömberg, U.; Schlums, H.; Guilhot, J.; Brück, O.; Lähteenmäki, H.; Kasanen, T.; Koskenvesa, P.; Söderlund, S.; Höglund, M.; et al. Increased proportion of mature NK cells is associated with successful imatinib discontinuation in chronic myeloid leukemia. Leukemia 2017, 31, 1108–1116. [Google Scholar] [CrossRef]
- Schütz, C.; Inselmann, S.; Sausslele, S.; Dietz, C.T.; Müller, M.C.; Eigendorff, E.; Brendel, A.C.; Metzelder, S.K.; Brümmendorf, T.H.; Waller, C. Expression of the CTLA-4 ligand CD86 on plasmacytoid dendritic cells (pDC) predicts risk of disease recurrence after treatment discontinuation in CML. Leukemia 2017, 31, 829–836. [Google Scholar] [CrossRef]
- Kumagai, T.; Nakaseko, C.; Nishiwaki, K.; Yoshida, C.; Ohashi, K.; Takezako, N.; Takano, H.; Kouzai, Y.; Murase, T.; Matsue, K.; et al. Dasatinib cessation after deep molecular response exceeding 2 years and natural killer cell transition during dasatinib consolidation. Cancer Sci. 2017, 109, 182–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irani, Y.D.; Hughes, A.; Clarson, J.; Kok, C.H.; Shanmuganathan, N.; White, D.L.; Yeung, D.T.; Ross, D.M.; Hughes, T.P.; Yong, A.S. Successful treatment-free remission in chronic myeloid leukaemia and its association with reduced immune suppressors and increased natural killer cells. Br. J. Haematol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Nishiwaki, K.; Nakaseko, C.; Aotsuka, N.; Sano, K.; Ohwada, C.; Kuroki, J.; Kimura, H.; Tokuhira, M.; Mitani, K.; et al. Treatment-free remission after two-year consolidation therapy with nilotinib in patients with chronic myeloid leukemia: STAT2 trial in Japan. Haematologica 2018, 103, 1835–1842. [Google Scholar] [CrossRef] [PubMed]
- Hattori, A.; Tsunoda, M.; Konuma, T.; Kobayashi, M.; Nagy, T.; Glushka, J.; Tayyari, F.; McSkimming, D.; Kannan, N.; Tojo, A. Cancer progression by reprogrammed BCAA me-tabolism in myeloid leukaemia. Nature 2017, 545, 500–504. [Google Scholar] [CrossRef] [Green Version]
- Naka, K.; Jomen, Y.; Ishihara, K.; Kim, J.; Ishimoto, T.; Bae, E.-J.; Mohney, R.P.; Stirdivant, S.M.; Oshima, H.; Oshima, M.; et al. Dipeptide species regulate p38MAPK–Smad3 signalling to maintain chronic myelogenous leukaemia stem cells. Nat. Commun. 2015, 6, 8039. [Google Scholar] [CrossRef] [Green Version]
- Yuan, H.; Wang, Z.; Li, L.; Zhang, H.; Modi, H.; Horne, D.; Bhatia, R.; Chen, W. Activation of stress response gene SIRT1 by BCR-ABL promotes leukemogenesis. Blood 2012, 119, 1904–1914. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Wang, L.; Wang, Z.; Ho, Y.; McDonald, T.; Holyoake, T.L.; Chen, W.; Bhatia, R. Activation of p53 by SIRT1 Inhibition Enhances Elimination of CML Leukemia Stem Cells in Combination with Imatinib. Cancer Cell 2012, 21, 266–281. [Google Scholar] [CrossRef] [Green Version]
- Abraham, A.; Qiu, S.; Chacko, B.K.; Li, H.; Paterson, A.; He, J.; Agarwal, P.; Shah, M.; Welner, R.; Darley-Usmar, V.M.; et al. SIRT1 regulates metabolism and leukemogenic potential in CML stem cells. J. Clin. Investig. 2019, 129, 2685–2701. [Google Scholar] [CrossRef]
- Wang, Y.-H.; Israelsen, W.J.; Lee, D.; Yu, V.W.; Jeanson, N.T.; Clish, C.B.; Cantley, L.C.; Heiden, M.G.V.; Scadden, D.T. Cell-State-Specific Metabolic Dependency in Hematopoiesis and Leukemogenesis. Cell 2014, 158, 1309–1323. [Google Scholar] [CrossRef] [Green Version]
- Tong, L.; Xu, N.; Zhou, X.; Huang, J.; Wan-Er, W.; Chen, C.; Liang, L.; Liu, Q.; Xiaoli, L. PKM2 Mediates Chronic Myeloid Leukemia Imatinib Resistance By Regulating Glycolysis Energy Metabolism. Blood 2018, 132, 1724. [Google Scholar] [CrossRef]
- Kumar, R.; Godavarthy, P.S.; Krause, D.S. The bone marrow microenvironment in health and disease at a glance. J. Cell Sci. 2018, 131, jcs201707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, J.A.; Ferraro, F.; Roussakis, E.; Klein, A.; Wu, J.; Runnels, J.M.; Zaher, W.; Mortensen, L.J.; Alt, C.; Turcotte, R.; et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nat. Cell Biol. 2014, 508, 269–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Li, H.; Xi, H.S.; Li, S. HIF1α is required for survival maintenance of chronic myeloid leukemia stem cells. Blood 2012, 119, 2595–2607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatia, R.; Munthe, H.A.; Verfaillie, C.M. Role of abnormal integrin-cytoskeletal interactions in impaired beta1 integrin function in chronic myelogenous leukemia hematopoietic progenitors. Exp. Hematol. 1999, 27. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, R.; Wayner, E.A.; McGlave, P.B.; Verfaillie, C. Interferon-alpha restores normal adhesion of chronic myelogenous leukemia hematopoietic progenitors to bone marrow stroma by correcting impaired beta 1 integrin receptor function. J. Clin. Investig. 1994, 94, 384–391. [Google Scholar] [CrossRef] [Green Version]
- Hazlehurst, L.A.; Argilagos, R.F.; Dalton, W.S. Beta1 integrin mediated adhesion increases Bim protein degradation and con-tributes to drug resistance in leukaemia cells. Br. J. Haematol. 2007, 136, 269–275. [Google Scholar] [CrossRef]
- Kumar, R.; Pereira, R.S.; Zanetti, C.; Minciacchi, V.R.; Merten, M.; Meister, M.; Niemann, J.; Dietz, M.S.; Rüssel, N.; Schnütgen, F. Specific, targetable interactions with the mi-croenvironment influence imatinib-resistant chronic myeloid leukemia. Leukemia 2020, 34, 2087–2101. [Google Scholar] [CrossRef]
- Rothe, K.; Babaian, A.; Nakamichi, N.; Chen, M.; Chafe, S.C.; Watanabe, A.; Forrest, D.L.; Mager, D.L.; Eaves, C.J.; Dedhar, S.; et al. Integrin-Linked Kinase Mediates Therapeutic Resistance of Quiescent CML Stem Cells to Tyrosine Kinase Inhibitors. Cell Stem Cell 2020, 27, 110–124.e9. [Google Scholar] [CrossRef]
- Lundell, B.I.; McCarthy, J.B.; Kovach, N.L.; Verfaillie, C.M. Activation of beta1 integrins on CML progenitors reveals cooper-ation between beta1 integrins and CD44 in the regulation of adhesion and proliferation. Leukemia 1997, 11, 822–829. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.; Tabe, Y.; Konoplev, S.; Xu, Y.; Leysath, C.E.; Lu, H.; Kimura, S.; Ohsaka, A.; Rios, M.-B.; Calvert, L.; et al. CXCR4 up-regulation by imatinib induces chronic myelogenous leukemia (CML) cell migration to bone marrow stroma and promotes survival of quiescent CML cells. Mol. Cancer Ther. 2008, 7, 48–58. [Google Scholar] [CrossRef] [Green Version]
- Beider, K.; Darash-Yahana, M.; Blaier, O.; Koren-Michowitz, M.; Abraham, M.; Wald, H.; Wald, O.; Galun, E.; Eizenberg, O.; Peled, A.; et al. Combination of imatinib with CXCR4 antagonist BKT140 overcomes the protective effect of stroma and targets CML in vitro and in vivo. Mol. Cancer Ther. 2014, 13, 1155–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisberg, E.; Azab, A.K.; Manley, P.W.; Kung, A.L.; Christie, A.L.; Bronson, R.; Ghobrial, I.M.; Griffin, J.D. Inhibition of CXCR4 in CML cells disrupts their interaction with the bone marrow microenvironment and sensitizes them to nilotinib. Leukemia 2011, 26, 985–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, A.; Fleischman, A.G.; Petersen, C.L.; MacKenzie, R.; Luty, S.; Loriaux, M.; Druker, B.J.; Woltjer, R.L.; Deininger, M.W. Effects of plerixafor in combination with BCR-ABL kinase inhibition in a murine model of CML. Blood 2012, 120, 2658–2668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Ho, Y.W.; Huang, Q.; Maeda, T.; Lin, A.; Lee, S.-U.; Hair, A.; Holyoake, T.L.; Huettner, C.; Bhatia, R. Altered Microenvironmental Regulation of Leukemic and Normal Stem Cells in Chronic Myelogenous Leukemia. Cancer Cell 2012, 21, 577–592. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, P.; Isringhausen, S.; Li, H.; Paterson, A.J.; He, J.; Gomariz, A.; Nagasawa, T.; Nombela-Arriet, C.; Bhatia, R. Mesenchymal Niche-Specific Expression of Cxcl12 Controls Quiescence of Treatment-Resistant Leukemia Stem Cells. Cell Stem Cell. 2019, 24, 769–784.e6. [Google Scholar] [CrossRef]
- Reynaud, D.; Pietras, E.; Barry-Holson, K.; Mir, A.; Binnewies, M.; Jeanne, M.; Sala-Torra, O.; Radich, J.P.; Passegué, E. IL-6 Controls Leukemic Multipotent Progenitor Cell Fate and Contributes to Chronic Myelogenous Leukemia Development. Cancer Cell 2011, 20, 661–673. [Google Scholar] [CrossRef] [Green Version]
- Welner, R.S.; Amabile, G.; Bararia, D.; Czibere, A.; Yang, H.; Zhang, H.; Pontes, L.L.D.F.; Ye, M.; Levantini, E.; Di Ruscio, A.; et al. Treatment of Chronic Myelogenous Leukemia by Blocking Cytokine Alterations Found in Normal Stem and Progenitor Cells. Cancer Cell 2015, 27, 671–681. [Google Scholar] [CrossRef] [Green Version]
- Estrov, Z.; Kurzrock, R.; Wetzler, M.; Kantarjian, H.; Blake, M.; Harris, D.; Gutterman, J.U.; Talpaz, M. Suppression of chronic myelogenous leukemia colony growth by interleukin-1 (IL-1) receptor antagonist and soluble IL-1 receptors: A novel application for inhibitors of IL-1 activity. Blood 1991, 78, 1476–1484. [Google Scholar] [CrossRef] [Green Version]
- Estrov, Z.; Markowitz, A.B.; Kurzrock, R.; Wetzler, M.; Kantarjian, H.M.; Ferrajoli, A.; Gutterman, J.U.; Talpaz, M. Suppression of chronic myelogenous leukemia colony growth by interleukin-4. Leukemia 1993, 7, 214–220. [Google Scholar]
- Zhang, X.; Tu, H.; Yang, Y.; Jiang, X.; Hu, X.; Luo, Q.; Li, J. Bone marrow-derived mesenchymal stromal cells promote resistance to tyrosine kinase inhibitors in chronic myeloid leukemia via the IL-7/JAK1/STAT5 pathway. J. Biol. Chem. 2019, 294, 12167–12179. [Google Scholar] [CrossRef]
- Dorsey, J.F.; Cunnick, J.M.; Lanehart, R.; Huang, M.; Kraker, A.J.; Bhalla, K.N.; Jove, R.; Wu, J. Interleukin-3 protects Bcr-Abl-transformed hem-atopoietic progenitor cells from apoptosis induced by Bcr-Abl tyrosine kinase inhibitors. Leukemia 2002, 16, 1589–1595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naka, K.; Hoshii, T.; Muraguchi, T.; Tadokoro, Y.; Ooshio, T.; Kondo, Y.; Nakao, S.; Motoyama, N.; Hirao, A. TGF-beta-FOXO signalling maintains leukae-mia-initiating cells in chronic myeloid leukaemia. Nature 2010, 463, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Krause, D.S.; Fulzele, K.; Catic, A.; Sun, C.C.; Dombkowski, D.; Hurley, M.P.; Lezeau, S.; Attar, E.; Wu, J.Y.; Lin, H.Y.; et al. Differential regulation of myeloid leukemias by the bone marrow microenvironment. Nat. Med. 2013, 19, 1513–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, T.; Kharabi Masouleh, B.; Loges, S.; Cauwenberghs, S.; Fraisl, P.; Maes, C.; Jonckx, B.; De Keersmaecker, K.; Kleppe, M.; Tjwa, M.; et al. Loss or inhibition of stromal-derived PlGF prolongs survival of mice with imatinib-resistant Bcr-Abl1(+) leukemia. Cancer Cell. 2011, 19, 740–753. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R. The biology and function of exosomes in cancer. J. Clin. Investig. 2016, 126, 1208–1215. [Google Scholar] [CrossRef]
- Taverna, S.; Flugy, A.; Saieva, L.; Kohn, E.C.; Santoro, A.; Meraviglia, S.; De Leo, G.; Alessandro, R. Role of exosomes released by chronic myelogenous leukemia cells in angiogenesis. Int. J. Cancer 2011, 130, 2033–2043. [Google Scholar] [CrossRef] [Green Version]
- Corrado, C.; Raimondo, S.; Saieva, L.; Flugy, A.M.; De Leo, G.; Alessandro, R. Exosome-mediated crosstalk between chronic myelogenous leukemia cells and human bone marrow stromal cells triggers an Interleukin 8-dependent survival of leukemia cells. Cancer Lett. 2014, 348, 71–76. [Google Scholar] [CrossRef]
- Gao, X.; Wan, Z.; Wei, M.; Dong, Y.; Zhao, Y.; Chen, X.; Li, Z.; Qin, W.; Yang, G.; Liu, L. Chronic myelogenous leukemia cells remodel the bone marrow niche via exosome-mediated transfer of miR-320. Theranostics 2019, 9, 5642–5656. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Minciacchi, V.R.; Kumar, R.; Krause, D.S. Chronic Myeloid Leukemia: A Model Disease of the Past, Present and Future. Cells 2021, 10, 117. https://doi.org/10.3390/cells10010117
Minciacchi VR, Kumar R, Krause DS. Chronic Myeloid Leukemia: A Model Disease of the Past, Present and Future. Cells. 2021; 10(1):117. https://doi.org/10.3390/cells10010117
Chicago/Turabian StyleMinciacchi, Valentina R., Rahul Kumar, and Daniela S. Krause. 2021. "Chronic Myeloid Leukemia: A Model Disease of the Past, Present and Future" Cells 10, no. 1: 117. https://doi.org/10.3390/cells10010117
APA StyleMinciacchi, V. R., Kumar, R., & Krause, D. S. (2021). Chronic Myeloid Leukemia: A Model Disease of the Past, Present and Future. Cells, 10(1), 117. https://doi.org/10.3390/cells10010117