The Impact of HIV- and ART-Induced Mitochondrial Dysfunction in Cellular Senescence and Aging


Abstract
:1. Introduction
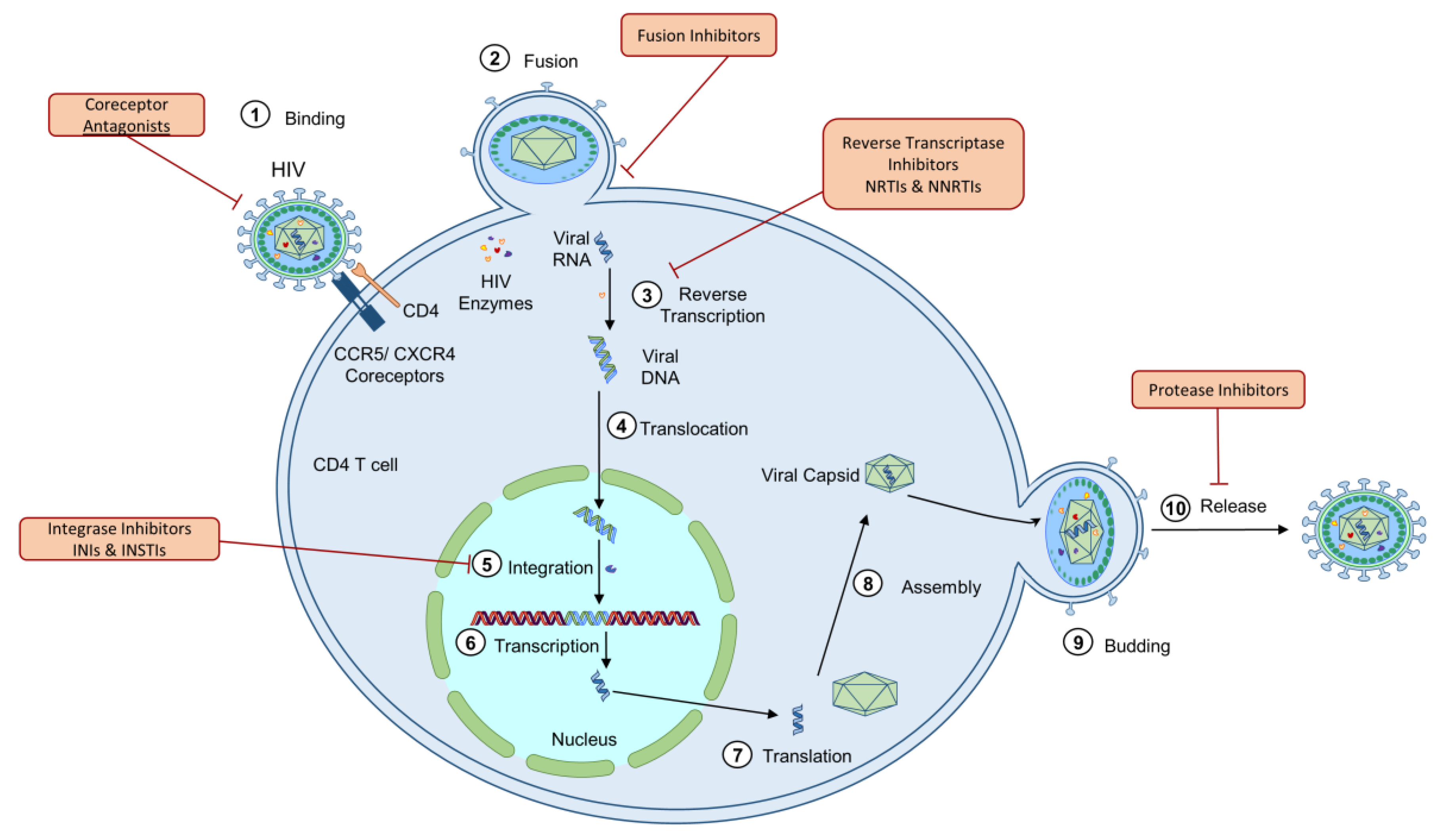
1.1. Human Immunodeficiency Virus (HIV)
1.2. Mitochondria Function in Cellular Energetics and Homeostasis
1.3. ART-Suppressed HIV and Mitochondrial Dysfunction in Cellular Senescence and Aging
2. Current Outlook
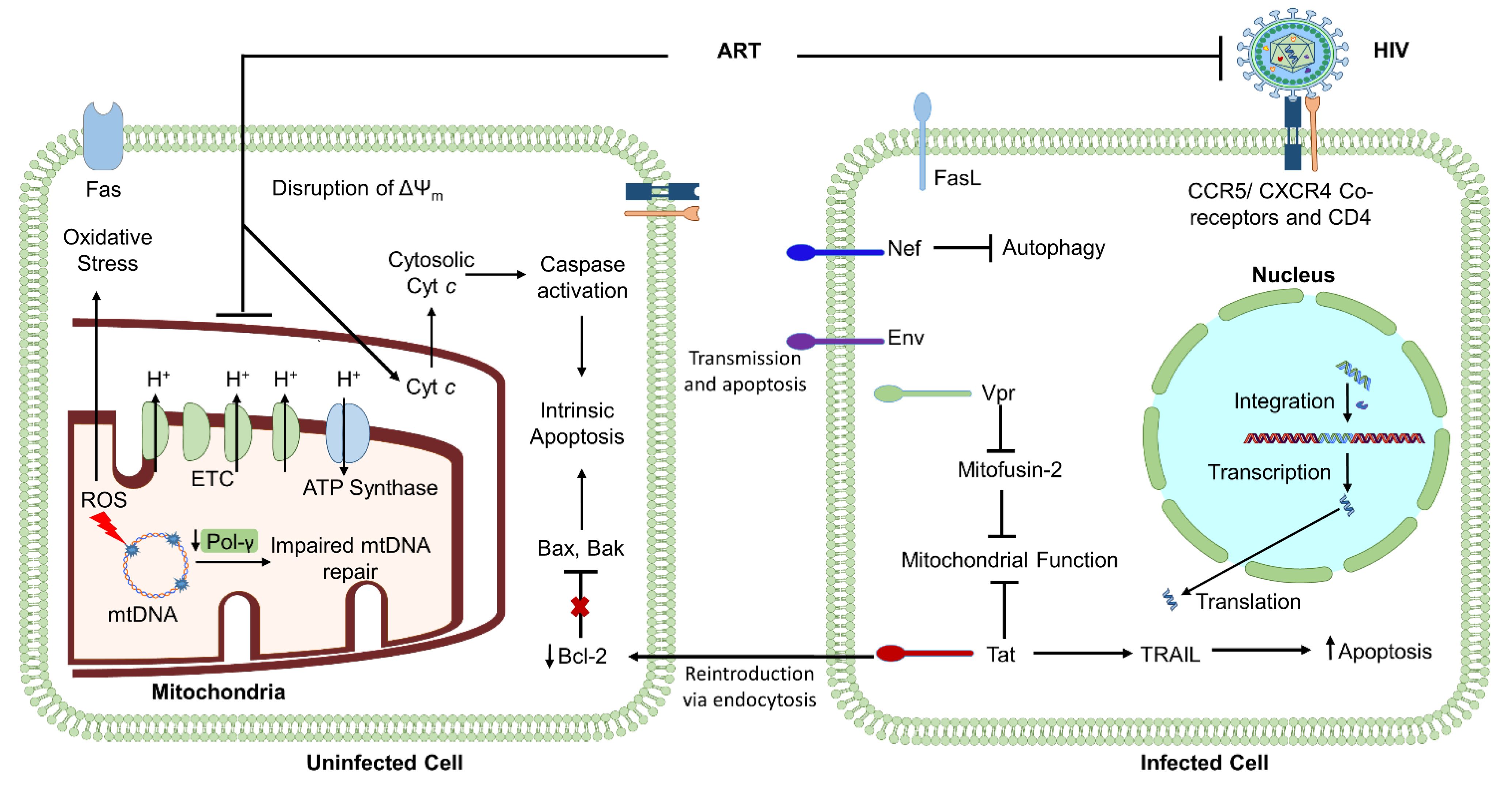
2.1. HIV-Induced Mitochondrial Dysfunction: The Influence of Virally Encoded Proteins
2.2. HIV-Encoded Env: A Regulator of Viral Infection, Apoptosis, and Mitochondria
2.3. HIV-Encoded Vpr: A Regulator of Apoptosis and Mitochondrial Function
2.4. HIV-Encoded Tat: A Regulator of Apoptosis and DNA Damage Repair
2.5. HIV-Encoded Nef: A Regulator of Apoptosis and Mitophagy
2.6. HIV-Mediated Mitochondrial Compromise
2.7. ART-Induced Mitochondrial Dysfunction
2.8. NRTIs
2.9. NNRTIs
2.10. PIs
2.11. INSTIs
3. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fanales-Belasio, E.; Raimondo, M.; Suligoi, B.; Buttò, S. HIV virology and pathogenetic mechanisms of infection: A brief overview. Ann. Ist. Super. Sanita 2010, 46, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Arhel, N. Revisiting HIV-1 uncoating. Retrovirology 2010, 7, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seitz, R. Human Immunodeficiency Virus (HIV). Transfus. Med. Hemotherapy 2016, 43, 203–222. [Google Scholar]
- Fuentes, G.M.; Fay, P.J.; Bambara, R.A. Relationship between plus strand DNA synthesis and removal of downstream segments of RNA by human immunodeficiency virus, murine leukemia virus and avian myeloblastoma virus reverse transcriptases. Nucleic Acids Res. 1996, 24, 1719–1726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craigie, R. HIV Integrase, a Brief Overview from Chemistry to Therapeutics. J. Biol. Chem. 2001, 276, 23213–23216. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.; Yu, F.; DU, L.-Y.; Xu, W.; Jiang, S.-B. Tactics used by HIV-1 to evade host innate, adaptive, and intrinsic immunities. Chin. Med. J. (Engl.) 2013, 126, 2374–2379. [Google Scholar]
- Yu, F.; Hao, Y.; Zhao, H.; Xiao, J.; Han, N.; Zhang, Y.; Dai, G.; Chong, X.; Zeng, H.; Zhang, F. Distinct mitochondrial disturbance in CD4+T and CD8+T cells from HIV-infected patients. J. Acquir. Immune Defic. Syndr. 2017, 74, 206–212. [Google Scholar] [CrossRef]
- Palmer, C.S.; Henstridge, D.C.; Yu, D.; Singh, A.; Balderson, B.; Duette, G.; Cherry, C.L.; Anzinger, J.J.; Ostrowski, M.; Crowe, S.M. Emerging Role and Characterization of Immunometabolism: Relevance to HIV Pathogenesis, Serious Non-AIDS Events, and a Cure. J. Immunol. 2016, 196, 4437–4444. [Google Scholar] [CrossRef] [Green Version]
- Virgin, H.W.; Wherry, E.J.; Ahmed, R. Redefining Chronic Viral Infection. Cell 2009, 138, 30–50. [Google Scholar] [CrossRef] [Green Version]
- Cossarizza, A.; Mussini, C.; Mongiardo, N.; Borghi, V.; Sabbatini, A.; De Rienzo, B.; Franceschi, C. Mitochondria alterations and dramatic tendency to undergo apoptosis in peripheral blood lymphocytes during acute HIV syndrome. AIDS 1997, 11, 19–26. [Google Scholar] [CrossRef]
- Vijayan, K.V.; Karthigeyan, K.P.; Tripathi, S.P.; Hanna, L.E. Pathophysiology of CD4+ T-Cell depletion in HIV-1 and HIV-2 infections. Front. Immunol. 2017, 8, 580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okoye, A.A.; Picker, L.J. CD4+ T cell depletion in HIV infection: Mechanisms of immunological failure. Immunol Rev 2013, 254, 54–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catalfamo, M.; Le Saout, C.; Lane, H.C. The role of cytokines in the pathogenesis and treatment of HIV infection. Cytokine Growth Factor Rev. 2012, 23, 207–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cummins, N.W.; Badley, A.D. Anti-apoptotic mechanisms of HIV: Lessons and novel approaches to curing HIV. Cell. Mol. Life Sci. 2013, 70, 3355–3363. [Google Scholar] [CrossRef] [Green Version]
- Eisele, E.; Siliciano, R.F. Redefining the Viral Reservoirs That Prevent HIV-1 Eradication. Immunity 2012, 37, 377–388. [Google Scholar] [CrossRef] [Green Version]
- Vanhamel, J.; Bruggemans, A.; Debyser, Z. Establishment of latent HIV-1 reservoirs: What do we really know? J. Virus Erad. 2019, 5, 3–9. [Google Scholar] [CrossRef]
- Castellano, P.; Prevedel, L.; Valdebenito, S.; Eugenin, E.A. HIV infection and latency induce a unique metabolic signature in human macrophages. Sci. Rep. 2019, 9, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Hiscott, J.; Kwon, H.; Génin, P. Hostile takeovers: Viral appropriation of the NF-κB pathway. J. Clin. Invest. 2001, 107, 143–151. [Google Scholar] [CrossRef]
- Murray, J.M.; Kelleher, A.D.; Cooper, D.A. Timing of the Components of the HIV Life Cycle in Productively Infected CD4+ T Cells in a Population of HIV-Infected Individuals. J. Virol. 2011, 85, 10798–10805. [Google Scholar] [CrossRef] [Green Version]
- Hima Bindu, A.; Naga Anusha, P. Adverse effects of highly Active Anti-Retroviral Therapy (HAART). J. Antivirals Antiretrovir. 2011, 3, 060–064. [Google Scholar]
- Arts, E.J.; Hazuda, D.J. HIV-1 antiretroviral drug therapy. Cold Spring Harb. Perspect. Med. 2012, 2, a007161. [Google Scholar] [CrossRef] [PubMed]
- Buzón, M.J.; Massanella, M.; Llibre, J.M.; Esteve, A.; Dahl, V.; Puertas, M.C.; Gatell, J.M.; Domingo, P.; Paredes, R.; Sharkey, M.; et al. HIV-1 replication and immune dynamics are affected by raltegravir intensification of HAART-suppressed subjects. Nat. Med. 2010, 16, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, M.; Bukrinsky, M. Human immunodeficiency virus (HIV) latency: The major hurdle in HIV eradication. Mol. Med. 2012, 18, 1096–1098. [Google Scholar] [CrossRef] [PubMed]
- Chomont, N.; El-far, M.; Ancuta, P.; Trautmann, L.; Procopio, F.A.; Yassine-diab, B.; Boucher, G.; Boulassel, R.; Ghattas, G.; Brenchley, J.M.; et al. HIV persistence is driven by homeostatic proliferation. Nat. Med. 2010, 15, 893–900. [Google Scholar] [CrossRef]
- Chavez, L.; Calvanese, V.; Verdin, E. HIV Latency Is Established Directly and Early in Both Resting and Activated Primary CD4 T Cells. PLoS Pathog. 2015, 11, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Moore RD, C.R. Moore RD, Chaisson RE. Natural history of HIV infection in the era of combination antiretroviral therapy. AIDS 1999, 13, 1933–1942. [Google Scholar] [CrossRef]
- Piconi, S.; Trabattoni, D.; Gori, A.; Parisotto, S.; Magni, C.; Meraviglia, P.; Bandera, A.; Capetti, A.; Rizzardini, G.; Clerici, M. Immune activation, apoptosis, and treg activity are associated with persistently reduced CD4+ T-cell counts during antiretroviral therapy. AIDS 2010, 24, 1991–2000. [Google Scholar] [CrossRef]
- Lederman, M.M.; Calabrese, L.; Funderburg, N.T.; Clagett, B.; Medvik, K.; Bonilla, H.; Gripshover, B.; Salata, R.A.; Taege, A.; Lisgaris, M.; et al. Immunologic failure despite suppressive antiretroviral therapy is related to activation and turnover of memory CD4 cells. J. Infect. Dis. 2011, 204, 1217–1226. [Google Scholar] [CrossRef] [Green Version]
- Chan, D.C. Mitochondria: Dynamic Organelles in Disease, Aging, and Development. Cell 2006, 125, 1241–1252. [Google Scholar] [CrossRef] [Green Version]
- Barile, M.; Valenti, D.; Hobbs, G.A.; Abruzzese, M.F.; Keilbaugh, S.A.; Passarella, S.; Quagliariello, E.; Simpson, M.V. Mechanisms of toxicity of 3′-azido-3′- deoxythymidine. Biochem. Pharmacol. 1994, 48, 1405–1412. [Google Scholar] [CrossRef]
- Meiliana, A.; Dewi, N.M.; Wijaya, A. Mitochondria in health and disease. Indones. Biomed. J. 2019, 11, 1–15. [Google Scholar] [CrossRef]
- de Souza Breda, C.N.; Davanzo, G.G.; Basso, P.J.; Saraiva Câmara, N.O.; Moraes-Vieira, P.M.M. Mitochondria as central hub of the immune system. Redox Biol. 2019, 26, 101255. [Google Scholar] [CrossRef] [PubMed]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal Biochem 2018, 552, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Youle, R.J. The role of mitochondria in apoptosis. BMB Rep. 2008, 41, 11–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.; Syed, G.H.; Kim, S.J.; Siddiqui, A. Mitochondrial dynamics and viral infections: A close nexus. Biochim. Biophys. Acta Mol. Cell Res. 2015, 1853, 2822–2833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Fang, P.; Mai, J.; Choi, E.T.; Wang, H.; Yang, X.F. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J. Hematol. Oncol. 2013, 6, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanza, I.R.; Nair, K.S. Mitochondrial metabolic function assessed in vivo and in vitro. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 511–517. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, S.; Guha, M.; Kashina, A.; Avadhani, N.G. Mitochondrial dysfunction and mitochondrial dynamics-The cancer connection. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 602–614. [Google Scholar] [CrossRef]
- Keogh, M.J.; Chinnery, P.F. Mitochondrial DNA mutations in neurodegeneration. Biochim. Biophys. Acta Bioenerg. 2015, 1847, 1401–1411. [Google Scholar] [CrossRef] [Green Version]
- Trewin, A.J.; Bahr, L.L.; Almast, A.; Berry, B.J.; Wei, A.Y.; Foster, T.H.; Wojtovich, A.P. Mitochondrial Reactive Oxygen Species Generated at the Complex-II Matrix or Intermembrane Space Microdomain Have Distinct Effects on Redox Signaling and Stress Sensitivity in Caenorhabditis elegans. Antioxid. Redox Signal. 2019, 31, 594–607. [Google Scholar] [CrossRef]
- Conley, K.E.; Marcinek, D.J.; Villarin, J. Mitochondrial dysfunction and age. Curr. Opin. Clin. Nutr. Metab. Care 2007, 10, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Guarente, L. Mitochondria-A Nexus for Aging, Calorie Restriction, and Sirtuins? Cell 2008, 132, 171–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Nguyen, L.N.T.; Nguyen, L.N.; Dang, X.; Cao, D.; Khanal, S.; Schank, M.; Thakuri, B.K.C.; Ogbu, S.C.; Morrison, Z.D.; et al. ATM Deficiency Accelerates DNA Damage, Telomere Erosion, and Premature T Cell Aging in HIV-Infected Individuals on Antiretroviral Therapy. Front. Immunol. 2019, 10, 2531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, J.A.; Daniel, R. Following the path of the virus: The exploitation of host DNA repair mechanisms by retroviruses. ACS Chem. Biol. 2006, 1, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Druzhyna, N.M.; Wilson, G.L.; LeDoux, S.P. Mitochondrial DNA repair in aging and disease. Mech Ageing Dev. 2008, 129, 383–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, Y.; Dang, X.; Nguyen, L.N.T.; Nguyen, L.N.; Zhao, J.; Cao, D.; Khanal, S.; Schank, M.; Wu, X.Y.; Morrison, Z.D.; et al. Topological DNA damage, telomere attrition and T cell senescence during chronic viral infections. Immun. Ageing 2019, 16, 12. [Google Scholar] [CrossRef] [PubMed]
- Dang, X.; Ogbu, S.C.; Zhao, J.; Nguyen, L.N.T.; Cao, D.; Nguyen, L.N.; Khanal, S.; Schank, M.; Thakuri, B.K.C.; Wu, X.Y.; et al. Inhibition of topoisomerase IIA (Top2α) induces telomeric DNA damage and T cell dysfunction during chronic viral infection. Cell Death Dis. 2020, 19, 1–2. [Google Scholar] [CrossRef] [Green Version]
- Piekna-Przybylska, D.; Sharma, G.; Maggirwar, S.B.; Bambara, R.A. Deficiency in DNA damage response, a new characteristic of cells infected with latent HIV-1. Cell Cycle 2017, 16, 968–978. [Google Scholar] [CrossRef] [Green Version]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Picard, M.; Wallace, D.C.; Burelle, Y. The rise of mitochondria in medicine. Mitochondrion 2016, 30, 105–116. [Google Scholar] [CrossRef]
- Van Epps, P.; Kalayjian, R.C. Human Immunodeficiency Virus and Aging in the Era of Effective Antiretroviral Therapy. Infect. Dis. Clin. North Am. 2017, 31, 791–810. [Google Scholar] [CrossRef] [PubMed]
- Sahin, E.; DePinho, R.A. Axis of ageing: Telomeres, p53 and mitochondria. Nat. Rev. Mol. Cell Biol. 2012, 13, 397–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco, J.R.; Jarrin, I.; Martinez, A.; Siles, E.; Larrayoz, I.M.; Canuelo, A.; Gutierrez, F.; Gonzalez-Garcia, J.; Vidal, F.; Moreno, S. Shorter telomere length predicts poorer immunological recovery in virologically suppressed hiv-1-infected patients treated with combined antiretroviral therapy. J. Acquir. Immune Defic. Syndr. 2015, 68, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Nelson, G.; Wordsworth, J.; Wang, C.; Jurk, D.; Lawless, C.; Martin-Ruiz, C.; von Zglinicki, T. A senescent cell bystander effect: Senescence-induced senescence. Aging Cell 2012, 11, 345–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Correia-Melo, C.; Marques, F.D.; Anderson, R.; Hewitt, G.; Hewitt, R.; Cole, J.; Carroll, B.M.; Miwa, S.; Birch, J.; Merz, A.; et al. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 2016, 35, 724–742. [Google Scholar] [CrossRef] [PubMed]
- Correia-Melo, C.; Passos, J.F. Mitochondria: Are they causal players in cellular senescence? Biochim. Biophys. Acta Bioenerg. 2015, 1847, 1373–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphreys, D.; Elghazaly, M.; Frisan, T. Senescence and Host–Pathogen Interactions. Cells 2020, 9, 1747. [Google Scholar] [CrossRef] [PubMed]
- Baz-Martínez, M.; Da Silva-Álvarez, S.; Rodríguez, E.; Guerra, J.; El Motiam, A.; Vidal, A.; Garciá-Caballero, T.; González-Barcia, M.; Sánchez, L.; Munõz-Fontela, C.; et al. Cell senescence is an antiviral defense mechanism. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef]
- Kaplan-Lewis, E.; Aberg, J.A.; Lee, M. Aging with HIV in the ART era. Semin. Diagn. Pathol. 2017, 34, 384–397. [Google Scholar] [CrossRef]
- Appay, V.; Sauce, D. Assessing immune aging in HIV-infected patients. Virulence 2017, 8, 529–538. [Google Scholar] [CrossRef]
- Jimnez, V.C.; Wit, F.W.N.M.; Joerink, M.; Maurer, I.; Harskamp, A.M.; Schouten, J.; Prins, M.; Van Leeuwen, E.M.M.; Booiman, T.; Deeks, S.G.; et al. T-Cell Activation Independently Associates with Immune Senescence in HIV-Infected Recipients of Long-term Antiretroviral Treatment. J. Infect. Dis. 2016, 214, 216–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caron, M.; Auclair, M.; Vissian, A.; Vigouroux, C.; Capeau, J. Contribution of mitochondrial dysfunction and oxidative stress to cellular premature senescence induced by antiretroviral thymidine analogues. Antivir. Ther. 2008, 13, 27–38. [Google Scholar] [PubMed]
- Sternfeld, T.; Tischleder, A.; Schuster, M.; Bogner, J.R. Mitochondrial membrane potential and apoptosis of blood mononuclear cells in untreated HIV-1 infected patients. HIV Med. 2009, 10, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Korencak, M.; Byrne, M.; Richter, E.; Schultz, B.T.; Juszczak, P.; Ake, J.A.; Ganesan, A.; Okulicz, J.F.; Robb, M.L.; de Los Reyes, B.; et al. Effect of HIV infection and antiretroviral therapy on immune cellular functions. JCI Insight 2019, 4, e126675. [Google Scholar] [CrossRef] [PubMed]
- Garg, H.; Mohl, J.; Joshi, A. HIV-1 induced bystander apoptosis. Viruses 2012, 4, 3020–3043. [Google Scholar] [CrossRef]
- Shedlock, D.J.; Hwang, D.; Choo, A.Y.; Chung, C.W.; Muthumani, K.; Weiner, D.B. HIV-1 viral genes and mitochondrial apoptosis. Apoptosis 2008, 13, 1088–1099. [Google Scholar] [CrossRef]
- Arnoult, D.; Petit, F.; Lelièvre, J.D.; Estaquier, J. Mitochondria in HIV-1-induced apoptosis. Biochem. Biophys. Res. Commun. 2003, 304, 561–574. [Google Scholar] [CrossRef]
- Roda, R.H.; Hoke, A. Mitochondrial dysfunction in HIV-induced peripheral neuropathy. In International Review of Neurobiology; Academic Press Inc.: Cambridge, MA, USA, 2019; Volume 145, pp. 67–82. ISBN 9780128172247. [Google Scholar]
- Arrildt, K.T.; Joseph, S.B.; Swanstrom, R. The HIV-1 Env protein: A coat of many colors. Curr. HIV/AIDS Rep. 2012, 9, 52–63. [Google Scholar] [CrossRef] [Green Version]
- Law, K.M.; Komarova, N.L.; Yewdall, A.W.; Lee, R.K.; Herrera, O.L.; Wodarz, D.; Chen, B.K. In Vivo HIV-1 Cell-to-Cell Transmission Promotes Multicopy Micro-compartmentalized Infection. Cell Rep. 2016, 15, 2771–2783. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Izadmehr, S.; Kamau, E.; Kong, X.P.; Chen, B.K. Sequential trafficking of Env and Gag to HIV-1 T cell virological synapses revealed by live imaging. Retrovirology 2019, 16, 1–16. [Google Scholar] [CrossRef]
- Heiskanen, K.M.; Bhat, M.B.; Wang, H.W.; Ma, J.; Nieminen, A.L. Mitochondrial depolarization accompanies cytochrome c release during apoptosis in PC6 cells. J. Biol. Chem. 1999, 274, 5654–5658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakai, K.; Dimas, J.; Lenardo, M.J. The Vif and Vpr accessory proteins independently cause HIV-1-induced T cell cytopathicity and cell cycle arrest. Proc. Natl. Acad. Sci. USA 2006, 103, 3369–3374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.Y.; Chiang, S.F.; Lin, T.Y.; Chiou, S.H.; Chow, K.C. HIV-1 Vpr triggers mitochondrial destruction by impairing Mfn2-mediated ER-mitochondria interaction. PLoS ONE 2012, 7, e33657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kogan, M.; Rappaport, J. HIV-1 Accessory Protein Vpr: Relevance in the pathogenesis of HIV and potential for therapeutic intervention. Retrovirology 2011, 8, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muthumani, K.; Choo, A.Y.; Hwang, D.S.; Chattergoon, M.A.; Dayes, N.N.; Zhang, D.; Lee, M.D.; Duvvuri, U.; Weiner, D.B. Mechanism of HIV-1 viral protein R-induced apoptosis. Biochem. Biophys. Res. Commun. 2003, 304, 583–592. [Google Scholar] [CrossRef]
- Filadi, R.; Pendin, D.; Pizzo, P. Mitofusin 2: From functions to disease. Cell Death Dis. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Giacca, M. HIV-1 Tat, apoptosis and the mitochondria: A tubulin link? Retrovirology 2005, 2, 4–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gougeon, M.L. To kill or be killed: How HIV exhausts the immune system. Cell Death Differ. 2005, 12, 845–854. [Google Scholar] [CrossRef]
- Comandini, A.; Naro, C.; Adamo, R.; Akbar, A.N.; Lanna, A.; Bonmassar, E.; Franzese, O. Molecular mechanisms involved in HIV-1-Tat mediated inhibition of telomerase activity in human CD4+ T lymphocytes. Mol. Immunol. 2013, 54, 181–192. [Google Scholar] [CrossRef]
- Ariumi, Y.; Kaida, A.; Hatanaka, M.; Shimotohno, K. Functional cross-talk of HIV-1 tat with p53 through its C-terminal domain. Biochem. Biophys. Res. Commun. 2001, 287, 556–561. [Google Scholar] [CrossRef]
- Li, C.J.; Wang, C.; Friedman, D.J.; Pardee, A.B. Reciprocal modulations between p53 and Tat of human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. USA 1995, 92, 5461–5464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miro, O.; Lopez, S.; Martinez, E.; Pedrol, E.; Milinkovic, A.; Deig, E.; Garrabou, G.; Casademont, J.; Gatell, J.M.; Cardellach, F. Mitochondrial Effects of HIV Infection on the Peripheral Blood Mononuclear Cells of HIV-Infected Patients Who Were Never Treated with Antiretrovirals. Clin. Infect. Dis. 2004, 39, 710–716. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Matute, P.; Pérez-Martínez, L.; Blanco, J.R.; Oteo, J.A. Role of mitochondria in HIV infection and associated metabolic disorders: Focus on nonalcoholic fatty liver disease and lipodystrophy syndrome. Oxid. Med. Cell. Longev. 2013. [Google Scholar] [CrossRef] [PubMed]
- Das, S.R.; Jameel, S. Biology of the HIV Nef protein. Indian J. Med. Res. 2005, 121, 315–332. [Google Scholar] [PubMed]
- Ding, W.X.; Yin, X.M. Mitophagy: Mechanisms, pathophysiological roles, and analysis. Biol. Chem. 2012, 393, 547–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apostolova, N.; Blas-Garcia, A.; V Esplugues, J. Mitochondria Sentencing About Cellular Life and Death: A Matter of Oxidative Stress. Curr. Pharm. Des. 2012, 17, 4047–4060. [Google Scholar] [CrossRef]
- Garrabou, G.; López, S.; Morén, C.; Martínez, E.; Fontdevila, J.; Cardellach, F.; Gatell, J.M.; Miró, Ò. Mitochondrial damage in adipose tissue of untreated HIV-infected patients. AIDS 2011, 25, 165–170. [Google Scholar] [CrossRef]
- Hulgan, T.; Gerschenson, M. HIV and mitochondria: More than just drug toxicity. J. Infect. Dis. 2012, 205, 1769–1771. [Google Scholar] [CrossRef] [Green Version]
- Sahin, E.; Colla, S.; Liesa, M.; Moslehi, J.; Müller, F.L.; Cooper, M.; Kotton, D.; Fabian, A.J.; Walkey, C.; Richard, S.; et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature 2011, 470, 359–365. [Google Scholar] [CrossRef] [Green Version]
- Tedone, E.; Huang, E.; O’Hara, R.; Batten, K.; Ludlow, A.T.; Lai, T.P.; Arosio, B.; Mari, D.; Wright, W.E.; Shay, J.W. Telomere length and telomerase activity in T cells are biomarkers of high-performing centenarians. Aging Cell 2019, 18, e12859. [Google Scholar] [CrossRef]
- Ron-Harel, N.; Sharpe, A.H.; Haigis, M.C. Mitochondrial metabolism in T cell activation and senescence: A mini-review. Gerontology 2015, 61, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Somasundaran, M.; Zapp, M.L.; Beattie, L.K.; Pang, L.; Byron, K.S.; Bassell, G.J.; Sullivan, J.L.; Singer, R.H. Localization of HIV RNA in mitochondria of infected cells: Potential role in cytopathogenicity. J. Cell Biol. 1994, 126, 1353–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee-Huang, S.; Lin Huang, P.; Lee Huang, P. Live-cell real-time imaging reveals role of mitochondria in cell-to-cell transmission of HIV-1. Biochem. Biophys. Res. Commun. 2011, 415, 384–389. [Google Scholar] [CrossRef]
- Peraire, J.; Miro, O.; Saumoy, M.; Domingo, P.; Pedrol, E.; Villarroya, F.; Martinez, E.; Lopez-Dupla, M.; Garrabou, G.; Sambeat, M.A.; et al. HIV-1-Infected Long-Term Non-Progressors have Milder Mitochondrial Impairment and Lower Mitochondrially-Driven Apoptosis in Peripheral Blood Mononuclear Cells than Typical Progressors. Curr. HIV Res. 2007, 5, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.L.; Kelly, B.; O’Neill, L.A.J. Mitochondria are the powerhouses of immunity. Nat. Immunol. 2017, 18, 488–498. [Google Scholar] [CrossRef]
- Brinkman, K.; Ter Hofstede, H.J.M.; Burger, D.M.; Smeitink, J.A.M.; Koopmans, P.P. Adverse effects of reverse transcriptase inhibitors: Mitochondrial toxicity as common pathway. AIDS 1998, 12, 1735–1744. [Google Scholar] [CrossRef]
- Apostolova, N.; Blas-Garcia, A.; V Esplugues, J. Mitochondrial Toxicity in HAART: An Overview of In Vitro Evidence. Curr. Pharm. Des. 2011, 17, 2130–2144. [Google Scholar] [CrossRef]
- Brinkman, K.; Kakuda, T.N. Mitochondrial toxicity of nucleoside analogue reverse transcriptase inhibitors: A looming obstacle for long-term antiretroviral therapy? Curr. Opin. Infect. Dis. 2000, 13, 5–11. [Google Scholar] [CrossRef]
- White, A.J. Mitochondrial toxicity and HIV therapy. Sex. Transm. Infect. 2001, 77, 158–173. [Google Scholar] [CrossRef] [Green Version]
- Pau, A.K.; George, J.M. Antiretroviral therapy: Current drugs. Infect. Dis. Clin. North Am. 2014, 28, 371–402. [Google Scholar] [CrossRef] [Green Version]
- Pinti, M.; Salomoni, P.; Cossarizza, A. Anti-HIV drugs and the mitochondria. Biochim. Biophys. Acta Bioenerg. 2006, 1757, 700–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrin, S.; Cremer, J.; Roll, P.; Faucher, O.; Ménard, A.; Reynes, J.; Dellamonica, P.; Naqvi, A.; Micallef, J.; Jouve, E.; et al. Hiv-1 infection and first line art induced differential responses in mitochondria from blood lymphocytes and monocytes: The anrs ep45 “aging” study. PLoS ONE 2012, 7, e41129. [Google Scholar] [CrossRef] [PubMed]
- Fiala, M.; Murphy, T.; MacDougall, J.; Yang, W.; Luque, A.; Iruela-Arispe, L.; Cashman, J.; Buga, G.; Byrns, R.E.; Barbaro, G.; et al. HAART drugs induce mitochondrial damage and intercellular gaps and gp120 causes apoptosis. Cardiovasc. Toxicol. 2004, 4, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Margolis, A.M.; Heverling, H.; Pham, P.A.; Stolbach, A. A Review of the Toxicity of HIV Medications. J. Med. Toxicol. 2014, 10, 26–39. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Foli, Y.; Liu, Z.; Wang, G.; Hu, Y.; Lu, Q.; Selvaraj, S.; Lam, W.; Paintsil, E. High frequency of mitochondrial DNA mutations in HIV-infected treatment-experienced individuals. HIV Med. 2017, 18, 45–55. [Google Scholar] [CrossRef] [Green Version]
- Barroso, S.; Morén, C.; González-Segura, À.; Riba, N.; Arnaiz, J.A.; Manriquez, M.; Santana, G.; Blanco, J.L.; Larousse, M.; Loncà, M.; et al. Metabolic, mitochondrial, renal and hepatic safety of enfuvirtide and raltegravir antiretroviral administration: Randomized crossover clinical trial in healthy volunteers. PLoS ONE 2019, 14, 1–14. [Google Scholar] [CrossRef]
- Blas-García, A.; Polo, M.; Alegre, F.; Funes, H.A.; Martínez, E.; Apostolova, N.; Esplugues, J.V. Lack of mitochondrial toxicity of darunavir, raltegravir and rilpivirine in neurons and hepatocytes: A comparison with efavirenz. J. Antimicrob. Chemother. 2014, 69, 2995–3000. [Google Scholar] [CrossRef] [Green Version]
- Apostolova, N.; Gomez-Sucerquia, L.J.; Moran, A.; Alvarez, A.; Blas-Garcia, A.; Esplugues, J.V. Enhanced oxidative stress and increased mitochondrial mass during Efavirenz-induced apoptosis in human hepatic cells. Br. J. Pharmacol. 2010, 160, 2069–2084. [Google Scholar] [CrossRef] [Green Version]
- Pilon, A.A.; Lum, J.J.; Sanchez-Dardon, J.; Phenix, B.N.; Douglas, R.; Badley, A.D. Induction of apoptosis by a nonnucleoside human immunodeficiency virus type 1 reverse transcriptase inhibitor. Antimicrob. Agents Chemother. 2002, 46, 2687–2691. [Google Scholar] [CrossRef] [Green Version]
- Graziewicz, M.A.; Day, B.J.; Copeland, W.C. The mitochondrial DNA polymerase as a target of oxidative damage. Nucleic Acids Res. 2002, 30, 2817–2824. [Google Scholar] [CrossRef] [Green Version]
- Apostolova, N.; Blas-García, A.; Esplugues, J.V. Mitochondrial interference by anti-HIV drugs: Mechanisms beyond Pol-γ inhibition. Trends Pharmacol. Sci. 2011, 32, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Setzer, B.; Schlesier, M.; Thomas, A.K.; Walker, U.A. Mitochondrial toxicity of nucleoside analogues in primary human lymphocytes. Antivir. Ther. 2005, 10, 327–334. [Google Scholar] [PubMed]
- Barile, M.; Valenti, D.; Passarella, S.; Quagliariello, E. 3’-Azido-3’-deoxythymidine uptake into isolated rat liver mitochondria and impairment of ADP/ATP translocator. Biochem. Pharmacol. 1997, 53, 913–920. [Google Scholar] [CrossRef]
- Karamchand, L.; Dawood, H.; Chuturgoon, A.A. Lymphocyte mitochondrial depolarization and apoptosis in HIV-1-infected HAART patients. J. Acquir. Immune Defic. Syndr. 2008, 48, 381–388. [Google Scholar] [CrossRef] [Green Version]
- Jamaluddina, M.S.; Lin, P.H.; Yao, Q.; Chen, C. Non-nucleoside reverse transcriptase inhibitor efavirenz increases monolayer permeability of human coronary artery endothelial cells. Atherosclerosis 2010, 208, 104–111. [Google Scholar] [CrossRef] [Green Version]
- Ganta, K.K.; Mandal, A.; Chaubey, B. Depolarization of mitochondrial membrane potential is the initial event in non-nucleoside reverse transcriptase inhibitor efavirenz induced cytotoxicity. Cell Biol. Toxicol. 2017, 33, 69–82. [Google Scholar] [CrossRef]
- Reyskens, K.M.S.E.; Essop, M.F. HIV protease inhibitors and onset of cardiovascular diseases: A central role for oxidative stress and dysregulation of the ubiquitin-proteasome system. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 256–268. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Mu, H.; Chai, H.; Liao, D.; Yao, Q.; Chen, C. Human immunodeficiency virus protease inhibitor ritonavir inhibits cholesterol efflux from human macrophage-derived foam cells. Am. J. Pathol. 2007, 171, 304–314. [Google Scholar] [CrossRef] [Green Version]
- Apostolova, N.; Gomez-Sucerquia, L.J.; Alegre, F.; Funes, H.A.; Victor, V.M.; Barrachina, M.D.; Blas-Garcia, A.; Esplugues, J.V. ER stress in human hepatic cells treated with Efavirenz: Mitochondria again. J. Hepatol. 2013, 59, 780–789. [Google Scholar] [CrossRef]
- Ganta, K.K.; Chaubey, B. Endoplasmic reticulum stress leads to mitochondria-mediated apoptosis in cells treated with anti-HIV protease inhibitor ritonavir. Cell Biol. Toxicol. 2019, 35, 189–204. [Google Scholar] [CrossRef]
- Blas-Garcia, A.; Apostolova, N.V.; Esplugues, J. Oxidative Stress and Mitochondrial Impairment After Treatment with Anti-HIV Drugs: Clinical Implications. Curr. Pharm. Des. 2012, 17, 4076–4086. [Google Scholar] [CrossRef] [PubMed]
- Day, B.J.; Lewis, W. Oxidative stress in NRTI-induced toxicity: Evidence from clinical experience and experiments in vitro and in vivo. Cardiovasc. Toxicol. 2004, 4, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Holeca, A.D.; Mandal, S.; Prathipati, P.K.; Destache, C.J. Nucleotide Reverse Transcriptase Inhibitors: A Thorough Review, Present Status and Future Perspective as HIV Therapeutics. Physiol. Behav. 2017, 15, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Lewis, W.; Copeland, W.C.; Day, B.J. Mitochondrial DNA depletion, oxidative stress, and mutation: Mechanisms of dysfunction from nucleoside reverse transcriptase inhibitors. Lab. Investig. 2001, 81, 777–790. [Google Scholar] [CrossRef] [Green Version]
- Lewis, W.; Dalakas, M.C. Mitochondrial toxicity of antiviral drugs. Nat. Med. 1995, 1, 417–422. [Google Scholar] [CrossRef]
- Lewis, W.; Day, B.J.; Copeland, W.C. Mitochondrial toxicity of NRTI antiviral drugs: An integrated cellular perspective. Nat. Rev. Drug Discov. 2003, 2, 812–822. [Google Scholar] [CrossRef]
- Petit, F.; Fromenty, B.; Owen, A.; Estaquier, J. Mitochondria are sensors for HIV drugs. Trends Pharmacol. Sci. 2005, 26, 258–264. [Google Scholar] [CrossRef]
- Côté, H.C.F.; Brumme, Z.L.; Craib, K.J.P.; Alexander, C.S.; Wynhoven, B.; Ting, L.; Wong, H.; Harris, M.; Harrigen, P.R.; O’Shaughnessy, M.V.; et al. Changes in Mitochondrial Dna As a Marker of Nucleoside Toxicity in Hiv-Infected Patients. N. Engl. J. Med. 2002, 346, 811–820. [Google Scholar] [CrossRef]
- Dagan, T.; Sable, C.; Bray, J.; Gerschenson, M. Mitochondrial dysfunction and antiretroviral nucleoside analog toxicities: What is the evidence? Mitochondrion 2002, 1, 397–412. [Google Scholar] [CrossRef]
- Hukezalie, K.R.; Thumati, N.R.; Côté, H.C.F.; Wong, J.M.Y. In Vitro and Ex Vivo Inhibition of Human Telomerase by Anti-HIV Nucleoside Reverse Transcriptase Inhibitors (NRTIs) but Not by Non-NRTIs. PLoS ONE 2012, 7, e47505. [Google Scholar] [CrossRef] [Green Version]
- Mandas, A.; Iorio, E.L.; Congiu, M.G.; Balestrieri, C.; Mereu, A.; Cau, D.; Dessì, S.; Curreli, N. Oxidative imbalance in HIV-1 infected patients treated with antiretroviral therapy. J. Biomed. Biotechnol. 2009, 2009, 749575. [Google Scholar] [CrossRef] [PubMed]
- Benito, J.M.; López, M.; Martín, J.C.; Lozano, S.; Martínez, P.; González-Lahoz, J.; Soriano, V. Differences in cellular activation and apoptosis in HIV-infected patients receiving protease inhibitors or nonnucleoside reverse transcriptase inhibitors. AIDS Res. Hum. Retrovir. 2002, 18, 1379–1388. [Google Scholar] [CrossRef] [PubMed]
- Deeks, S.G.; Lewin, S.R.; Havlir, D.V. The end of AIDS: HIV infection as a chronic disease. Lancet 2013, 382, 1525–1533. [Google Scholar] [CrossRef] [Green Version]
- Estrada, V.; De Villar, N.G.P.; de Villar, M.T.; Martínez Larrad, A.; González López, C.; Fernández, M.S.-R. Long-Term Metabolic Consequences of Switching from Protease Inhibitors to Efavirenz in Therapy for Human Immunodeficiency Virus-Infected Patients with Lipoatrophy. Infect. Dis. Clin. Pract. 2002, 11, 267. [Google Scholar] [CrossRef] [PubMed]
- Gibellini, L.; De Biasi, S.; Pinti, M.; Nasi, M.; Riccio, M.; Carnevale, G.; Cavallini, G.M.; Sala De Oyanguren, F.J.; O’Connor, J.E.; Mussini, C.; et al. The protease inhibitor atazanavir triggers autophagy and mitophagy in human preadipocytes. AIDS 2012, 26, 2017–2026. [Google Scholar] [CrossRef]
- Phenix, B.N.; Angel, J.B.; Mandy, F.; Kravcik, S.; Parato, K.; Chambers, K.A.; Gallicano, K.; Hawley-Foss, N.; Cassol, S.; William Cameron, D.; et al. Decreased HIV-associated T cell apoptosis by HIV protease inhibitors. AIDS Res. Hum. Retrovir. 2000, 16, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Sloand, E.M.; Kumar, P.N.; Kim, S.; Chaudhuri, A.; Weichold, F.F.; Young, N.S. Human immunodeficiency virus type 1 protease inhibitor modulates activation of peripheral blood CD4+ T cells and decreases their susceptibility to apoptosis in vitro and in vivo. Blood 1999, 94, 1021–1027. [Google Scholar] [CrossRef]
- Estaquier, J.; Lelièvre, J.; Petit, F.; Brunner, T.; Parseval, L.M.; Richman, D.D.; Ameisen, J.C.; Corbeil, J. Effects of Antiretroviral Drugs on Human Immunodeficiency Virus. Virology 2002, 76, 5966–5973. [Google Scholar] [CrossRef] [Green Version]
- Badley, A.D. In vitro and in vivo effects of HIV protease inhibitors on apoptosis. Cell Death Differ. 2005, 12, 924–931. [Google Scholar] [CrossRef] [Green Version]
- Atta, M.G.; De Seigneux, S.; Lucas, G.M. Clinical pharmacology in HIV therapy. Clin. J. Am. Soc. Nephrol. 2019, 14, 435–444. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Craigen, W.J.; Scaglia, F. Mitochondrial DNA maintenance defects. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1539–1555. [Google Scholar] [CrossRef] [PubMed]
- Shokolenko, I.N.; Wilson, G.L.; Alexeyev, M.F. Aging: A mitochondrial DNA perspective, critical analysis and an update. World J. Exp. Med. 2014, 4, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.; Moraes, C.T. Mechanisms Linking mtDNA Damage and Aging. Free Radic Biol Med 2015, 85, 250–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, J.B.; Chinnery, P.F. The dynamics of mitochondrial DNA heteroplasmy: Implications for human health and disease. Nat. Rev. Genet. 2015, 16, 530–542. [Google Scholar] [CrossRef] [PubMed]
- Hileman, C.O.; Funderburg, N.T. Inflammation, Immune Activation, and Antiretroviral Therapy in HIV. Curr. HIV/AIDS Rep. 2017, 14, 93–100. [Google Scholar] [CrossRef]
- Ahmed, D.; Roy, D.; Cassol, E. Examining relationships between metabolism and persistent inflammation in HIV patients on antiretroviral therapy. Mediat. Inflamm. 2018, 2018, 6238978. [Google Scholar] [CrossRef]
- Sena, L.A.; Li, S.; Jairaman, A.; Prakriya, M.; Ezponda, T.; David, A.; Wang, C.; Schumacker, P.T.; Licht, J.D.; Perlman, H.; et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 2013, 38, 225–236. [Google Scholar] [CrossRef] [Green Version]
- Bonomini, F.; Rodella, L.F.; Rezzani, R. Metabolic syndrome, aging and involvement of oxidative stress. Aging Dis. 2015, 6, 109–120. [Google Scholar] [CrossRef] [Green Version]
- Kirkwood, T.B.L. Understanding the odd science of aging. Cell 2005, 120, 137–147. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.C. A Mitochondrial Paradigm of Metabolic and Degenerative Diseases, Aging, and Cancer: A Dawn for Evolutionary Medicine. Annu. Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef] [Green Version]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.N.; Zhao, J.; Cao, D.; Dang, X.; Wang, L.; Lian, J.; Zhang, Y.; Jia, Z.; Wu, X.Y.; Morrison, Z.; et al. Inhibition of TRF2 accelerates telomere attrition and DNA damage in naïve CD4 T cells during HCV infection. Cell Death Dis. 2018, 9, 900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finley, L.W.S.; Haigis, M.C. The coordination of nuclear and mitochondrial communication during aging and calorie restriction. Ageing Res Rev. 2009, 8, 173–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derbré, F.; Gomez-Cabrera, M.C.; Nascimento, A.L.; Sanchis-Gomar, F.; Martinez-Bello, V.E.; Tresguerres, J.A.F.; Fuentes, T.; Gratas-Delamarche, A.; Monsalve, M.; Viña, J. Age associated low mitochondrial biogenesis may be explained by lack of response of PGC-1α to exercise training. Age (Omaha) 2012, 34, 669–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schank, M.; Zhao, J.; Wang, L.; Li, Z.; Cao, D.; Nguyen, L.N.; Dang, X.; Khanal, S.; Ngoc, L.; Nguyen, T.; et al. Telomeric injury by KML001 in human T cells induces mitochondrial dysfunction through the p53-PGC-1 α pathway. Cell Death Dis. 2020, 11. [Google Scholar] [CrossRef]
- Marchi, S.; Giorgi, C.; Suski, J.M.; Agnoletto, C.; Bononi, A.; Bonora, M.; De Marchi, E.; Missiroli, S.; Patergnani, S.; Poletti, F.; et al. Mitochondria-Ros Crosstalk in the Control of Cell Death and Aging. J. Signal Transduct. 2012, 2012, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Gindin, Y.; Gaggar, A.; Lok, A.S.; Janssen, H.L.A.; Ferreri, C.; Subramanian, G.M.; Jiang, Z.; Masur, H.; Emmanual, B.; Poonia, B.; et al. DNA Methylation and Immune Cell Markers Demonstrate Evidence of Accelerated Aging in Patients with Chronic HBV or HCV, with or without HIV Co-Infection. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Cao, D.; Zhao, J.; Nguyan, L.N.; Nguyen, L.N.T.; Khanal, S.; Dang, X.; Schank, M.; Chand Thakuri, B.K.; Wu, X.Y.; Morrison, Z.D.; et al. Disruption of telomere integrity and DNA repair machineries by KML001 induces T cell senescence, apoptosis, and cellular dysfunctions. Front. Immunol. 2019, 10, 1152. [Google Scholar] [CrossRef] [Green Version]
- Kelley, W.J.; Zemans, R.L.; Goldstein, D.R. Cellular Senescence: Friend or Foe to Respiratory Viral Infections? Eur. Respir. J. 2020, 56, 2002708. [Google Scholar] [CrossRef]
- Persson, B.D.; Jaffe, A.B.; Fearns, R.; Danahay, H. Respiratory syncytial virus can infect basal cells and alter human airway epithelial differentiation. PLoS ONE 2014, 9, 1–15. [Google Scholar] [CrossRef]
- Kim, J.A.; Seong, R.K.; Shin, O.S. Enhanced viral replication by cellular replicative senescence. Immune Netw. 2016, 16, 286–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shenoy, S. Coronavirus (Covid-19) sepsis: Revisiting mitochondrial dysfunction in pathogenesis, aging, inflammation, and mortality. Inflamm. Res. 2020, 69, 1077–1085. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.E.; Fazal, F.M.; Parker, K.R.; Zou, J.; Chang, H.Y. RNA-GPS Predicts SARS-CoV-2 RNA Residency to Host Mitochondria and Nucleolus. Cell Syst. 2020, 11, 102–108.e3. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Arkus, N. A mathematical model of cellular apoptosis and senescence through the dynamics of telomere loss. J. Theor. Biol. 2005, 235, 13–32. [Google Scholar] [CrossRef]
- Bowman, E.R.; Cameron, C.; Richardson, B.; Kulkarni, M.; Gabriel, J.; Kettelhut, A.; Hornsby, L.; Kwiek, J.J.; Turner, A.N.; Malvestutto, C.; et al. In vitro exposure of leukocytes to HIV pre-exposure prophylaxis (PrEP) decreases mitochondrial function and alters gene expression profiles. Antimicrob. Agents Chemother. 2020, 65, 1–13. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Drug Class | Mechanism of Action | Mitochondrial Dysfunction | Species and Cell Type Models |
|---|---|---|---|
| NRTIs (Abacavir, Tenofovir) | Prevents viral replication by inhibiting HIV reverse transcriptase | Inhibition of Pol-γ [111] | Human fibroblasts [111,112], PBMCs [106], CD4, and CD8 cells [113], and rat liver cells [30,114] |
| Reduction of mtDNA copy number/mitochondrial encoded proteins [106] | |||
| Reduced lymphocyte proliferation Respiratory chain deficiency Inhibition of ETC complexes [113] | |||
| ATP reduction [30,114] | |||
| Increased oxidative stress Decrease in Ψm [112] | |||
| NNRTIs (Rilpivirine, Efavirenz, Nevirapine) | Prevents viral replication by noncompetitively binding to HIV reverse transcriptase | Respiratory chain deficiency ATP reduction Increased oxidative stress [109] | Human hepatic cells [109], PBMCs [110,115], coronary artery endothelial cells [116], and hepatoma cells [117], and Jurkat T cell line [115] |
| Decrease in Ψm Apoptosis [110,115,116,117] | |||
| PIs (Ritonavir, Darunavir, Atazanavir, Indinavir, Saquinavir) | Prevents viral replication by inhibiting HIV protease | Increased oxidative stress [118] | Human CD4, CD8 [64], macrophage-derived foam cells [119], endothelial [118], hepatoma [117], and hepatic cells [120], and Huh-7.5, 293T, HeLa, and Hepa RG cell lines [121] |
| Reduced mtDNA copy number Respiratory chain deficiency Reduced ATP [64,119] | |||
| Apoptosis [119,122,123] | |||
| INIs (Raltegravir, Dolutegravir, Elvitegravir) | Prevents integration of viral DNA into the host genome by inhibiting HIV integrase enzyme | Respiratory chain deficiency Increased oxidative stress Increased cytoplasmic mtDNA copy number [64] | Human CD4 and CD8 [64] |
| Fusion Inhibitors (Leronlimab, Ibalizumab, Enfuvirtide) | Prevents viral fusion with target cell membrane by binding to the viral envelope protein gp41 | Not identified | N/A |
| Coreceptor Antagonists (Aplaviroc, Maraviroc, Vicriviroc) | Prevent viral infection by interfering with viral entrance into the cell by blocking the coreceptors, such as CCR5 or CXCR4, on the surface of target immune cells | Not identified | N/A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schank, M.; Zhao, J.; Moorman, J.P.; Yao, Z.Q. The Impact of HIV- and ART-Induced Mitochondrial Dysfunction in Cellular Senescence and Aging. Cells 2021, 10, 174. https://doi.org/10.3390/cells10010174
Schank M, Zhao J, Moorman JP, Yao ZQ. The Impact of HIV- and ART-Induced Mitochondrial Dysfunction in Cellular Senescence and Aging. Cells. 2021; 10(1):174. https://doi.org/10.3390/cells10010174
Chicago/Turabian StyleSchank, Madison, Juan Zhao, Jonathan P. Moorman, and Zhi Q. Yao. 2021. "The Impact of HIV- and ART-Induced Mitochondrial Dysfunction in Cellular Senescence and Aging" Cells 10, no. 1: 174. https://doi.org/10.3390/cells10010174
APA StyleSchank, M., Zhao, J., Moorman, J. P., & Yao, Z. Q. (2021). The Impact of HIV- and ART-Induced Mitochondrial Dysfunction in Cellular Senescence and Aging. Cells, 10(1), 174. https://doi.org/10.3390/cells10010174