Neuronal Metabolism and Neuroprotection: Neuroprotective Effect of Fingolimod on Menadione-Induced Mitochondrial Damage

 , , , and
, , , and 
Abstract
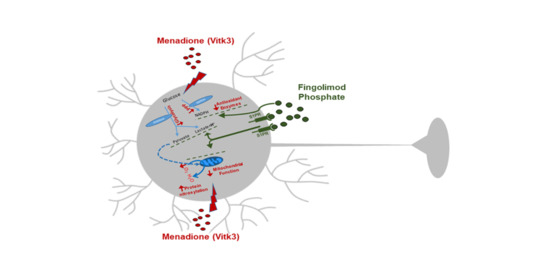
:
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatments
2.2. Antioxidant Enzyme Activity
2.3. Protein Nitrosylation
2.4. Determination of Aldolase
2.5. Glucose-6-Phosphate-Dehydrogenase Activity
2.6. Extracellular Acidification Rate (ECAR) and Mitochondrial Oxygen Consumption Rate (OCR)
2.7. Immunocytochemical Staining
2.8. Statistical Analysis
3. Results and Discussion
3.1. Mitochondrial Function Studies
3.2. Antioxidant Enzymes Studies
3.3. Glucose Metabolism Studies
3.4. Influence of S1P Receptors
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dienel, G.A. Brain glucose metabolism: Integration of energetics with function. Physiol. Rev. 2019, 99, 949–1045. [Google Scholar] [CrossRef] [PubMed]
- Yellen, G. Fueling thought: Management of glycolysis and oxidative phosphorylation in neuronal metabolism. J. Cell Biol. 2018, 217, 2235–2246. [Google Scholar] [CrossRef] [PubMed]
- Camandola, S.; Mattson, M.P. Brain metabolism in health, aging, and neurodegeneration. Embo J. 2017, 36, 1474–1492. [Google Scholar] [CrossRef]
- Błaszczyk, J.W. Energy metabolism decline in the aging brain; pathogenesis of neurodegenerative disorders. Preprints 2020, 2020090539. [Google Scholar] [CrossRef]
- Muddapu, V.R.; Dharshini, S.A.P.; Chakravarthy, V.S.; Gromiha, M.M. Neurodegenerative Diseases—Is Metabolic Deficiency the Root Cause? Front. Neurosci. 2020, 14, 213. [Google Scholar] [CrossRef] [PubMed]
- Barnham, K.J.; Masters, C.L.; Bush, A.I. Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 2004, 3, 205–214. [Google Scholar] [CrossRef]
- Elfawy, H.A.; Das, B. Crosstalk between mitochondrial dysfunction, oxidative stress, and age related neurodegenerative disease: Etiologies and therapeutic strategies. Life Sci. 2019, 218, 165–184. [Google Scholar] [CrossRef]
- Devine, M.J.; Kittler, J.T. Mitochondria at the neuronal presynapse in health and disease. Nat. Rev. Neurosci. 2018, 19, 63–80. [Google Scholar] [CrossRef]
- Rangaraju, V.; Calloway, N.; Ryan, T.A. Activity-driven local ATP synthesis is required for synaptic function. Cell 2014, 156, 825–835. [Google Scholar] [CrossRef] [Green Version]
- Uttara, B.; Singh, A.V.; Zamboni, P.; Mahajan, R.T. Oxidative stress and neurodegenerative diseases: A review of upstream and downstream antioxidant therapeutic options. Curr. Neuropharmacol. 2009, 7, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Zhang, N.; Ren, L.; Yan, Y.; Sun, N.; Li, Y.-J.; Han, W.; Xue, R.; Liu, Q.; Hao, J.; et al. Impact of an immune modulator fingolimod on acute ischemic stroke. Proc. Natl. Acad. Sci. USA 2014, 111, 18315–18320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miguez, A.; García-Díaz Barriga, G.; Brito, V.; Straccia, M.; Giralt, A.; Ginés, S.; Canals, J.M.; Alberch, J. Fingolimod (FTY720) enhances hippocampal synaptic plasticity and memory in Huntington’s disease by preventing p75 NTR up-regulation and astrocyte-mediated inflammation. Hum. Mol. Genet. 2015, 24, 4958–4970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asle-Rousta, M.; Kolahdooz, Z.; Dargahi, L.; Ahmadiani, A.; Nasoohi, S. Prominence of Central Sphingosine-1-Phosphate Receptor-1 in Attenuating Aβ-Induced Injury by Fingolimod. J. Mol. Neurosci. 2014, 54, 698–703. [Google Scholar] [CrossRef] [PubMed]
- White, E.J.; Clark, J.B. Menadione-treated synaptosomes as a model for post-ischaemic neuronal damage. Biochem. J. 1988, 253, 425–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, K.; Saito, H. Menadione toxicity in cultured rat cortical astrocytes. J. Pharm. Sci. 1996, 72, 299–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kesari, K.K.; Dhasmana, A.; Shandilya, S.; Prabhakar, N.; Shaukat, A.; Dou, J.; Rosenholm, J.M.; Vuorinen, T.; Ruokolainen, J. Plant-Derived Natural Biomolecule Picein Attenuates Menadione Induced Oxidative Stress on Neuroblastoma Cell Mitochondria. Antioxidants 2020, 9, 552. [Google Scholar] [CrossRef]
- Martín-Montañez, E.; Pavia, J.; Valverde, N.; Boraldi, F.; Lara, E.; Oliver, B.; Hurtado-Guerrero, I.; Fernandez, O.; Garcia-Fernandez, M. The S1P mimetic fingolimod phosphate regulates mitochondrial oxidative stress in neuronal cells. Free Radic. Biol. Med. 2019, 137, 116–130. [Google Scholar] [CrossRef] [Green Version]
- Milkovic, L.; Cipak Gasparovic, A.; Cindric, M.; Mouthuy, P.-A.; Zarkovic, N. Short Overview of ROS as Cell Function Regulators and Their Implications in Therapy Concepts. Cells 2019, 8, 793. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, J.A.; Cash, N.J.; Ouyang, Y.; Morton, J.C.; Chvanov, M.; Latawiec, D.; Awais, M.; Tepikin, A.V.; Sutton, R.; Criddle, D.N. Oxidative stress alters mitochondrial bioenergetics and modifies pancreatic cell death independently of cyclophilin D, resulting in an apoptosis-to-necrosis shift. J. Biol. Chem. 2018, 293, 8032–8047. [Google Scholar] [CrossRef] [Green Version]
- Son, J.H.; Chun, H.S.; Joh, T.H.; Cho, S.; Conti, B.; Lee, J.W. Neuroprotection and neuronal differentiation studies using substantia nigra dopaminergic cells derived from transgenic mouse embryos. J. Neurosci. 1999, 19, 10–20. [Google Scholar] [CrossRef] [Green Version]
- Pasquali-Ronchetti, I.; Garcia-Fernandez, M.I.; Boraldi, F.; Quaglino, D.; Gheduzzi, D.; De Vincenzi Paolinelli, C.; Tiozzo, R.; Bergamini, S.; Ceccarelli, D.; Muscatello, U. Oxidative stress in fibroblasts from patients with pseudoxanthoma elasticum: Possible role in the pathogenesis of clinical manifestations. J. Pathol. 2006, 208, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Jammer, A.; Gasperl, A.; Luschin-Ebengreuth, N.; Heyneke, E.; Chu, H.; Cantero-Navarro, E.; Großkinsky, D.K.; Albacete, A.A.; Stabentheiner, E.; Franzaring, J.; et al. Simple and robust determination of the activity signature of key carbohydrate metabolism enzymes for physiological phenotyping in model and crop plants. J. Exp. Bot. 2015, 66, 5531–5542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.; Neilson, A.; Swift, A.L.; Moran, R.; Tamagnine, J.; Parslow, D.; Armistead, S.; Lemire, K.; Orrell, J.; Teich, J.; et al. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am. J. Physiol. Cell Physiol. 2007, 292, 125–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, J.Y.; Park, J.H.; Kim, S.J.; Seo, K.S.; Han, J.S.; Lee, S.H.; Kim, J.M.; Park, J., II; Park, S.K.; Lim, K.; et al. DJ-1 Null Dopaminergic Neuronal Cells Exhibit Defects in Mitochondrial Function and Structure: Involvement of Mitochondrial Complex I Assembly. PLoS ONE 2012, 7, e32629. [Google Scholar] [CrossRef]
- Lofaro, F.D.; Boraldi, F.; Garcia-Fernandez, M.; Estrella, L.; Valdivielso, P.; Quaglino, D. Relationship between mitochondrial structure and bioenergetics in Pseudoxanthoma elasticum dermal fibroblasts. Front. Cell Dev. Biol. 2020, 8, 1532. [Google Scholar] [CrossRef]
- Tang, B.L. Glucose, glycolysis, and neurodegenerative diseases. J. Cell. Physiol. 2020, 235, 7653–7662. [Google Scholar] [CrossRef]
- Zilberter, Y.; Zilberter, M. The vicious circle of hypometabolism in neurodegenerative diseases: Ways and mechanisms of metabolic correction. J. Neurosci. Res. 2017, 95, 2217–2235. [Google Scholar] [CrossRef] [Green Version]
- Monteiro, J.P.; Martins, A.F.; Nunes, C.; Morais, C.M.; Lúcio, M.; Reis, S.; Pinheiro, T.J.T.; Geraldes, C.F.G.C.; Oliveira, P.J.; Jurado, A.S. A biophysical approach to menadione membrane interactions: Relevance for menadione-induced mitochondria dysfunction and related deleterious/therapeutic effects. Biochim. Biophys. Acta Biomembr. 2013, 1828, 1899–1908. [Google Scholar] [CrossRef] [Green Version]
- Lakhter, A.J.; Hamilton, J.; Dagher, P.C.; Mukkamala, S.; Hato, T.; Dong, X.C.; Mayo, L.D.; Harris, R.A.; Shekhar, A.; Ivan, M.; et al. Ferroxitosis: A cell death from modulation of oxidative phosphorylation and PKM2-dependent glycolysis in melanoma. Oncotarget 2014, 5, 12694–12703. [Google Scholar] [CrossRef] [Green Version]
- Bai, B.; Lunn, S.; Avila, R.; Wang, J.; Chmura, D.; Benson, E.; Kidd, G.; Medicetty, S.; Trapp, B. Fingolimod reduces axonal transection during demyelination (P1.153). Neurology 2015, 84. [Google Scholar]
- Brand, M.D.; Nicholls, D.G. Assessing mitochondrial dysfunction in cells. Biochem. J. 2011, 435, 297–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tavakol, S.; Hoveizi, E.; Tavakol, B.; Azedi, F.; Ebrahimi-Barough, S.; Keyhanvar, P.; Joghataei, M.T. Small molecule of sphingosine as a rescue of dopaminergic cells: A cell therapy approach in neurodegenerative diseases therapeutics. J. Cell. Physiol. 2019, 234, 11401–11410. [Google Scholar] [CrossRef] [PubMed]
- Desler, C.; Hansen, T.L.; Frederiksen, J.B.; Marcker, M.L.; Singh, K.K.; Juel Rasmussen, L. Is There a Link between Mitochondrial Reserve Respiratory Capacity and Aging? J. Aging Res. 2012, 2012, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancini, A.; Gaetani, L.; Gentili, L.; Di Filippo, M. Finding a way to preserve mitochondria: New pathogenic pathways in experimental multiple sclerosis. Neural Regen. Res. 2019, 14, 77–78. [Google Scholar] [CrossRef] [PubMed]
- Franco-Iborra, S.; Vila, M.; Perier, C. Mitochondrial Quality Control in Neurodegenerative Diseases: Focus on Parkinson’s Disease and Huntington’s Disease. Front. Neurosci. 2018, 12, 342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajwa, A.; Rosin, D.L.; Chroscicki, P.; Lee, S.; Dondeti, K.; Ye, H.; Kinsey, G.R.; Stevens, B.K.; Jobin, K.; Kenwood, B.M.; et al. Sphingosine 1-phosphate receptor-1 enhances mitochondrial function and reduces cisplatin-induced tubule injury. J. Am. Soc. Nephrol. 2015, 26, 908–925. [Google Scholar] [CrossRef]
- Varga, B.; Markó, K.; Hádinger, N.; Jelitai, M.; Demeter, K.; Tihanyi, K.; Vas, Á.; Madarász, E. Translocator protein (TSPO 18 kDa) is expressed by neural stem and neuronal precursor cells. Neurosci. Lett. 2009, 462, 257–262. [Google Scholar] [CrossRef]
- Weinert, M.; Cowley, S.A.; Alavian, K.N.; Matthews, P.M.; Owen, D.R. Exploring the mitochondrial TSPO protein as a possible immunometabolic modulatory target for treatment of multiple sclerosis. Mult. Scler. J. 2019, 25, 515. [Google Scholar]
- Strub, G.M.; Paillard, M.; Liang, J.; Gomez, L.; Allegood, J.C.; Hait, N.C.; Maceyka, M.; Price, M.M.; Chen, Q.; Simpson, D.C.; et al. Sphingosine-1-phosphate produced by sphingosine kinase 2 in mitochondria interacts with prohibitin 2 to regulate complex IV assembly and respiration. Faseb J. 2011, 25, 600–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, S.W.; Lee, J.; Kwon, H.; Park, S.E.; Rhee, E.J.; Park, C.Y.; Oh, K.W.; Park, S.W.; Lee, W.Y. Deficiency of Sphingosine-1-Phosphate Reduces the Expression of Prohibitin and Causes β-Cell Impairment via Mitochondrial Dysregulation. Endocrinol. Metab. 2018, 33, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Anwar, M.R.; Saldana-Caboverde, A.; Garcia, S.; Diaz, F. The organization of mitochondrial supercomplexes is modulated by oxidative stress in vivo in mouse models of mitochondrial encephalopathy. Int. J. Mol. Sci. 2018, 19, 1582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fugio, L.B.; Coeli-Lacchini, F.B.; Leopoldino, A.M. Sphingolipids and Mitochondrial Dynamic. Cells 2020, 9, 581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.M.; Shih, A.Y.; Murphy, T.H.; Johnson, J.A. NF-E2-related factor-2 mediates neuroprotection against mitochondrial complex I inhibitors and increased concentrations of intracellular calcium in primary cortical neurons. J. Biol. Chem. 2003, 278, 37948–37956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cadenas, E.; Davies, K.J.A. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Benhar, M. Roles of mammalian glutathione peroxidase and thioredoxin reductase enzymes in the cellular response to nitrosative stress. Free Radic. Biol. Med. 2018, 127, 160–164. [Google Scholar] [CrossRef]
- Wu, H.; Wang, X.; Gao, J.; Liang, S.; Hao, Y.; Sun, C.; Xia, W.; Cao, Y.; Wu, L. Fingolimod (FTY720) attenuates social deficits, learning and memory impairments, neuronal loss and neuroinflammation in the rat model of autism. Life Sci. 2017, 173, 43–54. [Google Scholar] [CrossRef]
- Bajpai, V.K.; Alam, M.B.; Quan, K.T.; Kwon, K.R.; Ju, M.K.; Choi, H.J.; Lee, J.S.; Yoon, J.I.; Majumder, R.; Rather, I.A.; et al. Antioxidant efficacy and the upregulation of Nrf2-mediated HO-1 expression by (+)-lariciresinol, a lignan isolated from Rubia philippinensis, through the activation of p38. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Beckman, J.S.; Koppenol, W.H. Nitric oxide, superoxide, and peroxynitrite: The good, the bad, and ugly. Am. J. Physiol. Physiol. 1996, 271, C1424–C1437. [Google Scholar] [CrossRef] [Green Version]
- Chang, A.H.K.; Sancheti, H.; Garcia, J.; Kaplowitz, N.; Cadenas, E.; Han, D. Respiratory substrates regulate S-nitrosylation of mitochondrial proteins through a thiol-dependent pathway. Chem. Res. Toxicol. 2014, 27, 794–804. [Google Scholar] [CrossRef]
- Moncada, S.; Bolanos, J.P. Nitric oxide, cell bioenergetics and neurodegeneration. J. Neurochem. 2006, 97, 1676–1689. [Google Scholar] [CrossRef]
- Efstathopoulos, P.; Kourgiantaki, A.; Karali, K.; Sidiropoulou, K.; Margioris, A.N.; Gravanis, A.; Charalampopoulos, I. Fingolimod induces neurogenesis in adult mouse hippocampus and improves contextual fear memory. Transl. Psychiatry 2015, 5, e685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardingham, N.; Dachtler, J.; Fox, K. The role of nitric oxide in pre-synaptic plasticity and homeostasis. Front. Cell. Neurosci. 2013, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Díaz-García, C.M.; Mongeon, R.; Lahmann, C.; Koveal, D.; Zucker, H.; Yellen, G. Neuronal Stimulation Triggers Neuronal Glycolysis and Not Lactate Uptake. Cell Metab. 2017, 26, 361–374.e4. [Google Scholar] [CrossRef]
- Lee, H.J.; Jung, Y.H.; Choi, G.E.; Kim, J.S.; Chae, C.W.; Lim, J.R.; Kim, S.Y.; Lee, J.E.; Park, M.C.; Yoon, J.H.; et al. O-cyclic phytosphingosine-1-phosphate stimulates HIF1α-dependent glycolytic reprogramming to enhance the therapeutic potential of mesenchymal stem cells. Cell Death Dis. 2019, 10, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, G.; Ryan, T.A. Glucose metabolism in nerve terminals. Curr. Opin. Neurobiol. 2017, 45, 156–161. [Google Scholar] [CrossRef]
- Hardas, S.S.; Sultana, R.; Clark, A.M.; Beckett, T.L.; Szweda, L.I.; Murphy, M.P.; Butterfield, D.A. Oxidative modification of lipoic acid by HNE in Alzheimer disease brain $. Redox Biol. 2013, 1, 80–85. [Google Scholar] [CrossRef] [Green Version]
- Trapp, B.D.; Stys, P.K. Virtual hypoxia and chronic necrosis of demyelinated axons in multiple sclerosis. Lancet Neurol. 2009, 8, 280–291. [Google Scholar] [CrossRef]
- Furst, A.J.; Rabinovici, G.D.; Rostomian, A.H.; Steed, T.; Alkalay, A.; Racine, C.; Miller, B.L.; Jagust, W.J.; Jagust, W. Cognition, glucose metabolism and amyloid burden in Alzheimer’s disease. Neurobiol Aging 2012, 33, 215–225. [Google Scholar] [CrossRef] [Green Version]
- Rone, M.B.; Cui, Q.-L.; Fang, J.; Wang, X.L.-C.; Zhang, J.; Khan, D.; Bedard, M.; Almazan, G.; Ludwin, S.K.; Jones, R.; et al. Neurobiology of Disease Oligodendrogliopathy in Multiple Sclerosis: Low Glycolytic Metabolic Rate Promotes Oligodendrocyte Survival. J. Neurosci. 2016, 36, 4698–4707. [Google Scholar] [CrossRef] [Green Version]
- Geffin, R.; Martinez, R.; de las Pozas, A.; Issac, B.; McCarthy, M. Fingolimod induces neuronal-specific gene expression with potential neuroprotective outcomes in maturing neuronal progenitor cells exposed to HIV. J. Neurovirol. 2017, 23, 808–824. [Google Scholar] [CrossRef] [Green Version]
- Evangelopoulos, M.E.; Miclea, A.; Schrewe, L.; Briner, M.; Salmen, A.; Engelhardt, B.; Huwiler, A.; Chan, A.; Hoepner, R. Frequency and clinical characteristics of Multiple Sclerosis rebounds after withdrawal of Fingolimod. Cns Neurosci. Ther. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, R.; Zhang, Y.; Simmering, J.E.; Schultz, J.L.; Li, Y.; Fernandez-Carasa, I.; Consiglio, A.; Raya, A.; Polgreen, P.M.; Narayanan, N.S.; et al. Enhancing glycolysis attenuates Parkinson’s disease progression in models and clinical databases. J. Clin. Investig. 2019, 129, 4539–4549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manzo, E.; Lorenzini, I.; Barrameda, D.; O’Conner, A.G.; Barrows, J.M.; Starr, A.; Kovalik, T.; Rabichow, B.E.; Lehmkuhl, E.M.; Shreiner, D.D.; et al. Glycolysis upregulation is neuroprotective as a compensatory mechanism in ALS. Elife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Herrero-Mendez, A.; Almeida, A.; Fernández, E.; Maestre, C.; Moncada, S.; Bolaños, J.P. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat. Cell Biol. 2009, 11, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Nóbrega-Pereira, S.; Fernandez-Marcos, P.J.; Brioche, T.; Gomez-Cabrera, M.C.; Salvador-Pascual, A.; Flores, J.M.; Viña, J.; Serrano, M. G6PD protects from oxidative damage and improves healthspan in mice. Nat. Commun. 2016, 7, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunn, L.; Allen, G.F.G.; Mamais, A.; Ling, H.; Li, A.; Duberley, K.E.; Hargreaves, I.P.; Pope, S.; Holton, J.L.; Lees, A.; et al. Dysregulation of glucose metabolism is an early event in sporadic Parkinson’s disease. Neurobiol. Aging 2014, 35, 1111–1115. [Google Scholar] [CrossRef] [Green Version]
- Heiss, E.H.; Schachner, D.; Zimmermann, K.; Dirsch, V.M. Glucose availability is a decisive factor for Nrf2-mediated gene expression. Redox Biol. 2013, 1, 359–365. [Google Scholar] [CrossRef] [Green Version]
- Hayes, J.D.; Ashford, M.L.J. Nrf2 Orchestrates Fuel Partitioning for Cell Proliferation. Cell Metab. 2012, 16, 139–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, O.J.; Kovarik, J.M.; Schmouder, R.L. Clinical pharmacokinetics of fingolimod. Clin. Pharm. 2012, 51, 15–28. [Google Scholar] [CrossRef]
- Brait, V.H.; Tarrasón, G.; Gavaldà, A.; Godessart, N.; Planas, A.M. Selective Sphingosine 1-Phosphate Receptor 1 Agonist Is Protective Against Ischemia/Reperfusion in Mice. Stroke 2016, 47, 3053–3056. [Google Scholar] [CrossRef] [Green Version]
- Kodydková, J.; Vávrová, L.; Kocík, M.; Žák, A.; Kodydková, J. Human Catalase, Its Polymorphisms, Regulation and Changes of Its Activity in Different Diseases. Folia Biol. 2014, 60, 153–167. [Google Scholar]
- Cantalupo, A.; Gargiulo, A.; Dautaj, E.; Liu, C.; Zhang, Y.; Hla, T.; Di Lorenzo, A. S1PR1 (Sphingosine-1-Phosphate Receptor 1) Signaling Regulates Blood Flow and Pressure. Hypertension 2017, 70, 426–434. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SOD2 U/106 cells | GPX U/106 cells | CAT KU/106 cells | Nit-Prot pmol/106 cells | SOD2/GPX | SOD2/CAT | |
|---|---|---|---|---|---|---|
| CO | 43.1 ± 3.0 | 21.9 ± 4.6 | 27.9 ± 3.1 | 0.76 ± 0.17 | 2 | 0.15 |
| VitK3 | 76.4 ± 4.0 (a) | 4.5 ± 0.8 (a) | 27.1 ± 6.3 | 4.5 ± 0.35 (a) | 17 (a) | 0.28 |
| VitK3+FP | 40.3 ± 3.0 (b) | 10.6 ± 0.9 (a)(b) | 27.8 ± 4.2 | 0.45 ± 0.15 (b) | 5 (a)(b) | 0.14 |
| VitK3+FP+W | 79.9 ± 6.0 (a) | 27.6 ± 4.8 (b)(c) | 14.4 ± 2.5 (a)(b)(c) | 3.75 ± 0.20 (a)(c) | 3 (b)(c) | 0.55 (a)(b)(c) |
| FP | 50.1 ± 3.0 (b)(c)(d) | 23.9 ± 4.0 (b)(c) | 44.1 ± 8.5 (a)(b)(c)(d) | 1.87 ± 0.5 (a)(b)(c)(d) | 2 (b)(c) | 0.11 (b)(d) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gil, A.; Martín-Montañez, E.; Valverde, N.; Lara, E.; Boraldi, F.; Claros, S.; Romero-Zerbo, S.-Y.; Fernández, O.; Pavia, J.; Garcia-Fernandez, M. Neuronal Metabolism and Neuroprotection: Neuroprotective Effect of Fingolimod on Menadione-Induced Mitochondrial Damage. Cells 2021, 10, 34. https://doi.org/10.3390/cells10010034
Gil A, Martín-Montañez E, Valverde N, Lara E, Boraldi F, Claros S, Romero-Zerbo S-Y, Fernández O, Pavia J, Garcia-Fernandez M. Neuronal Metabolism and Neuroprotection: Neuroprotective Effect of Fingolimod on Menadione-Induced Mitochondrial Damage. Cells. 2021; 10(1):34. https://doi.org/10.3390/cells10010034
Chicago/Turabian StyleGil, Antonio, Elisa Martín-Montañez, Nadia Valverde, Estrella Lara, Federica Boraldi, Silvia Claros, Silvana-Yanina Romero-Zerbo, Oscar Fernández, Jose Pavia, and Maria Garcia-Fernandez. 2021. "Neuronal Metabolism and Neuroprotection: Neuroprotective Effect of Fingolimod on Menadione-Induced Mitochondrial Damage" Cells 10, no. 1: 34. https://doi.org/10.3390/cells10010034
APA StyleGil, A., Martín-Montañez, E., Valverde, N., Lara, E., Boraldi, F., Claros, S., Romero-Zerbo, S. -Y., Fernández, O., Pavia, J., & Garcia-Fernandez, M. (2021). Neuronal Metabolism and Neuroprotection: Neuroprotective Effect of Fingolimod on Menadione-Induced Mitochondrial Damage. Cells, 10(1), 34. https://doi.org/10.3390/cells10010034