NMDA and AMPA Receptor Autoantibodies in Brain Disorders: From Molecular Mechanisms to Clinical Features

 ,
,  , , and
, , and
Abstract
:1. Introduction
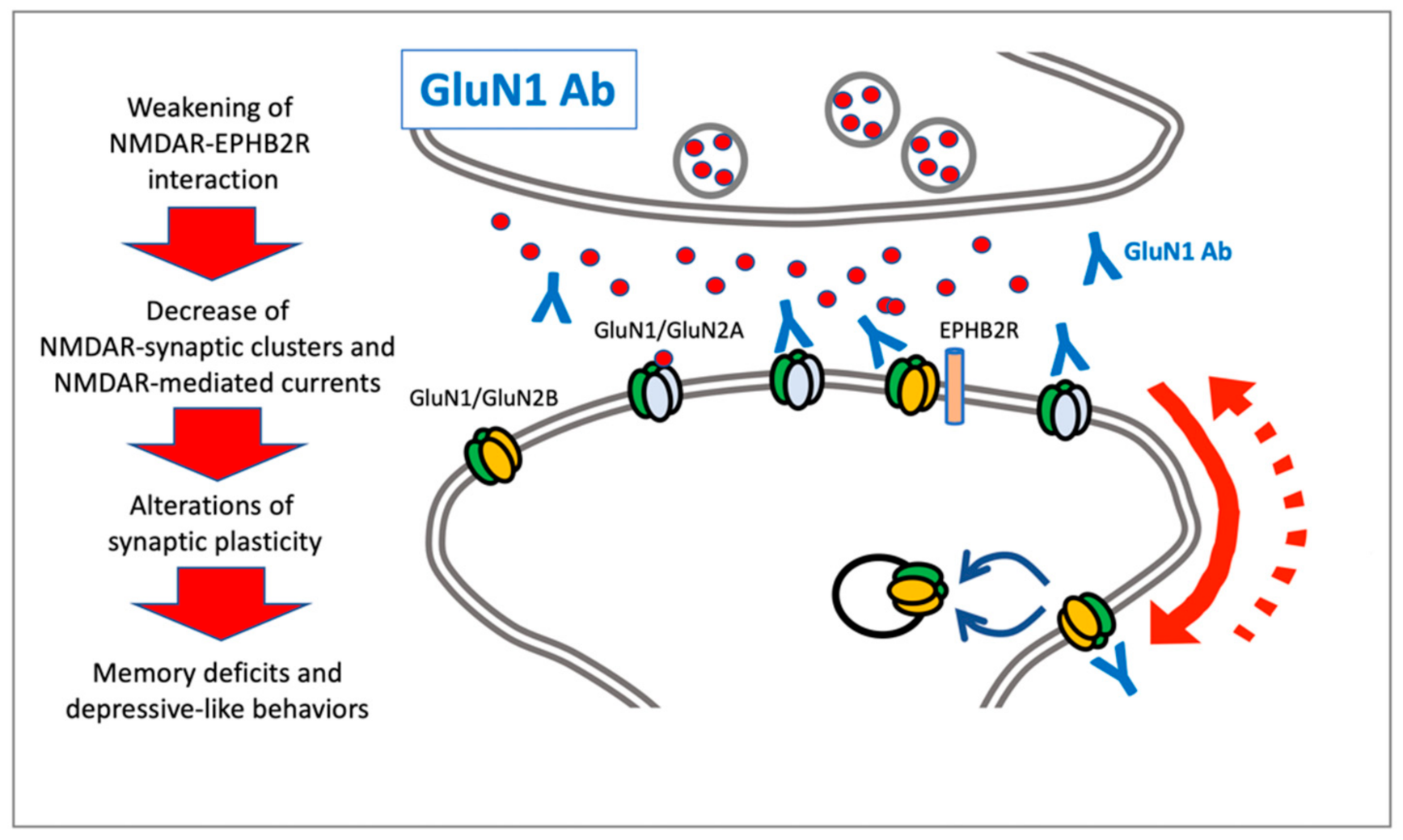
2. NMDAR Autoantibodies: Anti-GluN1
3. NMDAR Autoantibodies: Anti-GluN2B
4. AMPAR Autoantibodies: Anti-GluA1 and Anti-GluA2
5. AMPAR Autoantibodies: Anti-GluA3
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Palese, F.; Bonomi, E.; Nuzzo, T.; Benussi, A.; Mellone, M.; Zianni, E.; Cisani, F.; Casamassa, A.; Alberici, A.; Scheggia, D.; et al. Anti-GluA3 antibodies in frontotemporal dementia: Effects on glutamatergic neurotransmission and synaptic failure. Neurobiol. Aging 2020, 86, 143–155. [Google Scholar]
- Panzer, J.A.; Gleichman, A.J.; Lynch, D.R. Glutamatergic autoencephalitides: An emerging field. J. Neural Transm. 2014, 121, 957–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kayser, M.S.; Dalmau, J. Anti-NMDA receptor encephalitis, autoimmunity, and psychosis. Schizophr. Res. 2016, 176, 36–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levite, M. Glutamate receptor antibodies in neurological diseases: Anti-AMPA-GluR3 antibodies, anti-NMDA-NR1 anti-bodies, anti-NMDA-NR2A/B antibodies, anti-mGluR1 antibodies or anti-mGluR5 antibodies are present in subpopulations of patients with either: Epilepsy, encephalitis, cerebellar ataxia, systemic lupus erythematosus (SLE) and neuropsychiatric SLE, Sjogren’s syndrome, schizophrenia, mania or stroke. These autoimmune anti-glutamate receptor antibodies can bind neurons in few brain regions, activate glutamate receptors, decrease glutamate receptor’s expression, impair gluta-mate-induced signaling and function, activate blood brain barrier endothelial cells, kill neurons, damage the brain, induce behavioral/psychiatric/cognitive abnormalities and ataxia in animal models, and can be removed or silenced in some pa-tients by immunotherapy. J. Neural Transm. 2014, 121, 1029–1075. [Google Scholar] [PubMed]
- Hunter, D.; Jamet, Z.; Groc, L. Autoimmunity and NMDA receptor in brain disorders: Where do we stand? Neurobiol. Dis. 2021, 147, 105161. [Google Scholar] [CrossRef]
- Graus, F.; Titulaer, M.J.; Balu, R.; Benseler, S.; Bien, C.G.; Cellucci, T.; Cortese, I.; Dale, R.C.; Gelfand, J.M.; Geschwind, M.; et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol. 2016, 15, 391–404. [Google Scholar] [CrossRef] [Green Version]
- Franchini, L.; Carrano, N.; Di Luca, M.; Gardoni, F. Synaptic GluN2A-Containing NMDA Receptors: From Physiology to Pathological Synaptic Plasticity. Int. J. Mol. Sci. 2020, 21, 1538. [Google Scholar] [CrossRef] [Green Version]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef]
- Gleichman, A.J.; Spruce, L.A.; Dalmau, J.; Seeholzer, S.H.; Lynch, D.R. Anti-NMDA Receptor Encephalitis Antibody Binding Is Dependent on Amino Acid Identity of a Small Region within the GluN1 Amino Terminal Domain. J. Neurosci. 2012, 32, 11082–11094. [Google Scholar] [CrossRef] [Green Version]
- Dalmau, J.; Armangué, T.; Planagumà, J.; Radosevic, M.; Mannara, F.; Leypoldt, F.; Geis, C.; Lancaster, E.; Titulaer, M.J.; Rosenfeld, M.R.; et al. An update on anti-NMDA receptor encephalitis for neurologists and psychiatrists: Mechanisms and models. Lancet Neurol. 2019, 18, 1045–1057. [Google Scholar]
- Muñoz-Lopetegi, A.; Graus, F.; Dalmau, J.; Santamaria, J. Sleep disorders in autoimmune encephalitis. Lancet Neurol. 2020, 19, 1010–1022. [Google Scholar] [PubMed]
- Granerod, J.; Ambrose, H.E.; Davies, N.W.; Clewley, J.P.; Walsh, A.L.; Morgan, D.; Cunningham, R.; Zuckerman, M.; Mutton, K.J.; Solomon, T.; et al. Causes of encephalitis and differences in their clinical presentations in England: A multi-centre, population-based prospective study. Lancet Infect Dis. 2010, 10, 835–844. [Google Scholar] [PubMed] [Green Version]
- Armangue, T.; Spatola, M.; Vlagea, A.; Mattozzi, S.; Cárceles-Cordon, M.; Martinez-Heras, E.; Llufriu, S.; Muchart, J.; Erro, M.E.; Abraira, L.; et al. Frequency, symptoms, risk factors, and outcomes of autoimmune encephalitis after herpes simplex encephalitis: A prospective observational study and retrospective analysis. Lancet Neurol. 2018, 17, 760–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joubert, B.; Dalmau, J. The role of infections in autoimmune encephalitides. Rev. Neurol. 2019, 175, 420–426. [Google Scholar] [CrossRef]
- Monti, G.; Giovannini, G.; Marudi, A.; Bedin, R.; Melegari, A.; Simone, A.M.; Santangelo, M.; Pignatti, A.; Bertellini, E.; Trenti, T.; et al. Anti-NMDA receptor encephalitis presenting as new onset refractory status epilepticus in COVID-19. Seizure 2020, 81, 18–20. [Google Scholar] [CrossRef]
- Panariello, A.; Bassetti, R.; Radice, A.; Rossotti, R.; Puoti, M.; Corradin, M.; Moreno, M.; Percudani, M. Anti-NMDA receptor encephalitis in a psychiatric Covid-19 patient: A case report. Brain Behav. Immun. 2020, 87, 179–181. [Google Scholar] [CrossRef]
- Wang, H. Anti-NMDA Receptor Encephalitis, Vaccination and Virus. Curr. Pharm. Des. 2020, 25, 4579–4588. [Google Scholar] [CrossRef]
- Dalmau, J.; Tüzün, E.; Wu, H.Y.; Masjuan, J.; Rossi, J.E.; Voloschin, A.; Baehring, J.M.; Shimazaki, H.; Koide, R.; King, D.; et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann. Neurol. 2007, 61, 25–36. [Google Scholar]
- Busse, S.; Busse, M.; Brix, B.; Probst, C.; Genz, A.; Bogerts, B.; Stoecker, W.; Steiner, J. Seroprevalence of n-methyl-d-aspartate glutamate receptor (NMDA-R) autoantibodies in aging subjects without neuropsychiatric disorders and in dementia patients. Eur. Arch. Psychiatry Clin. Neurosci. 2014, 264, 545–550. [Google Scholar] [CrossRef]
- Doss, S.; Wandinger, K.-P.; Hyman, B.T.; Panzer, J.A.; Synofzik, M.; Dickerson, B.; Mollenhauer, B.; Scherzer, C.R.; Ivinson, A.J.; Finke, C.; et al. High prevalence of NMDA receptor IgA/IgM antibodies in different dementia types. Ann. Clin. Transl. Neurol. 2014, 1, 822–832. [Google Scholar] [CrossRef]
- Zerche, M.; Weissenborn, K.; Ott, C.; Dere, E.; Asif, A.R.; Worthmann, H.; Hassouna, I.; Rentzsch, K.; Tryc, A.B.; Dahm, L.; et al. Preexisting Serum Autoantibodies Against the NMDAR Subunit NR1 Modulate Evolution of Lesion Size in Acute Ischemic Stroke. Stroke 2015, 46, 1180–1186. [Google Scholar] [CrossRef] [PubMed]
- Hara, M.; Martinez-Hernandez, E.; Ariño, H.; Armangué, T.; Spatola, M.; Petit-Pedrol, M.; Saiz, A.; Rosenfeld, M.R.; Graus, F.; Dalmau, J. Clinical and pathogenic significance of IgG, IgA, and IgM antibodies against the NMDA receptor. Neurology 2018, 90, e1386–e1394. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Hernandez, E.; Guasp, M.; García-Serra, A.; Maudes, E.; Ariño, H.; Sepulveda, M.; Armangué, T.; Ramos, A.P.; Ben-Hur, T.; Iizuka, T.; et al. Clinical significance of anti-NMDAR concurrent with glial or neuronal surface antibodies. Neurology 2020, 94, e2302–e2310. [Google Scholar] [CrossRef] [PubMed]
- Titulaer, M.J.; Höftberger, R.; Iizuka, T.; Leypoldt, F.; McCracken, L.; Cellucci, T.; Benson, L.A.; Shu, H.; Irioka, T.; Hirano, M.; et al. Overlapping demyelinating syndromes and anti-N-methyl-D-aspartate receptor encephalitis. Ann. Neurol. 2014, 75, 411–428. [Google Scholar] [CrossRef]
- Dalmau, J.; Gleichman, A.J.; Hughes, E.G.; Rossi, E.J.; Peng, X.; Lai, M.; Dessain, S.K.; Rosenfeld, M.R.; Balice-Gordon, R.; Lynch, D.R. Anti-NMDA-receptor encephalitis: Case series and analysis of the effects of antibodies. Lancet Neurol. 2008, 7, 1091–1098. [Google Scholar] [CrossRef] [Green Version]
- Hughes, E.G.; Peng, X.; Gleichman, A.J.; Lai, M.; Zhou, L.; Tsou, R.; Parsons, T.D.; Lynch, D.R.; Dalmau, J.; Balice-Gordon, R.J. Cellular and Synaptic Mechanisms of Anti-NMDA Receptor Encephalitis. J. Neurosci. 2010, 30, 5866–5875. [Google Scholar] [CrossRef] [Green Version]
- Mikasova, L.; De Rossi, P.; Bouchet, D.; Georges, F.; Rogemond, V.; Didelot, A.; Meissirel, C.; Honnorat, J.; Groc, L. Dis-rupted surface cross-talk between NMDA and Ephrin-B2 receptors in anti-NMDA encephalitis. Brain 2012, 135, 1606–1621. [Google Scholar] [CrossRef] [Green Version]
- Moscato, E.H.; Peng, X.; Jain, A.; Parsons, T.D.; Dalmau, J.; Balice-Gordon, R.J. Acute mechanisms underlying antibody effects in anti-N-methyl-D-aspartate receptor encephalitis. Ann. Neurol. 2014, 76, 108–119. [Google Scholar] [CrossRef] [Green Version]
- Planagumà, J.; Leypoldt, F.; Mannara, F.; Gutiérrez-Cuesta, J.; Martín-García, E.; Aguilar, E.; Titulaer, M.J.; Petit-Pedrol, M.; Jain, A.; Balice-Gordon, R.; et al. Human N-methyl D-aspartate receptor antibodies alter memory and behaviour in mice. Brain 2015, 138, 94–109. [Google Scholar] [CrossRef]
- Planagumà, J.; Haselmann, H.; Mannara, F.; Petit-Pedrol, M.; Grünewald, B.; Aguilar, E.; Röpke, L.; Martín-García, E.; Titulaer, M.J.; Jercog, P.; et al. Ephrin-B2 prevents N-methyl-D-aspartate receptor antibody effects on memory and neu-roplasticity. Ann. Neurol. 2016, 80, 388–400. [Google Scholar] [CrossRef]
- Jézéquel, J.; Johansson, E.M.; Dupuis, J.P.; Rogemond, V.; Gréa, H.; Kellermayer, B.; Hamdani, N.; Le Guen, E.; Rabu, C.; Lepleux, M.; et al. Dynamic disorganization of synaptic NMDA receptors triggered by autoantibodies from psychotic pa-tients. Nat. Commun. 2017, 8, 1791. [Google Scholar] [CrossRef] [PubMed]
- Matute, C.; Palma, A.; Serrano-Regal, M.P.; Maudes, E.; Barman, S.; Sánchez-Gómez, M.V.; Domercq, M.; Goebels, N.; Dalmau, J. N-Methyl-D-Aspartate Receptor Antibodies in Autoimmune Encephalopathy Alter Oligodendrocyte Function. Ann. Neurol. 2020, 87, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Phillips, O.; Joshi, S.H.; Narr, K.L.; Shattuck, D.W.; Singh, M.; Di Paola, M.; Ploner, C.J.; Prüss, H.; Paul, F.; Finke, C. Superficial white matter damage in anti-NMDA receptor encephalitis. J. Neurol. Neurosurg. Psychiatry 2017, 89, 518–525. [Google Scholar] [CrossRef]
- Martinez-Hernandez, E.; Horvath, J.; Shiloh-Malawsky, Y.; Sangha, N.; Martinez-Lage, M.; Dalmau, J. Analysis of com-plement and plasma cells in the brain of patients with anti-NMDAR encephalitis. Neurology 2011, 77, 589–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagnon, I.; Hélie, P.; Bardou, I.; Regnauld, C.; Lesec, L.; Leprince, J.; Naveau, M.; Delaunay, B.; Toutirais, O.; Lemauff, B.; et al. Autoimmune encephalitis mediated by B-cell response against N-methyl-d-aspartate receptor. Brain 2020, 143, 2957–2972. [Google Scholar] [CrossRef]
- Takahashi, Y.; Mori, H.; Mishina, M.; Watanabè, M.; Fujiwara, T.; Shimomura, J.; Aiba, H.; Miyajima, T.; Saito, Y.; Nezu, A.; et al. Autoantibodies to NMDA receptor in patients with chronic forms of epilepsia partialis continua. Neurology 2003, 61, 891–896. [Google Scholar] [CrossRef]
- Fukuyama, T.; Takahashi, Y.; Kubota, Y.; Mogami, Y.; Imai, K.; Kondo, Y.; Sakuma, H.; Tominaga, K.; Oguni, H.; Nishimura, S. Semi-quantitative analyses of antibodies to N-methyl-d-aspartate type glutamate receptor subunits (GluN2B & GluN1) in the clinical course of Rasmussen syndrome. Epilepsy Res. 2015, 113, 34–43. [Google Scholar] [CrossRef]
- Mori, T.; Takahashi, Y.; Araya, N.; Oboshi, T.; Watanabe, H.; Tsukamoto, K.; Yamaguchi, T.; Yoshitomi, S.; Nasu, H.; Ikeda, H.; et al. Antibodies against peptides of NMDA-type GluR in cerebrospinal fluid of patients with epileptic spasms. Eur. J. Paediatr. Neurol. 2016, 20, 865–873. [Google Scholar] [CrossRef]
- Park, D.-J.; Takahashi, Y.; Kang, J.-H.; Yim, Y.-R.; Kim, J.-E.; Lee, J.-W.; Lee, K.-E.; Lee, J.-K.; Lee, S.-S. Anti-N-methyl-D-aspartate receptor antibodies are associated with fibromyalgia in patients with systemic lupus erythematosus: A case-control study. Clin. Exp. Rheumatol. 2017, 35, 54–60. [Google Scholar]
- Gon, J.; Takehisa, Y.; Yada, Y.; Kishi, Y.; Oshima, E.; Takahashi, Y.; Takaki, M. Encephalitis With Antibodies to GluN2B During Administration of Clozapine. Clin. Neuropharmacol. 2016, 39, 320–321. [Google Scholar] [CrossRef]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate Receptor Ion Channels: Structure, Regulation, and Function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gleichman, A.J.; Panzer, J.A.; Baumann, B.H.; Dalmau, J.; Lynch, D.R. Antigenic and mechanistic characterization of anti-AMPA receptor encephalitis. Ann. Clin. Transl. Neurol. 2014, 1, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.; Hughes, E.G.; Peng, X.; Zhou, L.; Gleichman, A.J.; Shu, H.; Matà, S.; Kremens, D.; Vitaliani, R.; Geschwind, M.D.; et al. AMPA receptor Human Autoantibodies in limbic encephalitis alter synaptic receptor location. Ann. Neurol. 2009, 65, 424–434. [Google Scholar] [CrossRef] [PubMed]
- Laurido-Soto, O.; Brier, M.R.; Simon, L.E.; McCullough, A.; Bucelli, R.C.; Day, G.S. Patient characteristics and outcome associations in AMPA receptor encephalitis. J. Neurol. 2019, 266, 450–460. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Hughes, E.G.; Moscato, E.H.; Parsons, T.D.; Dalmau, J.; Balice-Gordon, R.J. Cellular plasticity induced by an-ti-alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor encephalitis antibodies. Ann Neurol. 2015, 77, 381–398. [Google Scholar] [CrossRef] [Green Version]
- Höftberger, R.; Van Sonderen, A.; Leypoldt, F.; Houghton, D.; Geschwind, M.; Gelfand, J.; Paredes, M.; Sabater, L.; Saiz, A.; Albert, A.; et al. Encephalitis and AMPA receptor antibodies: Novel findings in a case series of 22 patients. Neurology 2015, 84, 2403–2412. [Google Scholar] [CrossRef] [Green Version]
- Haselmann, H.; Mannara, F.; Werner, C.; Planagumà, J.; Miguez-Cabello, F.; Schmidl, L.; Grünewald, B.; Petit-Pedrol, M.; Kirmse, K.; Classen, J.; et al. Human Autoantibodies against the AMPA Receptor Subunit GluA2 Induce Receptor Reor-ganization and Memory Dysfunction. Neuron 2018, 100, 91–105.e9. [Google Scholar] [CrossRef] [Green Version]
- Ganor, Y.; Goldberg-Stern, H.; Lerman-Sagie, T.; Teichberg, V.I.; Levite, M. Autoimmune epilepsy: Distinct subpopulations of epilepsy patients harbor serum autoantibodies to either glutamate/AMPA receptor GluR3, glutamate/NMDA receptor subunit NR2A or double-stranded DNA. Epilepsy Res. 2005, 65, 11–22. [Google Scholar] [CrossRef]
- Ganor, Y.; Goldberg-Stern, H.; Blank, M.; Shoenfeld, Y.; Dobrynina, L.A.; Kalashnikova, L.; Levite, M. Antibodies to glu-tamate receptor subtype 3 (GluR3) are found in some patients suffering from epilepsy as the main disease, but not in patients whose epilepsy accompanies antiphospholipid syndrome or Sneddon’s syndrome. Autoimmunity 2005, 38, 417–424. [Google Scholar] [CrossRef]
- Goldberg-Stern, H.; Ganor, Y.; Cohen, R.; Pollak, L.; Teichberg, V.; Levite, M. Glutamate receptor antibodies directed against AMPA receptors subunit 3 peptide B (GluR3B) associate with some cognitive/psychiatric/behavioral abnormalities in epilepsy patients. Psychoneuroendocrinology 2014, 40, 221–231. [Google Scholar] [CrossRef]
- Rogers, S.W.; Andrews, P.I.; Gahring, L.C.; Whisenand, T.; Cauley, K.; Crain, B.; Hughes, T.E.; Heinemann, S.F.; McNamara, J.O. Autoantibodies to glutamate receptor GluR3 in Rasmussen’s encephalitis. Science 1994, 265, 648–651. [Google Scholar] [CrossRef] [PubMed]
- Malina, K.C.-K.; Ganor, Y.; Levite, M.; Teichberg, V.I. Autoantibodies Against an Extracellular Peptide of the GluR3 Subtype of AMPA Receptors Activate Both Homomeric and Heteromeric AMPA Receptor Channels. Neurochem. Res. 2006, 31, 1181–1190. [Google Scholar] [CrossRef] [PubMed]
- Borroni, B.; Stanic, J.; Verpelli, C.; Mellone, M.; Bonomi, E.; Alberici, A.; Bernasconi, P.; Culotta, L.; Zianni, E.; Archetti, S.; et al. Anti-AMPA GluA3 antibodies in Frontotemporal dementia: A new molecular target. Sci. Rep. 2017, 7, 6723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberici, A.; Cristillo, V.; Gazzina, S.; Benussi, A.; Padovani, A.; Borroni, B. Autoimmunity and Frontotemporal Dementia. Curr. Alzheimer Res. 2018, 15, 602–609. [Google Scholar] [CrossRef]
- Benussi, A.; Alberici, A.; Buratti, E.; Ghidoni, R.; Gardoni, F.; Di Luca, M.; Padovani, A.; Borroni, B. Toward a Glutamate Hypothesis of Frontotemporal Dementia. Front. Neurosci. 2019, 13, 304. [Google Scholar] [CrossRef]
- Hodges, J.R.; Piguet, O. Progress and Challenges in Frontotemporal Dementia Research: A 20-Year Review. J. Alzheimer’s Dis. 2018, 62, 1467–1480. [Google Scholar] [CrossRef] [Green Version]
- Seelaar, H.; Rohrer, J.D.; Pijnenburg, Y.A.; Fox, N.C.; Van Swieten, J.C. Clinical, genetic and pathological heterogeneity of frontotemporal dementia: A review. J. Neurol. Neurosurg. Psychiatry 2011, 82, 476–486. [Google Scholar] [CrossRef] [Green Version]
- Gorno-Tempini, M.L.; Hillis, A.E.; Weintraub, S.; Kertesz, A.; Mendez, M.; Cappa, S.F.; Ogar, J.M.; Rohrer, J.D.; Black, S.; Boeve, B.F.; et al. Classification of primary progressive aphasia and its variants. Neurology 2011, 76, 1006–1014. [Google Scholar] [CrossRef] [Green Version]
- Rascovsky, K.; Hodges, J.R.; Knopman, D.; Mendez, M.F.; Kramer, J.H.; Neuhaus, J.; Van Swieten, J.C.; Seelaar, H.; Dopper, E.G.P.; Onyike, C.U.; et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011, 134, 2456–2477. [Google Scholar] [CrossRef]
- MacKenzie, I.R.; Neumann, M.; Bigio, E.H.; Cairns, N.J.; Alafuzoff, I.; Kril, J.; Kovacs, G.G.; Ghetti, B.; Halliday, G.; Holm, I.E.; et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: An update. Acta Neuropathol. 2009, 119, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Rohrer, J.D.; Guerreiro, R.; Vandrovcova, J.; Uphill, J.; Reiman, D.; Beck, J.; Isaacs, A.M.; Authier, A.; Ferrari, R.; Fox, N.C.; et al. The heritability and genetics of frontotemporal lobar degeneration. Neurology 2009, 73, 1451–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, Z.A.; Rankin, K.P.; Graff-Radford, N.R.; Takada, L.T.; Sturm, V.E.; Cleveland, C.M.; Criswell, L.A.; Jaeger, P.A.; Stan, T.; Heggeli, K.A.; et al. TDP-43 frontotemporal lobar degeneration and autoimmune disease. J. Neurol. Neurosurg. Psychiatry 2013, 84, 956–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, Z.A.; Sturm, V.E.; Camsari, G.B.; Karydas, A.; Yokoyama, J.S.; Grinberg, L.T.; Boxer, A.L.; Rosen, H.J.; Rankin, K.P.; Gorno-Tempini, M.L.; et al. Increased prevalence of autoimmune disease within C9 and FTD/MND cohorts: Completing the picture. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broce, I.; Karch, C.M.; Wen, N.; Fan, C.C.; Wang, Y.; Tan, C.H.; Kouri, N.; Ross, O.A.; Höglinger, G.U.; Muller, U.; et al. Immune-related genetic enrichment in frontotemporal dementia: An analysis of genome-wide association studies. PLoS Med. 2018, 15, e1002487. [Google Scholar]
- Cavazzana, I.; Alberici, A.; Bonomi, E.; Ottaviani, R.; Kumar, R.; Archetti, S.; Manes, M.; Cosseddu, M.; Buratti, E.; Padovani, A.; et al. Antinuclear antibodies in Frontotemporal Dementia: The tip’s of autoimmunity iceberg? J. Neuroimmunol. 2018, 325, 61–63. [Google Scholar] [CrossRef] [PubMed]
- Murley, A.G.; Rowe, J.B. Neurotransmitter deficits from frontotemporal lobar degeneration. Brain 2018, 141, 1263–1285. [Google Scholar] [CrossRef] [PubMed]
- Schwenk, J.; Baehrens, D.; Haupt, A.; Bildl, W.; Boudkkazi, S.; Roeper, J.; Fakler, B.; Schulte, U. Regional Diversity and Developmental Dynamics of the AMPA-Receptor Proteome in the Mammalian Brain. Neuron 2014, 84, 41–54. [Google Scholar] [CrossRef] [Green Version]
- Shi, S.-H.; Hayashi, Y.; Esteban, J.A.; Malinow, R. Subunit-Specific Rules Governing AMPA Receptor Trafficking to Synapses in Hippocampal Pyramidal Neurons. Cell 2001, 105, 331–343. [Google Scholar] [CrossRef]
- Diering, G.H.; Huganir, R.L. The AMPA Receptor Code of Synaptic Plasticity. Neuron 2018, 100, 314–329. [Google Scholar] [CrossRef] [Green Version]
- Benussi, A.; Di Lorenzo, F.; Dell’Era, V.; Cosseddu, M.; Alberici, A.; Caratozzolo, S.; Cotelli, M.S.; Micheli, A.; Rozzini, L.; Depari, A.; et al. Trans- cranial magnetic stimulation distinguishes Alzheimer disease from fronto- temporal dementia. Neurology 2017, 89, 665–672. [Google Scholar] [CrossRef]
- Benussi, A.; Gazzina, S.; Premi, E.; Cosseddu, M.; Archetti, S.; Dell’Era, V.; Cantoni, V.; Cotelli, M.S.; Alberici, A.; Micheli, A.; et al. Clinical and biomarker changes in presymptomatic genetic frontotemporal dementia. Neurobiol. Aging 2019, 76, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.H.; Sensi, S.L. Ca2+-Zn2+ permeable AMPA or kainate receptors: Possible key factors in selective neurodegener-ation. Trend Neurosci. 2000, 23, 365–371. [Google Scholar] [CrossRef]
- Gascon, E.; Lynch, K.; Ruan, H.; Almeida, S.; Verheyden, J.M.; Seeley, W.W.; Dickson, D.W.; Petrucelli, L.; Sun, D.; Jiao, J.; et al. Alterations in microRNA-124 and AMPA receptors contribute to social behavioral deficits in frontotemporal dementia. Nat. Med. 2014, 20, 1444–1451. [Google Scholar] [CrossRef] [PubMed]
- Pooler, A.M.; Phillips, E.C.; Lau, D.H.W.; Noble, W.; Hanger, D.P. Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 2013, 14, 389–394. [Google Scholar] [CrossRef]
- Gabilondo, I.; Saiz, A.; Galán, L.; González, V.; Jadraque, R.; Sabater, L.; Sans, A.; Sempere, A.; Vela, A.; Villalobos, F.; et al. Analysis of relapses in anti-NMDAR encephalitis. Neurology 2011, 77, 996–999. [Google Scholar] [CrossRef] [PubMed]
- Titulaer, M.J.; McCracken, L.; Gabilondo, I.; Armangue’, T.; Glaser, C.; Iizuka, T.; Honig, L.S.; Benseler, S.M.; Kawachi, I.; Martinez-Hernandez, E.; et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA re-ceptor encephalitis: An observational cohort study. Lancet Neurol. 2013, 12, 157–165. [Google Scholar] [CrossRef] [Green Version]
- Warikoo, N.; Brunwasser, S.J.; Benz, A.; Shu, H.J.; Paul, S.M.; Lewis, M.; Doherty, J.; Quirk, M.; Piccio, L.; Zorumski, C.F.; et al. Positive Allosteric Modulation as a Potential Therapeutic Strategy in Anti-NMDA Receptor En-cephalitis. J. Neurosci. 2018, 38, 3218–3229. [Google Scholar] [CrossRef] [Green Version]
- Koliaki, C.C.; Messini, C.; Tsolaki, M. Clinical efficacy of aniracetam, either as monotherapy or combined with cholines-terase inhibitors, in patients with cognitive impairment: A comparative open study. CNS Neurosci. Ther. 2012, 18, 302–312. [Google Scholar] [CrossRef]
- Reuillon, T.; Ward, S.E.; Beswick, P. AMPA Receptor Positive Allosteric Modulators: Potential for the Treatment of Neu-ropsychiatric and Neurological Disorders. Curr. Top. Med. Chem. 2016, 16, 3536–3565. [Google Scholar] [CrossRef]
- Bernard, K.; Gouttefangeas, S.; Bretin, S.; Galtier, S.; Robert, P.; Holthoff-Detto, V.; Cummings, J.; Pueyo, M. A 24-week double-blind placebo-controlled study of the efficacy and safety of the AMPA modulator S47445 in patients with mild to moderate Alzheimer’s disease and depressive symptoms. Alzheimer’s Dementia: Transl. Res. Clin. Interv. 2019, 5, 231–240. [Google Scholar] [CrossRef]
- Kim, J.-W.; Park, K.; Kang, R.J.; Gonzales, E.L.T.; Kim, D.G.; Oh, H.A.; Seung, H.; Ko, M.J.; Kwon, K.J.; Kim, K.C.; et al. Pharmacological modulation of AMPA receptor rescues social impairments in animal models of autism. Neuropsychopharmacology 2019, 44, 314–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Ionotropic Glutamate Receptor Subunit | GluN1 | GluN2B | GluA1 | GluA2 | GluA3 |
|---|---|---|---|---|---|
| Main clinical syndromes | Anti-NMDAR encephalitis | Rasmussen encephalitis, West syndrome | Limbic encephalitis, epilepsy | Rasmussen’s encephalitis, epilepsy, frontotemporal dementia | |
| Main clinical symptoms | Memory and behavioral disturbances, seizures, psychosis, sleep disorders | Seizures | Cognitive deficits, anxiety, sleep disturbances, mood disturbances and epilepsy | Cognitive and psychiatric symptoms, seizures | |
| Molecular-functional effects (in vitro) | Decreased NMDAR synaptic clusters and currents | Mostly unknown | Decreased AMPAR synaptic levels | Decreased AMPAR synaptic levels and currents | AMPAR internalization, spine loss |
| Molecular-functional effects (animal models; ex vivo) | Synaptic plasticity impairment, memory deficits, depressive-like behavior | Mostly unknown | Mostly unknown | AMPAR internalization, synaptic plasticity impairment, memory deficits, depressive-like behavior | AMPAR internalization, reduced intracortical facilitation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gardoni, F.; Stanic, J.; Scheggia, D.; Benussi, A.; Borroni, B.; Di Luca, M. NMDA and AMPA Receptor Autoantibodies in Brain Disorders: From Molecular Mechanisms to Clinical Features. Cells 2021, 10, 77. https://doi.org/10.3390/cells10010077
Gardoni F, Stanic J, Scheggia D, Benussi A, Borroni B, Di Luca M. NMDA and AMPA Receptor Autoantibodies in Brain Disorders: From Molecular Mechanisms to Clinical Features. Cells. 2021; 10(1):77. https://doi.org/10.3390/cells10010077
Chicago/Turabian StyleGardoni, Fabrizio, Jennifer Stanic, Diego Scheggia, Alberto Benussi, Barbara Borroni, and Monica Di Luca. 2021. "NMDA and AMPA Receptor Autoantibodies in Brain Disorders: From Molecular Mechanisms to Clinical Features" Cells 10, no. 1: 77. https://doi.org/10.3390/cells10010077
APA StyleGardoni, F., Stanic, J., Scheggia, D., Benussi, A., Borroni, B., & Di Luca, M. (2021). NMDA and AMPA Receptor Autoantibodies in Brain Disorders: From Molecular Mechanisms to Clinical Features. Cells, 10(1), 77. https://doi.org/10.3390/cells10010077