Estrogen and Glycemic Homeostasis: The Fundamental Role of Nuclear Estrogen Receptors ESR1/ESR2 in Glucose Transporter GLUT4 Regulation


Abstract
:1. Introduction
2. Early History of DM, Estrogen, and Their Relationship
3. The State of the Art in the Estrogen Regulation of Glycemic Homeostasis
3.1. Estrogen and Glycemic Homeostasis in Females
3.2. Estrogen and Glycemic Homeostasis in Males
3.3. Estrogen Receptors (ESRs)
3.4. Glucose Transporter GLUT4
4. SLC2A4/GLUT4 Expression and Glycemic Homeostasis
5. Esr1, Esr2 and Cytochrome P450 Subfamily A Member 1 (Cyp19a1) Gene Manipulation Contributions
5.1. Esr1, Esr2 and Cyp19a1 and Glycemic Homeostasis
5.2. Esr1, Esr2 and Cyp19a1 and GLUT4
6. Estrogen-Induced Regulation of Slc2a4/GLUT4 Expression
7. ESR1/ESR2-Mediated Regulation of SLC2A4/GLUT4
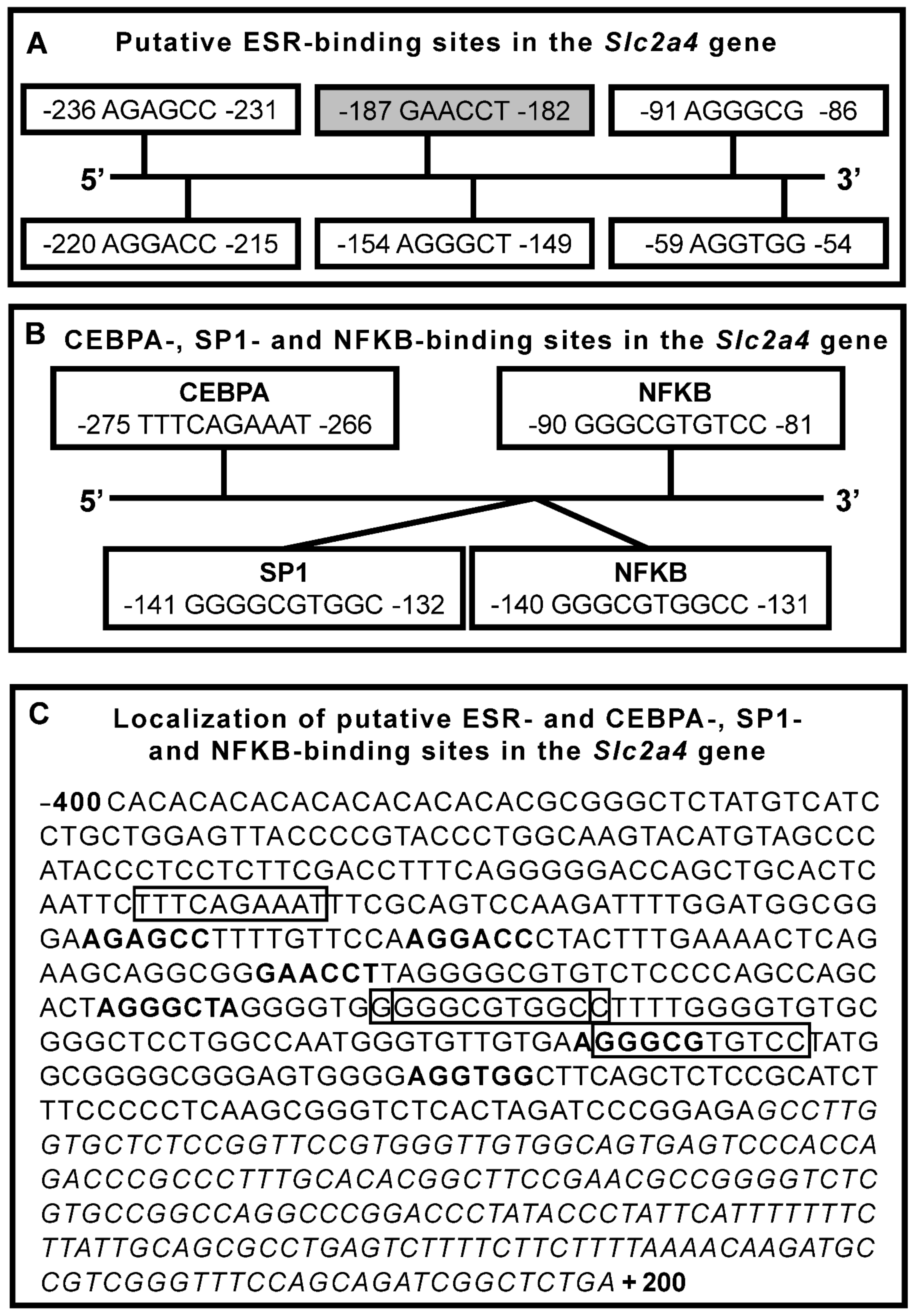
7.1. ESR1/ESR2 Nuclear Direct Regulation of SLC2A4 Gene
7.2. ESR1/ESR2 Nuclear Indirect Regulation of SLC2A4 Gene
7.2.1. Nuclear Factor NF-Kappa-B (NFKB)
7.2.2. Specific Protein 1 (SP1)
7.2.3. CCAAT/Enhancer-Binding Protein Alpha (CEBPA)
7.2.4. Peroxisome Proliferator-Activated Receptor Gamma (PPARG)
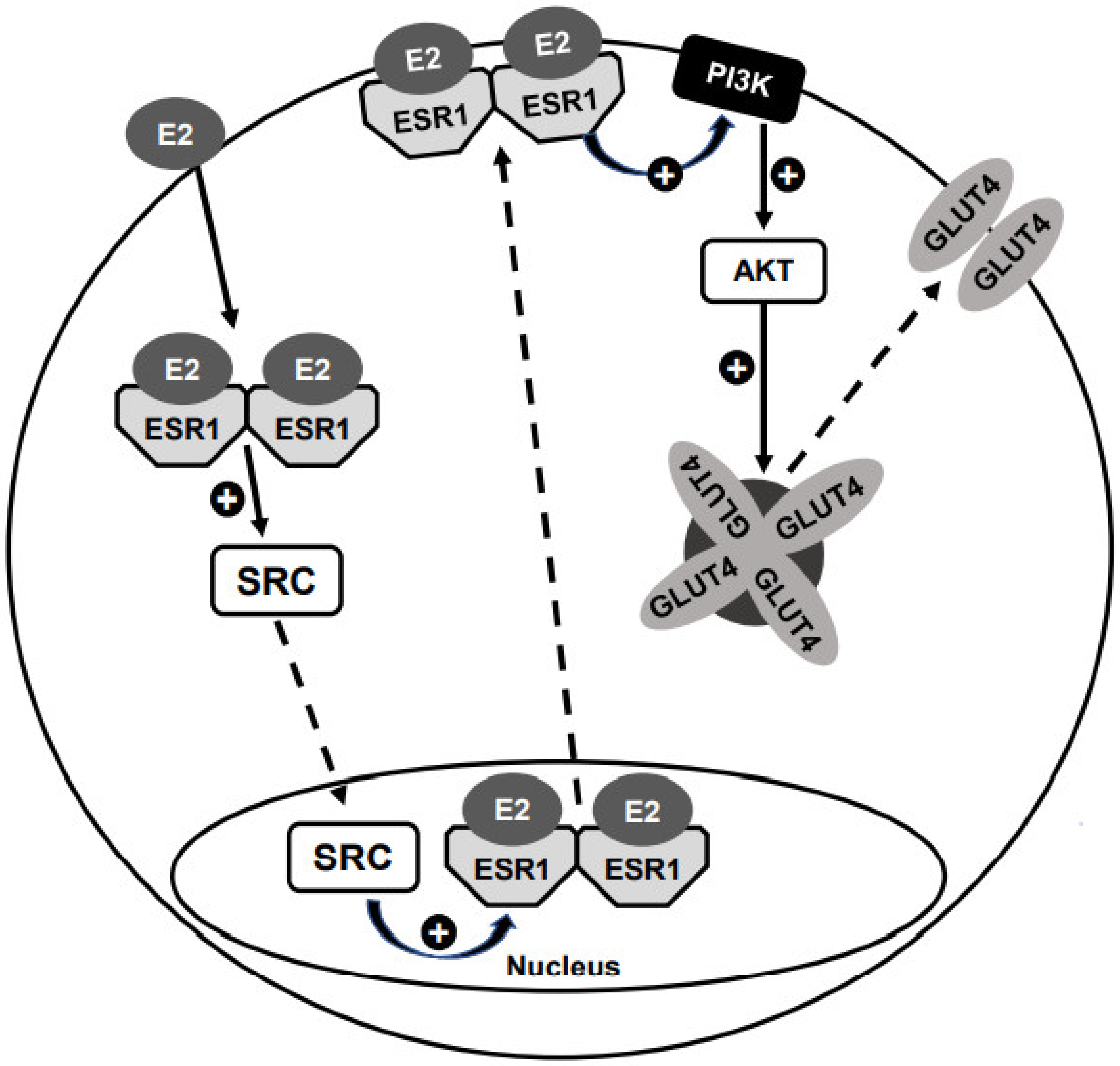
7.3. ESR1 Effects Associated to Its Plasma Membrane (PM) Localization
7.3.1. E2-Induced Translocation of ESR1 to the PM
7.3.2. E2-Induced GLUT4 Translocation to the PM
8. Phytoestrogens
9. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Barnes, B.O.; Regan, J.F.; Nelson, W.O. Improvement in experimental diabetes following the administration of amniotin. JAMA 1933, 101, 926–927. [Google Scholar] [CrossRef]
- American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care 2011, 34, S62–S69. [Google Scholar] [CrossRef] [Green Version]
- Klip, A.; McGraw, T.E.; James, D.E. Thirty sweet years of GLUT4. J. Biol. Chem. 2019, 294, 11369–11381. [Google Scholar] [CrossRef] [Green Version]
- Barnett, D.M.; Krall, L.P. The history of diabetes. In Joslins Diabetes Mellitus; Lippincott, Williams & Wilkins: Boston, MA, USA, 2005; pp. 1–17. [Google Scholar]
- Deanesly, R.; Parkes, A.S. Oestrogenic Action of Compounds of the Androsterone-Testosterone Series. BMJ 1936, 1, 257–258. [Google Scholar] [CrossRef] [Green Version]
- Jensen, E.V.; Jacobson, H.I. Basic guides to the mechanism of estrogen action. Recent Prog. Horm. Res. 1962, 18, 387–414. [Google Scholar]
- Jensen, E.V.; DeSombre, E.R.; Kawashima, T.; Suzuki, T.; Kyser, K.; Jungblut, P.W. Estrogen-Binding Substances of Target Tissues. Science 1967, 158, 529–530. [Google Scholar] [CrossRef]
- Wurtman, R.J.; Jensen, E.V. Estrogen Receptor: Ambiguities in the Use of This Term. Science 1968, 159, 1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Means, A.R.; O’Malley, B.W. Mechanism of estrogen action: Early transcriptional and translational events. Metabolism 1972, 21, 357–370. [Google Scholar] [CrossRef]
- Heldring, N.; Pike, A.; Andersson, S.; Matthews, J.; Cheng, G.; Hartman, J.; Tujague, M.; Ström, A.; Treuter, E.; Warner, M.; et al. Estrogen Receptors: How Do They Signal and What Are Their Targets. Physiol. Rev. 2007, 87, 905–931. [Google Scholar] [CrossRef] [Green Version]
- Rathery, F.; Rudolf, M. Folliculine, insuline et diabète. Bull. Mem. Soc. Med. 1928, 52, 741. [Google Scholar]
- Nelson, W.O.; Overholser, M.D. The effect of oestrogenic hormone on experimental pancreatic diabetes in the monkey. Endocrinology 1936, 20, 473–480. [Google Scholar] [CrossRef]
- Gessler, C.J.; Halsted, J.A.; Stetson, R.P. Effect of estrogenic substance on the blood sugar of female diabetics after the menopause. J. Clin. Investig. 1939, 18, 715–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasio, E.; Antaki, A.; Campenhout, J. Diabetes Mellitus in Gonadal Dysgenesis: Studies of Insulin and Growth Hormone Secretion. Eur. J. Clin. Investig. 1976, 6, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Polychronakos, C.; Letarte, J.; Collu, R.; Ducharme, J. Carbohydrate intolerance in children and adolescents with Turner syndrome. J. Pediatr. 1980, 96, 1009–1014. [Google Scholar] [CrossRef]
- Bakalov, V.K.; Cooley, M.M.; Quon, M.J.; Luo, M.L.; Yanovski, J.A.; Nelson, L.M.; Sullivan, G.; Bondy, C.A. Impaired Insulin Secretion in the Turner Metabolic Syndrome. J. Clin. Endocrinol. Metab. 2004, 89, 3516–3520. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Wang, Y.; Zhou, T.; Zhao, X.; Wang, Y.; Wang, G.; Gang, X. Glucose Metabolism in Turner Syndrome. Front. Endocrinol. 2019, 10, 49. [Google Scholar] [CrossRef]
- Saucedo, R.; Basurto, L.; Zárate, A.; Martínez, C.; Hernandez, M.; Galván, R. Effect of Estrogen Therapy on Insulin Resistance and Plasminogen Activator Inhibitor Type 1 Concentrations in Postmenopausal Women. Gynecol. Obstet. Investig. 2007, 64, 61–64. [Google Scholar] [CrossRef]
- Paschou, S.A.; Marina, L.V.; Spartalis, E.; Anagnostis, P.; Alexandrou, A.; Goulis, D.G.; Lambrinoudaki, I. Therapeutic strategies for type 2 diabetes mellitus in women after menopause. Maturitas 2019, 126, 69–72. [Google Scholar] [CrossRef]
- Dunaif, A.; Segal, K.R.; Futterweit, W.; Dobrjansky, A. Profound peripheral insulin resistance, independent of obesity, in polycystic ovary syndrome. Diabetes 1989, 38, 1165–1174. [Google Scholar] [CrossRef]
- Ditkoff, E.C.; Fruzzetti, F.; Chang, L.; Stancyzk, F.Z.; Lobo, R.A. The impact of estrogen on adrenal androgen sensitivity and secretion in polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 1995, 80, 603–607. [Google Scholar] [CrossRef]
- Livingstone, C.; Collison, M. Sex steroids and insulin resistance. Clin. Sci. 2002, 102, 151–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, X.; Xie, Y.J.; Liu, Y.T.; Long, S.L.; Mo, Z.C. Polycystic ovarian syndrome: Correlation between hyperandrogenism, insulin resistance and obesity. Clin. Chim. Acta 2020, 502, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, T.A.; Metzger, B.E.; Freinkel, N.; Bergman, R.N. Insulin sensitivity and B-cell responsiveness to glucose during late pregnancy in lean and moderately obese women with normal glucose tolerance or mild gestational diabetes. Am. J. Obstet. Gynecol. 1990, 162, 1008–1014. [Google Scholar] [CrossRef]
- Kaaja, R.J.; Greer, I.A. Manifestations of chronic disease during pregnancy. J. Am. Med. Assoc. 2005, 294, 2751–2757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reece, E.A.; Homko, C.; Wiznitzer, A. Metabolic Changes in Diabetic and Nondiabetic Subjects during Pregnancy. Obstet. Gynecol. Surv. 1994, 49, 64–71. [Google Scholar] [CrossRef]
- Case, A.M.; Reid, R.L. Menstrual cycle effects on common medical conditions. Compr. Ther. 2001, 27, 65–71. [Google Scholar] [CrossRef]
- Lopez, L.M.; Grimes, D.A.; Schulz, K.F. Steroidal contraceptives: Effect on carbohydrate metabolism in women without diabetes mellitus. Cochrane Database Syst. Rev. 2014, 2019, 31711271. [Google Scholar] [CrossRef]
- Cooke, P.S.; Nanjappa, M.K.; Ko, C.; Prin, G.S.; Hess, R.A. Estrogens in male physology. Physiol. Rev. 2017, 97, 995–1043. [Google Scholar] [CrossRef]
- Smith, E.P.; Boyd, J.; Frank, G.R.; Takahashi, H.; Cohen, R.M.; Specker, B.; Williams, T.C.; Lubahn, D.B.; Korach, K.S. Estrogen Resistance Caused by a Mutation in the Estrogen-Receptor Gene in a Man. N. Engl. J. Med. 1994, 331, 1056–1061. [Google Scholar] [CrossRef]
- Faustini-Fustini, M.; Rochira, V.; Carani, C. Oestrogen deficiency in men: Where are we today? Eur. J. Endocrinol. 1999, 140, 111–129. [Google Scholar] [CrossRef]
- Belgorosky, A.; Guercio, G.; Pepe, C.; Saraco, N.; Rivarola, M. Genetic and Clinical Spectrum of Aromatase Deficiency in Infancy, Childhood and Adolescence. Horm. Res. Paediatr. 2009, 72, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Elbers, J.M.H.; Giltay, E.J.; Teerlink, T.; Scheffer, P.G.; Asscheman, H.; Seidell, J.C.; Gooren, L.J.G. Effects of sex steroids on components of the insulin resistance syndrome in transsexual subjects. Clin. Endocrinol. 2003, 58, 562–571. [Google Scholar] [CrossRef] [Green Version]
- Meyer, M.R.; Clegg, D.J.; Prossnitz, E.R.; Barton, M. Obesity, insulin resistance and diabetes: Sex differences and role of oestrogen receptors. Acta Physiol. 2010, 203, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, S.; Mäkelä, S.; Treuter, E.; Tujague, M.; Thomsen, J.; Andersson, G.; Enmark, E.; Pettersson, K.; Warner, M.; Gustafsson, J.-Å. Mechanisms of Estrogen Action. Physiol. Rev. 2001, 81, 1535–1565. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, V.; Wang, X.; Safe, S. Estrogen receptor-Sp1 complexes mediate estrogen-induced cathepsin D gene expression in MCF-7 human breast cancer cells. J. Biol. Chem. 1994, 269, 15912–15917. [Google Scholar] [CrossRef]
- Eriksson, M.A.L.; Nilsson, L. Structural and dynamic differences of the estrogen receptor DNA-binding domain, binding as a dimer and as a monomer to DNA: Molecular dynamics simulation studies. Eur. Biophys. J. 1999, 28, 102–111. [Google Scholar] [CrossRef]
- Hewitt, S.C.; Li, Y.; Li, L.; Korach, K.S. Estrogen-mediated Regulation of Igf1 Transcription and Uterine Growth Involves Direct Binding of Estrogen Receptor α to Estrogen-responsive Elements. J. Biol. Chem. 2009, 285, 2676–2685. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Deschamps, A.; Korach, K.S.; Murphy, E. Estrogen-Enhanced Gene Expression of Lipoprotein Lipase in Heart Is Antagonized by Progesterone. Endocrinology 2007, 149, 711–716. [Google Scholar] [CrossRef] [Green Version]
- Safe, S.; Kim, K. Non-classical genomic estrogen receptor (ER)/specificity protein and ER/activating protein-1 signaling pathways. J. Mol. Endocrinol. 2008, 41, 263–275. [Google Scholar] [CrossRef]
- Dauvois, S.; White, R.; Parker, M.G. The antiestrogen ICI 182780 disrupts estrogen receptor nucleocytoplasmic shuttling. J. Cell Sci. 1993, 106, 1377–1388. [Google Scholar]
- Acconcia, F.; Ascenzi, P.; Bocedi, A.; Spisni, E.; Tomasi, V.; Trentalance, A.; Visca, P.; Marino, M. Palmitoylation-dependent Estrogen Receptor α Membrane Localization: Regulation by 17β-Estradiol. Mol. Biol. Cell 2005, 16, 231–237. [Google Scholar] [CrossRef]
- Adlanmerini, M.; Solinhac, R.; Abot, A.; Fabre, A.; Raymond-Letron, I.; Guihot, A.L.; Boudou, F.; Sautier, L.; Vessieres, E.; Kim, S.H.; et al. Mutation of the palmitoylation site of estrogen receptor a in vivo reveals tissue-specific roles for membrane versus nuclear actions. Proc. Natl. Acad. Sci. USA 2014, 111, E283–E290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castoria, G.; Giovannelli, P.; Lombardi, M.; De Rosa, C.; Giraldi, T.; De Falco, A.; Barone, M.V.; Abbondanza, C.; Migliaccio, A.; Auricchio, F. Tyrosine phosphorylation of estradiol receptor by Src regulates its hormone-dependent nuclear export and cell cycle progression in breast cancer cells. Oncogene 2012, 31, 4868–4877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chojnacka, K.; Mruk, D.D. The Src non-receptor tyrosine kinase paradigm: New insights into mammalian Sertoli cell biology. Mol. Cell. Endocrinol. 2015, 415, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Lucas, T.F.G.; Siu, E.R.; Esteves, C.A.; Monteiro, H.P.; Oliveira, C.A.; Porto, C.S.; Lazari, M.F.M. 17Beta-Estradiol Induces the Translocation of the Estrogen Receptors ESR1 and ESR2 to the Cell Membrane, MAPK3/1 Phosphorylation and Proliferation of Cultured Immature Rat Sertoli Cells. Biol. Reprod. 2008, 78, 101–114. [Google Scholar] [CrossRef] [Green Version]
- Simoncini, T.; Hafezi-Moghadam, A.; Brazil, D.P.; Ley, K.; Chin, W.W.; Liao, J.K. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nat. Cell Biol. 2000, 407, 538–541. [Google Scholar] [CrossRef]
- Fuentes, N.; Silveyra, P. Estrogen receptor signaling mechanisms. Adv. Protein Chem. Struct. Biol. 2019, 116, 135–170. [Google Scholar] [CrossRef]
- Luo, J.; Liu, D. Does GPER really function as a G protein-coupled estrogen receptor in vivo? Front. Endocrinol. 2020, 11, 148. [Google Scholar] [CrossRef]
- Mueckler, M.; Thorens, B. The SLC2 (GLUT) family of membrane transporters. Mol. Asp. Med. 2013, 34, 121–138. [Google Scholar] [CrossRef] [Green Version]
- Klip, A.; Sun, Y.; Chiu, T.T.; Foley, K.P. Signal transduction meets vesicle traffic: The software and hardware of GLUT4 translocation. Am. J. Physiol. Physiol. 2014, 306, C879–C886. [Google Scholar] [CrossRef] [Green Version]
- Im, S.-S.; Kwon, S.-K.; Kim, T.-H.; Kim, H.; Ahn, Y.-H. Regulation of glucose transporter type 4 isoform gene expression in muscle and adipocytes. IUBMB Life 2007, 59, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Karnieli, E.; Armoni, M. Transcriptional regulation of the insulin-responsive glucose transporter GLUT4 gene: From physiology to pathology. Am. J. Physiol. Metab. 2008, 295, E38–E45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannella, M.L.C.C.; Machado, U.F. SLC2A4gene: A promising target for pharmacogenomics of insulin resistance. Pharmacogenomics 2013, 14, 847–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado, U.F.; Shimizu, I.; Saito, M. Reduced content and preserved translocation of glucose transporter (GLUT 4) in white adipose tissue of obese mice. Physiol. Behav. 1994, 55, 621–625. [Google Scholar] [CrossRef]
- Gibbs, E.M.; Stock, J.L.; McCoid, S.C.; Stukenbrok, H.A.; Pessin, J.E.; Stevenson, R.W.; Milici, A.J.; McNeish, J.D. Glycemic improvement in diabetic db/db mice by overexpression of the human insulin-regulatable glucose transporter (GLUT4). J. Clin. Investig. 1995, 95, 1512–1518. [Google Scholar] [CrossRef] [Green Version]
- Zisman, A.; Peroni, O.D.; Abel, E.D.; Michael, M.D.; Mauvais-Jarvis, F.; Lowell, B.B.; Wojtaszewski, J.F.; Hirshman, M.F.; Virkamaki, A.; Goodyear, L.J.; et al. Targeted disruption of the glucose transporter 4 selectively in muscle causes insulin resistance and glucose intolerance. Nat. Med. 2000, 6, 924–928. [Google Scholar] [CrossRef]
- Esteves, J.V.; Yonamine, C.Y.; Pinto-Junior, D.C.; Gerlinger-Romero, F.; Enguita, F.J.; Machado, U.F. Diabetes Modulates MicroRNAs 29b-3p, 29c-3p, 199a-5p and 532-3p Expression in Muscle: Possible Role in GLUT4 and HK2 Repression. Front. Endocrinol. 2018, 9, 536. [Google Scholar] [CrossRef] [Green Version]
- Yonamine, C.Y.; Alves-Wagner, A.B.; Esteves, J.V.; Okamoto, M.M.; Correa-Giannella, M.L.; Giannella-Neto, D.; Machado, U.F. Diabetes induces tri-methylation at lysine 9 of histone 3 at Slc2a4 gene in skeletal muscle: A new target to improve glycemic control. Mol. Cell. Endocrinol. 2019, 481, 26–34. [Google Scholar] [CrossRef]
- Esteves, J.V.; Enguita, F.J.; Machado, U.F. MicroRNAs-Mediated Regulation of Skeletal Muscle GLUT4 Expression and Translocation in Insulin Resistance. J. Diabetes Res. 2017, 2017, 7267910. [Google Scholar] [CrossRef]
- Esteves, J.V.; Yonamine, C.Y.; Machado, U.F. SLC2A4 expression and its epigenetic regulation as biomarkers for insulin resistance treatment in diabetes mellitus. Biomark. Med. 2020, 14, 413–416. [Google Scholar] [CrossRef]
- Heine, P.A.; Taylor, J.A.; Iwamoto, G.A.; Lubahn, D.B.; Cooke, P.S. Increased adipose tissue in male and female estrogen receptor-α knockout mice. Proc. Natl. Acad. Sci. USA 2000, 97, 12729–12734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, K.; Toda, K.; Saibara, T.; Nakagawa, M.; Saika, K.; Onishi, T.; Sugiura, T.; Shizuta, Y. Progressive development of insulin resistance phenotype in male mice with complete aromatase (CYP19) deficiency. J. Endocrinol. 2003, 176, 237–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubinow, K.B.; Hartigh, L.J.D.; Goodspeed, L.; Wang, S.; Oz, O.K. Aromatase deficiency in hematopoietic cells improves glucose tolerance in male mice through skeletal muscle-specific effects. PLoS ONE 2020, 15, e0227830. [Google Scholar] [CrossRef] [PubMed]
- Foryst-Ludwig, A.; Clemenz, M.; Hohmann, S.; Hartge, M.; Sprang, C.; Frost, N.; Krikov, M.; Bhanot, S.; Barros, R.; Morani, A.; et al. Metabolic Actions of Estrogen Receptor Beta (ERβ) are Mediated by a Negative Cross-Talk with PPARγ. PLoS Genet. 2008, 4, e1000108. [Google Scholar] [CrossRef] [PubMed]
- Barros, R.P.A.; Machado, U.F.; Warner, M.N.; Gustafsson, J.-Å. Muscle GLUT4 regulation by estrogen receptors ERbeta and ER. Proc. Natl. Acad. Sci. USA 2006, 103, 1605–1608. [Google Scholar] [CrossRef] [Green Version]
- Fatima, L.A.; Campello, R.S.; Barreto-Andrade, J.N.; Passarelli, M.; Santos, R.S.; Clegg, D.J.; Machado, U.F. Estradiol stimulates adipogenesis and Slc2a4/GLUT4 expression via ESR1-mediated activation of CEBPA. Mol. Cell. Endocrinol. 2019, 498, 110447. [Google Scholar] [CrossRef]
- Barros, R.P.A.; Gabbi, C.; Morani, A.; Warner, M.; Gustafsson, J.-Å. Participation of ER and ER in glucose homeostasis in skeletal muscle and white adipose tissue. AJP Endocrinol. Metab. 2009, 297, E124–E133. [Google Scholar] [CrossRef]
- Barros, R.P.; Machado, U.F.; Gustafsson, J.-Å. Estrogen receptors: New players in diabetes mellitus. Trends Mol. Med. 2006, 12, 425–431. [Google Scholar] [CrossRef]
- Barros, R.P.; Gustafsson, J.-Å. Estrogen Receptors and the Metabolic Network. Cell Metab. 2011, 14, 289–299. [Google Scholar] [CrossRef] [Green Version]
- Barros, R.P.D.A.; Morani, A.; Moriscot, A.S.; Machado, U.F. Insulin resistance of pregnancy involves estrogen-induced repression of muscle GLUT4. Mol. Cell. Endocrinol. 2008, 295, 24–31. [Google Scholar] [CrossRef] [Green Version]
- Anhê, G.F.; Hirabara, S.M.; Turrer, T.C.; Caperuto, L.C.; Anhê, F.F.; Ribeiro, L.M.; Marçal, A.C.; Carvalho, C.R.O.; Curi, R.; Machado, U.F.; et al. Postpartum glycemic homeostasis in early lactating rats is accompanied by transient and specific increase of soleus insulin response through IRS2/AKT pathway. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R2225–R2233. [Google Scholar] [CrossRef] [Green Version]
- Okuno, S.; Akazawo, S.; Yasuhi, I.; Kawasaki, E.; Matsumoto, K.; Yamasaki, H.; Matsuo, H.; Yamaguchi, Y.; Nagataki, S. Decreased Expression of the GLUT4 Glucose Transporter Protein in Adipose Tissue During Pregnancy. Horm. Metab. Res. 1995, 27, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.; Khan, H.R.; Alex, N.S.; Puttaraju, M.; Chandrasekaran, T.T.; Rudraiah, M. Under-carboxylated osteocalcin regulates glucose and lipid metabolism during pregnancy and lactation in rats. J. Endocrinol. Investig. 2020, 43, 1081–1095. [Google Scholar] [CrossRef] [PubMed]
- Campello, R.S.; Fátima, L.A.; Barreto-Andrade, J.N.; Lucas, T.F.; Mori, R.C.; Porto, C.S.; Machado, U.F. Estradiol-induced regulation of GLUT4 in 3T3-L1 cells: Involvement of ESR1 and AKT activation. J. Mol. Endocrinol. 2017, 59, 257–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campello, R.S.; Alves-Wagner, A.B.; Lucas, T.F.; Mori, R.C.; Furuya, D.T.; Porto, C.S.; Machado, U.F. Estrogen receptor 1 agonist PPT stimulates Slc2a4 gene expression and improves insulin-induced glucose uptake in adipocytes. Curr. Top. Med. Chem. 2012, 12, 2059–2069. [Google Scholar] [CrossRef]
- Safe, S. Transcriptional activation of genes by 17β-estradiol through estrogen receptor-Sp1 interactions. Vitam. Horm. 2001, 62, 231–252. [Google Scholar] [CrossRef]
- Furuya, D.T.; Neri, E.A.; Poletto, A.C.; Anhê, G.F.; Freitas, H.S.; Campello, R.S.; Rebouças, N.A.; Machado, U.F. Identification of nuclear factor-κB sites in the Slc2a4 gene promoter. Mol. Cell. Endocrinol. 2013, 370, 87–95. [Google Scholar] [CrossRef]
- Moraes, P.A.; Yonamine, C.Y.; Junior, D.C.P.; Esteves, J.V.; Machado, U.F.; Mori, R.C. Insulin acutely triggers transcription of Slc2a4 gene: Participation of the AT-rich, E-box and NFKB-binding sites. Life Sci. 2014, 114, 36–44. [Google Scholar] [CrossRef]
- Poletto, A.C.; Furuya, D.T.; David-Silva, A.; Ebersbach-Silva, P.; Santos, C.L.; Correa-Giannella, M.L.; Passarelli, M.; Machado, U.F. Oleic and linoleic fatty acids downregulate Slc2a4/GLUT4 expression via NFKB and SREBP1 in skeletal muscle cells. Mol. Cell. Endocrinol. 2015, 401, 65–72. [Google Scholar] [CrossRef]
- Pinto-Junior, D.C.; Silva, K.S.; Michalani, M.L.; Yonamine, C.Y.; Esteves, J.V.; Fabre, N.T.; Thieme, K.; Catanozi, S.; Okamoto, M.M.; Seraphim, P.M.; et al. Advanced glycation end products-induced insulin resistance involves repression of skeletal muscle GLUT4 expression. Sci. Rep. 2018, 8, 8109. [Google Scholar] [CrossRef]
- Ebersbach-Silva, P.; Poletto, A.C.; David-Silva, A.; Seraphim, P.M.; Anhê, G.F.; Passarelli, M.; Furuya, D.T.; Machado, U.F. Palmitate-induced Slc2a4/GLUT4 downregulation in L6 muscle cells: Evidence of inflammatory and endoplasmic reticulum stress involvement. Lipids Health Dis. 2018, 17, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, J.L.; Giannocco, G.; Furuya, D.T.; Lima, G.A.; Moraes, P.A.; Nachef, S.; Bordin, S.; Britto, L.R.; Nunes, M.T.; Machado, U.F. NF-κB, MEF2A, MEF2D and HIF1-a involvement on insulin- and contraction-induced regulation of GLUT4 gene expression in soleus muscle. Mol. Cell. Endocrinol. 2005, 240, 82–93. [Google Scholar] [CrossRef]
- Galien, R.; Garcia, T. Estrogen receptor impairs interleukin-6 expression by preventing protein binding on the NF-κB site. Nucleic Acids Res. 1997, 25, 2424–2429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sas, L.; Lardon, F.; Vermeulen, P.; Hauspy, J.; Van Dam, P.; Pauwels, P.; Dirix, L.Y.; Van Laere, S.J. The interaction between ER and NFκB in resistance to endocrine therapy. Breast Cancer Res. 2012, 14, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pradhan, M.; Bembinster, L.A.; Baumgarten, S.C.; Frasor, J. Proinflammatory Cytokines Enhance Estrogen-dependent Expression of the Multidrug Transporter GeneABCG2through Estrogen Receptor and NFκB Cooperativity at Adjacent Response Elements. J. Biol. Chem. 2010, 285, 31100–31106. [Google Scholar] [CrossRef] [Green Version]
- Koizume, S.; Miyagi, Y. Diverse Mechanisms of Sp1-Dependent Transcriptional Regulation Potentially Involved in the Adaptive Response of Cancer Cells to Oxygen-Deficient Conditions. Cancers 2015, 8, 2. [Google Scholar] [CrossRef] [Green Version]
- Im, S.-S.; Kwon, S.-K.; Kang, S.-Y.; Kim, T.-H.; Kim, H.-I.; Hur, M.-W.; Kim, K.-S.; Ahn, Y.-H. Regulation of GLUT4 gene expression by SREBP-1c in adipocytes. Biochem. J. 2006, 399, 131–139. [Google Scholar] [CrossRef]
- Barreto-Andrade, J.N.; De Fátima, L.A.; Campello, R.S.; Guedes, J.A.C.; De Freitas, H.S.; Machado, M.M.F. Estrogen Receptor 1 (ESR1) Enhances Slc2a4/GLUT4 Expression by a SP1 Cooperative Mechanism. Int. J. Med. Sci. 2018, 15, 1320–1328. [Google Scholar] [CrossRef] [Green Version]
- Ramji, D.P.; Foka, P. CCAAT/enhancer-binding proteins: Structure, function and regulation. Biochem. J. 2002, 365, 561–575. [Google Scholar] [CrossRef] [Green Version]
- Kaestner, K.H.; Christy, R.J.; Lane, M.D. Mouse insulin-responsive glucose transporter gene: Characterization of the gene and trans-activation by the CCAAT/enhancer binding protein. Proc. Natl. Acad. Sci. USA 1990, 87, 251–255. [Google Scholar] [CrossRef] [Green Version]
- Pessler-Cohen, D.; Pekala, P.H.; Kovsan, J.; Bloch-Damti, A.; Rudich, A.; Bashan, N. GLUT4 repression in response to oxidative stress is associated with reciprocal alterations in C/EBP alpha and delta isoforms in 3T3-L1 adipocytes. Arch. Physiol. Biochem. 2006, 112, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Xie, Y.; Morrison, R.F.; Bucher, N.L.R.; Farmer, S.R. PPARγ induces the insulin-dependent glucose transporter GLUT4 in the absence of C/EBPα during the conversion of 3T3 fibroblasts into adipocytes. J. Clin. Investig. 1998, 101, 22–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, W.; Parra, M.; Centrella, M.; McCarthy, T.L. Interactions between CCAAT enhancer binding protein δ and estrogen receptor α control insulin-like growth factor I (igf1) and estrogen receptor-dependent gene expression in osteoblasts. Gene 2005, 345, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-H.; Lee, M.-J.; Chang, H.-H.; Hung, P.-F.; Kao, Y.-H. 17β-Estradiol Stimulates Resistin Gene Expression in 3T3-L1 Adipocytes via the Estrogen Receptor, Extracellularly Regulated Kinase, and CCAAT/Enhancer Binding Protein-α Pathways. Endocrinology 2006, 147, 4496–4504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R.; Su, D.; Zhu, W.; Huang, Q.; Liu, M.; Xue, Y.; Zhang, Y.; Li, D.; Zhao, A.; Liu, Y. Estrogen suppresses adipogenesis by inhibiting S100A16 expression. J. Mol. Endocrinol. 2014, 52, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Moseti, D.; Regassa, A.; Kim, W. Molecular Regulation of Adipogenesis and Potential Anti-Adipogenic Bioactive Molecules. Int. J. Mol. Sci. 2016, 17, 124. [Google Scholar] [CrossRef] [Green Version]
- Armoni, M.; Harel, C.; Karnieli, E. Transcriptional regulation of the GLUT4 gene: From PPAR-γ and FOXO1 to FFA and inflammation. Trends Endocrinol. Metab. 2007, 18, 100–107. [Google Scholar] [CrossRef]
- Armoni, M.; Kritz, N.; Harel, C.; Bar-Yoseph, F.; Chen, H.; Quon, M.J.; Karnieli, E. Peroxisome Proliferator-activated Receptor-γ RepressesGLUT4Promoter Activity in Primary Adipocytes, and Rosiglitazone Alleviates This Effect. J. Biol. Chem. 2003, 278, 30614–30623. [Google Scholar] [CrossRef] [Green Version]
- Chandra, V.; Huang, P.; Hamuro, Y.; Raghuram, S.; Wang, Y.; Burris, T.P.; Rastinejad, F. Structure of the intact PPAR-γ-RXR-α nuclear receptor complex on DNA. Nature 2008, 456, 350–356. [Google Scholar] [CrossRef]
- Dang, Z.C.; Van Bezooijen, R.L.; Karperien, M.; Papapoulos, S.E.; Löwik, C.W.G.M. Exposure of KS483 Cells to Estrogen Enhances Osteogenesis and Inhibits Adipogenesis. J. Bone Miner. Res. 2002, 17, 394–405. [Google Scholar] [CrossRef]
- Jeong, S.; Yoon, M. 17β-Estradiol inhibition of PPARγ-induced adipogenesis and adipocyte-specific gene expression. Acta Pharmacol. Sin. 2011, 32, 230–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Kilgore, M.W. Signal cross-talk between estrogen receptor alpha and beta and the peroxisome proliferator-activated receptor gamma1 in MDA-MB-231 and MCF-7 breast cancer cells. Mol. Cell. Endocrinol. 2002, 194, 123–133. [Google Scholar] [CrossRef]
- Bonofiglio, D.; Gabriele, S.; Aquila, S.; Catalano, S.; Gentile, M.; Middea, E.; Giordano, F.; Andò, S. Estrogen Receptor α Binds to Peroxisome Proliferator–Activated Receptor Response Element and Negatively Interferes with Peroxisome Proliferator–Activated Receptor γ Signaling in Breast Cancer Cells. Clin. Cancer Res. 2005, 11, 6139–6147. [Google Scholar] [CrossRef] [Green Version]
- Saczko, J.; Michel, O.; Chwiłkowska, A.; Sawicka, E.; Mączyńska, J.; Kulbacka, J. Estrogen receptors in cell membranes: Regulation and signaling. Adv. Anat. Embryol. Cell Biol. 2017, 227, 93–105. [Google Scholar]
- Collison, M.; Campbell, I.W.; Salt, I.P.; Dominiczak, A.F.; Connell, J.M.C.; Lyall, H.; Gould, G.W. Sex hormones induce insulin resistance in 3T3-L1 adipocytes by reducing cellular content of IRS proteins. Diabetologia 2000, 43, 1374–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muraki, K.; Okuya, S.; Tanizawa, Y. Estrogen Receptor α Regulates Insulin Sensitivity through IRS-1 Tyrosine Phosphorylation in Mature 3T3-L1 Adipocytes. Endocr. J. 2006, 53, 841–851. [Google Scholar] [CrossRef] [Green Version]
- Nagira, K.; Sasaoka, T.; Wada, T.; Fukui, K.; Ikubo, M.; Hori, S.; Tsuneki, H.; Saito, S.; Kobayashi, M. Altered Subcellular Distribution of Estrogen Receptor α Is Implicated in Estradiol-Induced Dual Regulation of Insulin Signaling in 3T3-L1 Adipocytes. Endocrinology 2006, 147, 1020–1028. [Google Scholar] [CrossRef] [Green Version]
- Gupta, C.; Prakash, D.; Gupta, S. Phytoestrogens as Pharma Foods. Adv. Food Technol. Nutr. Sci. Open J. 2016, 2, 19–31. [Google Scholar] [CrossRef]
- Kuiper, G.G.; Lemmen, J.G.; Carlsson, B.; Corton, J.C.; Safe, S.H.; van der Saag, P.T.; van der Burg, B.; Gustafsson, J.A. Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinology 1998, 139, 4252–4263. [Google Scholar] [CrossRef]
- Turner, J.V.; Glass, B.D.; Agatonovic-Kustrin, S. Molecular aspects of phytoestrogen selective binding at estrogen receptors. J. Pharm. Sci. 2007, 96, 1879–1885. [Google Scholar] [CrossRef]
- Anhê, G.F.; Okamoto, M.M.; Kinote, A.; Sollon, C.; Santos, C.D.L.; Anhê, F.F.; Lima, G.A.; Hirabara, S.M.; Velloso, L.A.; Bordin, S.; et al. Quercetin decreases inflammatory response and increases insulin action in skeletal muscle of ob/ob mice and in L6 myotubes. Eur. J. Pharmacol. 2012, 689, 285–293. [Google Scholar] [CrossRef]
- Yonamine, C.Y.; Pinheiro-Machado, E.; Michalani, M.L.; Alves-Wagner, A.B.; Esteves, J.V.; Freitas, H.S.; Machado, U.F. Resveratrol Improves Glycemic Control in Type 2 Diabetic Obese Mice by Regulating Glucose Transporter Expression in Skeletal Muscle and Liver. Molecules 2017, 22, 1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dresseno, L.P.; Lehnen, A.M.; Teló, G.; Silveira, A.; Markoski, M.M.; Machado, U.F.; Schaan, B.D. Impact of flaxseed and soy nuts as dietary supplements on lipid profile, insulin sensitivity, and GLUT4 expression in ovariectomized rats. Appl. Physiol. Nutr. Metab. 2018, 43, 1282–1287. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Expósito, M.J.; Martínez-Martos, J.M.; Cantón-Habas, V.; González, M.D.P.C. Putative Involvement of Endocrine Disruptors in the Alzheimer’s disease Via the Insulin-Regulated Aminopeptidase/GLUT4 Pathway. Curr. Neuropharmacol. 2020, 18, 1–17. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classes | Main Food Sources | Compounds | RBA * | |
|---|---|---|---|---|
| ESR1 | ESR2 | |||
| 17β-estradiol (E2) | 100 | 100 | ||
| Coumestans | Mung Beans, Soy Sprouts, Alfalfa Sprouts, Clover | Coumestrol | 20 | 140 |
| Isoflavones | Soy (milk, cheese, protein, tofu), Peanut, Clover, Sunflower, Seeds, Walnut | Genistein | 4 | 87 |
| Daidzein | 0.1 | 0.5 | ||
| Biochanin A | <0.01 | <0.01 | ||
| Flavones | Parsley, Celery, Capsicum, Citrus Peels, Pepper | Apigenin | 0.3 | 6 |
| Chrysin | <0.01 | <0.01 | ||
| Flavanols | Beans, Tea, Spinach, Broccoli | Kaempferol | 0.1 | 3 |
| Chalcones | Apple, Tea, Soy-based Foods | Phloretin | 0.2 | 0.7 |
| Stilbenes | Grape, Wine | Resveratrol | ND | ND |
| Lignans | Soybean, Peanut, Broccoli, Kiwi, Banana, Cashew Nut, Orange, Flaxseeds, Cereals, Onion, Garlic | ** Secoisolariciresinol | ND | |
| ** Matairesional | ND | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gregorio, K.C.R.; Laurindo, C.P.; Machado, U.F. Estrogen and Glycemic Homeostasis: The Fundamental Role of Nuclear Estrogen Receptors ESR1/ESR2 in Glucose Transporter GLUT4 Regulation. Cells 2021, 10, 99. https://doi.org/10.3390/cells10010099
Gregorio KCR, Laurindo CP, Machado UF. Estrogen and Glycemic Homeostasis: The Fundamental Role of Nuclear Estrogen Receptors ESR1/ESR2 in Glucose Transporter GLUT4 Regulation. Cells. 2021; 10(1):99. https://doi.org/10.3390/cells10010099
Chicago/Turabian StyleGregorio, Karen Cristina Rego, Caroline Pancera Laurindo, and Ubiratan Fabres Machado. 2021. "Estrogen and Glycemic Homeostasis: The Fundamental Role of Nuclear Estrogen Receptors ESR1/ESR2 in Glucose Transporter GLUT4 Regulation" Cells 10, no. 1: 99. https://doi.org/10.3390/cells10010099
APA StyleGregorio, K. C. R., Laurindo, C. P., & Machado, U. F. (2021). Estrogen and Glycemic Homeostasis: The Fundamental Role of Nuclear Estrogen Receptors ESR1/ESR2 in Glucose Transporter GLUT4 Regulation. Cells, 10(1), 99. https://doi.org/10.3390/cells10010099