
ACLY Nuclear Translocation in Human Macrophages Drives Proinflammatory Gene Expression by NF-κB Acetylation

 , , , ,
, , , , 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Isolation and Differentiation of Human Monocytes
2.2. Cell Culture and Treatments
2.3. Collection of Human Septic Samples
2.4. Quantitative Real-Time PCR
2.5. Western Blotting
2.6. Transient Transfections and RNA Interference
2.7. Immunocytochemistry
2.8. ACLY Activity
2.9. Cytosol Nucleus Fractionation
2.10. Co-Immunoprecipitation
2.11. ChIP-qPCR and ReChIP-qPCR
2.12. Mass Spectrometry Analysis
2.13. Citrate Lyase 3D Comparative Analyses
2.14. Quantification and Statistical Analysis
3. Results
3.1. Early ACLY Expression and Activity in LPS-Triggered Human Macrophages
3.2. ACLY Translocates from the Cytosol to the Nucleus after LPS Treatment
3.3. Nuclear ACLY Fosters NF-κB Acetylation and Its Activity
3.4. LTA and Sepsis in the Early Hyperinflammatory Phase Trigger ACLY-Mediated NF-κB Acetylation
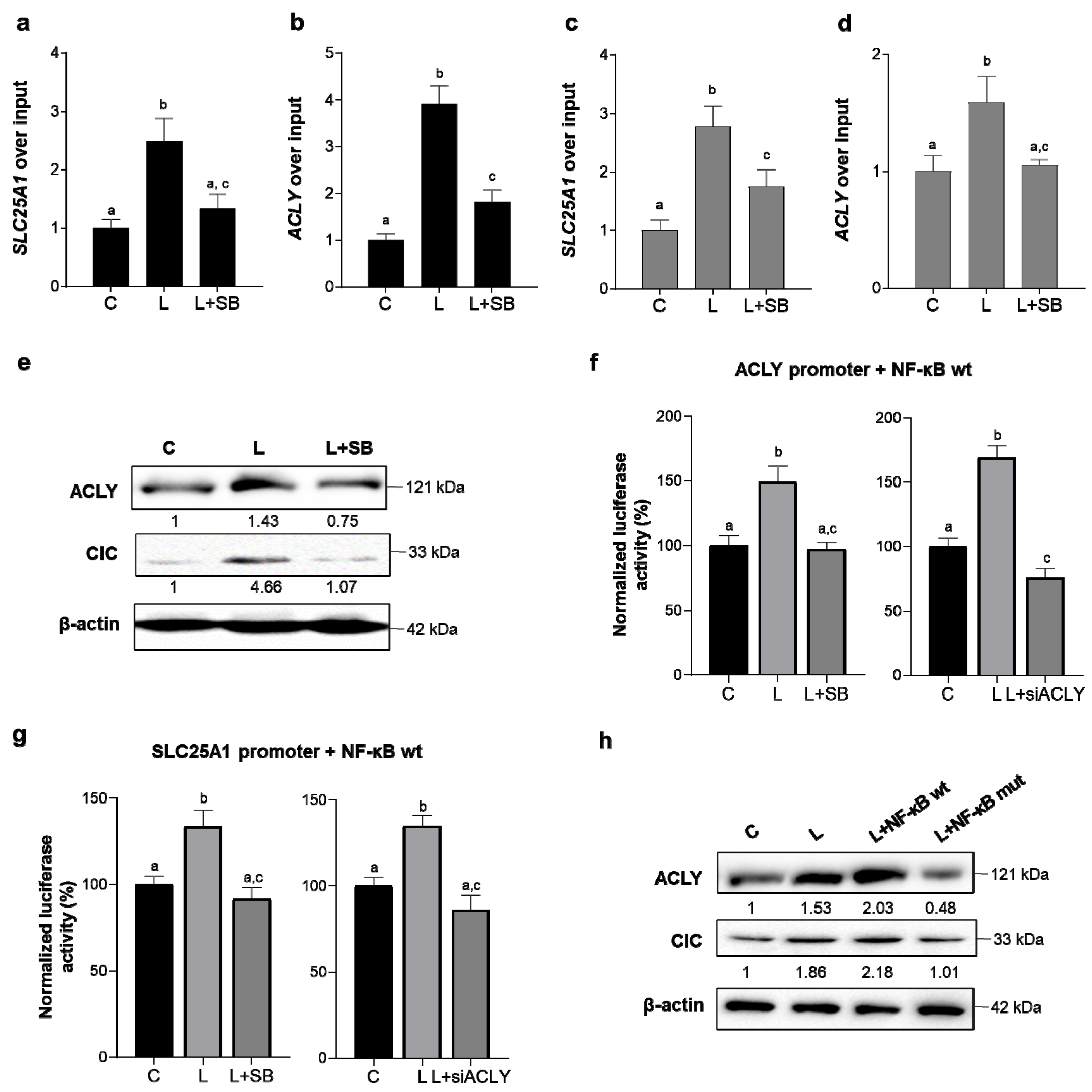
3.5. ACLY-Mediated NF-κB Full Activation Upregulates SLC25A1 and ACLY Genes and Thus Triggers a Positive Self-Regulated Loop
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cai, L.; Tu, B.P. On acetyl-CoA as a gauge of cellular metabolic state. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 195–202. [Google Scholar] [CrossRef]
- Wellen, K.E.; Hatzivassiliou, G.; Sachdeva, U.M.; Bui, T.V.; Cross, J.R.; Thompson, C.B. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 2009, 324, 1076–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauterbach, M.A.; Hanke, J.E.; Serefidou, M.; Mangan, M.S.J.; Kolbe, C.C.; Hess, T.; Rothe, M.; Kaiser, R.; Hoss, F.; Gehlen, J.; et al. Toll-like receptor signaling rewires macrophage metabolism and promotes histone acetylation via ATP-citrate lyase. Immunity 2019, 51, 997–1011. [Google Scholar] [CrossRef] [PubMed]
- Assmann, N.; O’Brien, K.L.; Donnelly, R.P.; Dyck, L.; Zaiatz-Bittencourt, V.; Loftus, R.M.; Heinrich, P.; Oefner, P.J.; Lynch, L.; Gardiner, C.M.; et al. Srebp-controlled glucose metabolism is essential for NK cell functional responses. Nat. Immunol. 2017, 18, 1197–1206. [Google Scholar] [CrossRef] [PubMed]
- Infantino, V.; Iacobazzi, V.; Palmieri, F.; Menga, A. ATP-citrate lyase is essential for macrophage inflammatory response. Biochem. Biophys. Res. Commun. 2013, 440, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Infantino, V.; Pierri, C.L.; Iacobazzi, V. Metabolic routes in inflammation: The citrate pathway and its potential as therapeutic target. Curr. Med. Chem. 2019, 26, 7104–7116. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.C.; Chang, J.H.; Jin, J. Regulation of nuclear factor-κB in autoimmunity. Trends Immunol. 2013, 34, 282–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal. Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayden, M.S.; Ghosh, S. NF-κB in immunobiology. Cell Res. 2011, 21, 223–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.F.; Mu, Y.; Greene, W.C. Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-kappaB. EMBO J. 2002, 21, 6539–6548. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Barozzi, I.; Termanini, A.; Prosperini, E.; Recchiuti, A.; Dalli, J.; Mietton, F.; Matteoli, G.; Hiebert, S.; Natoli, G. Requirement for the histone deacetylase Hdac3 for the inflammatory gene expression program in macrophages. Proc. Natl. Acad. Sci. USA 2012, 109, E2865–E2874. [Google Scholar] [CrossRef] [Green Version]
- Sivanand, S.; Rhoades, S.; Jiang, Q.; Lee, J.V.; Benci, J.; Zhang, J.; Yuan, S.; Viney, I.; Zhao, S.; Carrer, A.; et al. Nuclear Acetyl-CoA production by ACLY promotes homologous recombination. Mol. Cell 2017, 67, 252–265.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santarsiero, A.; Convertini, P.; Vassallo, A.; Santoro, V.; Todisco, S.; Iacobazzi, D.; Fondufe-Mittendorf, Y.; Martelli, G.; de Oliveira, M.R.; Montanaro, R.; et al. Phenolic compounds of red wine. Oxid. Med. Cell Longev. 2021, 2021, 5533793. [Google Scholar]
- Mills, E.L.; Ryan, D.G.; Prag, H.A.; Dikovskaya, D.; Menon, D.; Zaslona, Z.; Jedrychowski, M.P.; Costa, A.S.H.; Higgins, M.; Hams, E.; et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 2018, 556, 113–117. [Google Scholar] [CrossRef]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Infantino, V.; Convertini, P.; Cucci, L.; Panaro, M.A.; Di Noia, M.A.; Calvello, R.; Palmieri, F.; Iacobazzi, V. The mitochondrial citrate carrier: A new player in inflammation. Biochem. J. 2011, 438, 433–436. [Google Scholar] [CrossRef] [Green Version]
- Santarsiero, A.; Onzo, A.; Pascale, R.; Acquavia, M.A.; Coviello, M.; Convertini, P.; Todisco, S.; Marsico, M.; Pifano, C.; Iannece, P.; et al. Pistacia lentiscus Hydrosol: Untargeted Metabolomic Analysis and Anti-Inflammatory Activity Mediated by NF-κB and the Citrate Pathway. Oxid. Med. Cell Longev. 2020, 2020, 4264815. [Google Scholar] [CrossRef] [PubMed]
- Infantino, V.; Dituri, F.; Convertini, P.; Santarsiero, A.; Palmieri, F.; Todisco, S.; Mancarella, S.; Giannelli, G.; Iacobazzi, V. Epigenetic upregulation and functional role of the mitochondrial aspartate/glutamate carrier isoform 1 in hepatocellular carcinoma. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Linn, T.C.; Srere, P.A. Identification of ATP citrate lyase as a phosphoprotein. J. Biol. Chem. 1979, 254, 1691–1698. [Google Scholar] [CrossRef]
- Convertini, P.; Todisco, S.; De Santis, F.; Pappalardo, I.; Iacobazzi, D.; Castiglione Morelli, M.A.; Fondufe-Mittendorf, Y.N.; Martelli, G.; Palmieri, F.; Infantino, V. Transcriptional regulation factors of the human mitochondrial aspartate/glutamate carrier gene, isoform 2 (SLC25A13): USF1 as basal factor and FOXA2 as activator in liver cells. Int. J. Mol. Sci. 2019, 20, 1888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Komakula, A.; Fraser, M.E. Binding of hydroxycitrate to human ATP-citrate lyase. Acta Crystallogr. D Struct. Biol. 2017, 73, 660–671. [Google Scholar] [CrossRef] [PubMed]
- Pierri, C.L.; Parisi, G.; Porcelli, V. Computational approaches for protein function prediction: A combined strategy from multiple sequence alignment to molecular docking-based virtual screening. Biochim. Biophys. Acta 2010, 1804, 1695–1712. [Google Scholar] [CrossRef]
- Verschueren, K.H.G.; Blanchet, C.; Felix, J.; Dansercoer, A.; De Vos, D.; Bloch, Y.; Van Beeumen, J.; Svergun, D.; Gutsche, I.; Savvides, S.N.; et al. Structure of ATP citrate lyase and the origin of citrate synthase in the Krebs cycle. Nature 2019, 568, 571–575. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Joo, K.; Lee, J.; Raman, S.; Thompson, J.; Tyka, M.; Baker, D.; Karplus, K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins 2009, 77 (Suppl. 9), 114–122. [Google Scholar] [CrossRef] [Green Version]
- Panduro, A.; Shalaby, F.; Shafritz, D.A. Changing patterns of transcriptional and post-transcriptional control of liver-specific gene expression during rat development. Genes Dev. 1987, 1, 1172–1182. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Hayden, M.S. New regulators of NF-kappaB in inflammation. Nat. Rev. Immunol. 2008, 8, 837–848. [Google Scholar] [CrossRef]
- Chen, L.F.; Greene, W.C. Shaping the nuclear action of NF-kappaB. Nat. Rev. Mol. Cell Biol. 2004, 5, 392–401. [Google Scholar] [CrossRef]
- Pearce, N.J.; Yates, J.W.; Berkhout, T.A.; Jackson, B.; Tew, D.; Boyd, H.; Camilleri, P.; Sweeney, P.; Gribble, A.D.; Shaw, A.; et al. The role of ATP citrate-lyase in the metabolic regulation of plasma lipids. Hypolipidaemic effects of SB-204990, a lactone prodrug of the potent ATP citrate-lyase inhibitor SB-201076. Biochem. J. 1998, 334, 113–119. [Google Scholar] [CrossRef]
- Yamamoto, K.; Arakawa, T.; Ueda, N.; Yamamoto, S. Transcriptional roles of nuclear factor kappa B and nuclear factor-interleukin-6 in the tumor necrosis factor alpha-dependent induction of cyclooxygenase-2 in MC3T3-E1 cells. J. Biol. Chem. 1995, 270, 31315–31320. [Google Scholar] [CrossRef] [Green Version]
- Hiscott, J.; Marois, J.; Garoufalis, J.; D’Addario, M.; Roulston, A.; Kwan, I.; Pepin, N.; Lacoste, J.; Nguyen, H.; Bensi, G. Characterization of a functional NF-kappa B site in the human interleukin 1 beta promoter: Evidence for a positive autoregulatory loop. Mol. Cell Biol. 1993, 13, 6231–6240. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Tee, W.W. Enhancers and chromatin structures: Regulatory hubs in gene expression and diseases. Biosci. Rep. 2017, 37, BSR20160183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimura, A.; Lien, E.; Ingalls, R.R.; Tuomanen, E.; Dziarski, R.; Golenbock, D. Cutting edge: Recognition of Gram-positive bacterial cell wall components by the innate immune system occurs via Toll-like receptor 2. J. Immunol. 1999, 163, 1–5. [Google Scholar]
- Rahman, M.M.; McFadden, G. Modulation of NF-κB signalling by microbial pathogens. Nat. Rev. Microbiol. 2011, 9, 291–306. [Google Scholar] [CrossRef] [PubMed]
- Böhrer, H.; Qiu, F.; Zimmermann, T.; Zhang, Y.; Jllmer, T.; Männel, D.; Böttiger, B.W.; Stern, D.M.; Waldherr, R.; Saeger, H.D. and others. Role of NFkappaB in the mortality of sepsis. J. Clin. Investig. 1997, 100, 972–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.F.; Malik, A.B. NF-kappa B activation as a pathological mechanism of septic shock and inflammation. Am. J. Physiol. Lung. Cell Mol. Physiol. 2006, 290, L622–L645. [Google Scholar] [CrossRef]
- Iacobazzi, V.; Infantino, V. Citrate—New functions for an old metabolite. Biol. Chem. 2014, 395, 387–399. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Mantovani, A. Orchestration of metabolism by macrophages. Cell Metab. 2012, 15, 432–437. [Google Scholar] [CrossRef] [Green Version]
- Palsson-McDermott, E.M.; Curtis, A.M.; Goel, G.; Lauterbach, M.A.; Sheedy, F.J.; Gleeson, L.E.; van den Bosch, M.W.; Quinn, S.R.; Domingo-Fernandez, R.; Johnston, D.G.; et al. Pyruvate kinase M2 regulates Hif-1α activity and IL-1β induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. 2015, 21, 65–80. [Google Scholar] [CrossRef] [Green Version]
- Hooftman, A.; Angiari, S.; Hester, S.; Corcoran, S.E.; Runtsch, M.C.; Ling, C.; Ruzek, M.C.; Slivka, P.F.; McGettrick, A.F.; Banahan, K.; et al. The immunomodulatory metabolite itaconate modifies NLRP3 and inhibits inflammasome activation. Cell Metab. 2020, 32, 468–478.e7. [Google Scholar] [CrossRef] [PubMed]
- Vassallo, A.; Santoro, V.; Pappalardo, I.; Santarsiero, A.; Convertini, P.; De Luca, M.; Martelli, G.; Infantino, V.; Caddeo, C. Liposome-mediated inhibition of inflammation by hydroxycitrate. Nanomaterials 2020, 10, 2080. [Google Scholar] [CrossRef]
- Galván-Peña, S.; Carroll, R.G.; Newman, C.; Hinchy, E.C.; Palsson-McDermott, E.; Robinson, E.K.; Covarrubias, S.; Nadin, A.; James, A.M.; Haneklaus, M.; et al. Malonylation of GAPDH is an inflammatory signal in macrophages. Nat. Commun. 2019, 10, 338. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.; Li, C.; Xiao, S.; Tang, Y.; Huang, J.; Zhao, S.; Li, X.; Li, J.; Zhang, R.; Yu, W. Acetylation promotes TyrRS nuclear translocation to prevent oxidative damage. Proc. Natl. Acad. Sci. USA 2017, 114, 687–692. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Peng, L.; Seto, E.; Huang, S.; Qiu, Y. Modulation of histone deacetylase 6 (HDAC6) nuclear import and tubulin deacetylase activity through acetylation. J. Biol. Chem. 2012, 287, 29168–29174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, R.; Tao, R.; Gao, X.; Li, T.; Zhou, X.; Guan, K.L.; Xiong, Y.; Lei, Q.Y. Acetylation stabilizes ATP-citrate lyase to promote lipid biosynthesis and tumor growth. Mol. Cell 2013, 51, 506–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baardman, J.; Verberk, S.G.S.; van der Velden, S.; Gijbels, M.J.J.; van Roomen, C.P.P.A.; Sluimer, J.C.; Broos, J.Y.; Griffith, G.R.; Prange, K.H.M.; van Weeghel, M.; et al. Macrophage ATP citrate lyase deficiency stabilizes atherosclerotic plaques. Nat. Commun. 2020, 11, 6296. [Google Scholar] [CrossRef]
- Tak, P.P.; Firestein, G.S. NF-kappaB: A key role in inflammatory diseases. J. Clin. Investig. 2001, 107, 7–11. [Google Scholar] [CrossRef]
- Danning, C.L.; Illei, G.G.; Hitchon, C.; Greer, M.R.; Boumpas, D.T.; McInnes, I.B. Macrophage-derived cytokine and nuclear factor kappaB p65 expression in synovial membrane and skin of patients with psoriatic arthritis. Arthritis Rheum. 2000, 43, 1244–1256. [Google Scholar] [CrossRef]
- Kang, L.J.; Kwon, E.S.; Lee, K.M.; Cho, C.; Lee, J.I.; Ryu, Y.B.; Youm, T.H.; Jeon, J.; Cho, M.R.; Jeong, S.Y.; et al. 3′-Sialyllactose as an inhibitor of p65 phosphorylation ameliorates the progression of experimental rheumatoid arthritis. Br. J. Pharmacol. 2018, 175, 4295–4309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kircheis, R.; Haasbach, E.; Lueftenegger, D.; Heyken, W.T.; Ocker, M.; Planz, O. NF-κB Pathway as a Potential target for treatment of critical stage COVID-19 patients. Front. Immunol. 2020, 11, 598444. [Google Scholar] [CrossRef]
- Guisado-Vasco, P.; Cano-Megías, M.; Rodríguez-López, M.; de-Luna-Boquera, I.M.; Carnevali-Ruiz, D.; Immunosuppressants Against COVID-19 Working Team. COVID-19 and Metabolic Syndrome: NF-κB Activation. Crossroads. Trends Endocrinol. Metab. 2020, 31, 802–803. [Google Scholar] [CrossRef]
- Chen, L.F.; Fischle, W.; Verdin, E.; Greene, W.C. Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science 2001, 293, 1653–1657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishinaga, H.; Jono, H.; Lim, J.H.; Kweon, S.M.; Xu, H.; Ha, U.H.; Koga, T.; Yan, C.; Feng, X.H.; Chen, L.F.; et al. TGF-beta induces p65 acetylation to enhance bacteria-induced NF-kappaB activation. EMBO J. 2007, 26, 1150–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arts, R.J.; Gresnigt, M.S.; Joosten, L.A.; Netea, M.G. Cellular metabolism of myeloid cells in sepsis. J. Leukoc. Biol. 2017, 101, 151–164. [Google Scholar] [CrossRef] [Green Version]
- Badshah, S.L.; Faisal, S.; Muhammad, A.; Poulson, B.G.; Emwas, A.H.; Jaremko, M. Antiviral activities of flavonoids. Biomed. Pharmacother. 2021, 140, 111596. [Google Scholar] [CrossRef] [PubMed]
- Badshah, S.L.; Riaz, A.; Muhammad, A.; Tel Çayan, G.; Çayan, F.; Emin Duru, M.; Ahmad, N.; Emwas, A.H.; Jaremko, M. Isolation, characterization, and medicinal potential of polysaccharides of Morchella esculenta. Molecules 2021, 26, 1459. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Gordon, S.; Locati, M.; Mantovani, A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: New molecules and patterns of gene expression. J. Immunol. 2006, 177, 7303–7311. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pt | Source of Infection | Age | Sex | WBC (mmc) | Neutrophils (mmc) | CRP (mg/L) | Lactate (mmol/L) | PCT (ng/µL) | PLT (mmc) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Uroseptic syndrome with positive blood and urine culture for E. coli; multidrug resistant | 53 | M | 18.9 | 17.5 | 345 (<5) | 4.5 (0.5–2.2) | 6 (0–0.5) | 140.000 |
| 2 | Abdominal sepsis caused by perforated diverticulitis; negative blood culture | 73 | F | 21.3 | 18.5 | 240 (<5) | 4.8 (0.5–2.2) | 5 (0–0.5) | 170.000 |
| 3 | BSI (blood stream infection) caused by Klebsiella pneumoniae carbapenemase producer (KPC) | 78 | M | 17.4 | 14.5 | 85 (<5) | 5.1 (0.5–2.2) | 14 (0–0.5) | 120.000 |
| 4 | Pyonephrosis with a positive culture for VRE (Enterococcus faecium vancomycin resistant) from the nephrostomy sample | 59 | M | 13.4 | 11.5 | 157 (<5) | 5.1 (0.5–2.2) | 10 (0–0.5) | 120.000 |
| 5 | Methicillin-resistant Staphylococcus aureus (MRSA) bacteriemia | 20 | F | 18.2 | 15.5 | 253 (<5) | 5.1 (0.5–2.2) | 16 (0–0.5) | 190.000 |
| 6 | Methicillin susceptible Staphylococcus aureus (MSSA) prosthetic joint infection | 76 | M | 22.3 | 18.5 | 138 (<5) | 5.1 (0.5–2.2) | 8 (0–0.5) | 190.000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santarsiero, A.; Convertini, P.; Todisco, S.; Pierri, C.L.; De Grassi, A.; Williams, N.C.; Iacobazzi, D.; De Stefano, G.; O’Neill, L.A.J.; Infantino, V. ACLY Nuclear Translocation in Human Macrophages Drives Proinflammatory Gene Expression by NF-κB Acetylation. Cells 2021, 10, 2962. https://doi.org/10.3390/cells10112962
Santarsiero A, Convertini P, Todisco S, Pierri CL, De Grassi A, Williams NC, Iacobazzi D, De Stefano G, O’Neill LAJ, Infantino V. ACLY Nuclear Translocation in Human Macrophages Drives Proinflammatory Gene Expression by NF-κB Acetylation. Cells. 2021; 10(11):2962. https://doi.org/10.3390/cells10112962
Chicago/Turabian StyleSantarsiero, Anna, Paolo Convertini, Simona Todisco, Ciro L. Pierri, Anna De Grassi, Niamh C. Williams, Dominga Iacobazzi, Giulio De Stefano, Luke A. J. O’Neill, and Vittoria Infantino. 2021. "ACLY Nuclear Translocation in Human Macrophages Drives Proinflammatory Gene Expression by NF-κB Acetylation" Cells 10, no. 11: 2962. https://doi.org/10.3390/cells10112962
APA StyleSantarsiero, A., Convertini, P., Todisco, S., Pierri, C. L., De Grassi, A., Williams, N. C., Iacobazzi, D., De Stefano, G., O’Neill, L. A. J., & Infantino, V. (2021). ACLY Nuclear Translocation in Human Macrophages Drives Proinflammatory Gene Expression by NF-κB Acetylation. Cells, 10(11), 2962. https://doi.org/10.3390/cells10112962