
Ferroptosis: Cancer Stem Cells Rely on Iron until “to Die for” It


Abstract
:1. Introduction
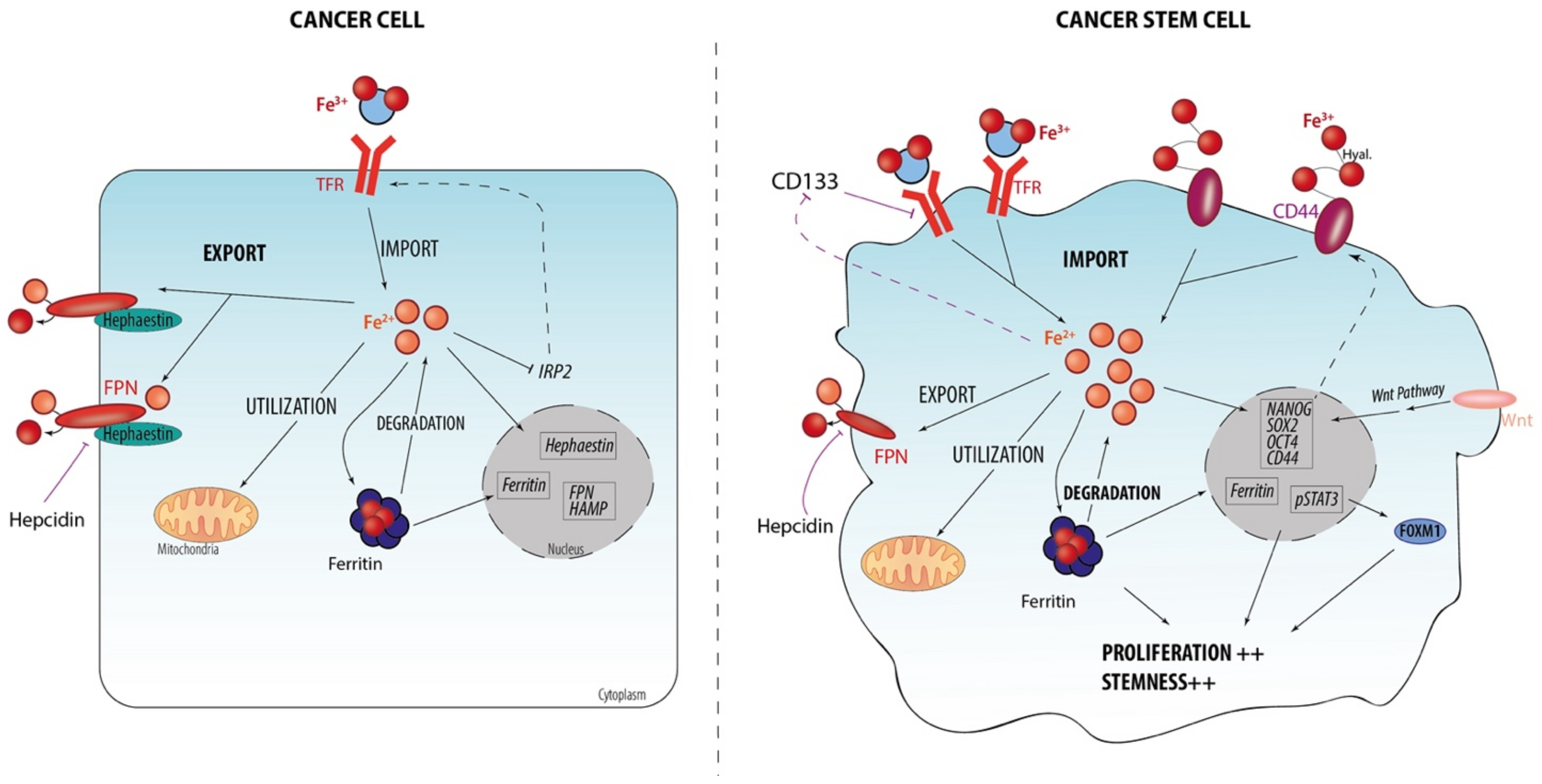
2. Iron and CSC
2.1. Iron Homeostasis at Cellular Level
2.2. Iron Metabolism Dysregulation as a Hallmark of CSCs
2.3. Iron-Related Stemness Features/Markers
2.4. Iron Regulation and Stemness Behaviors
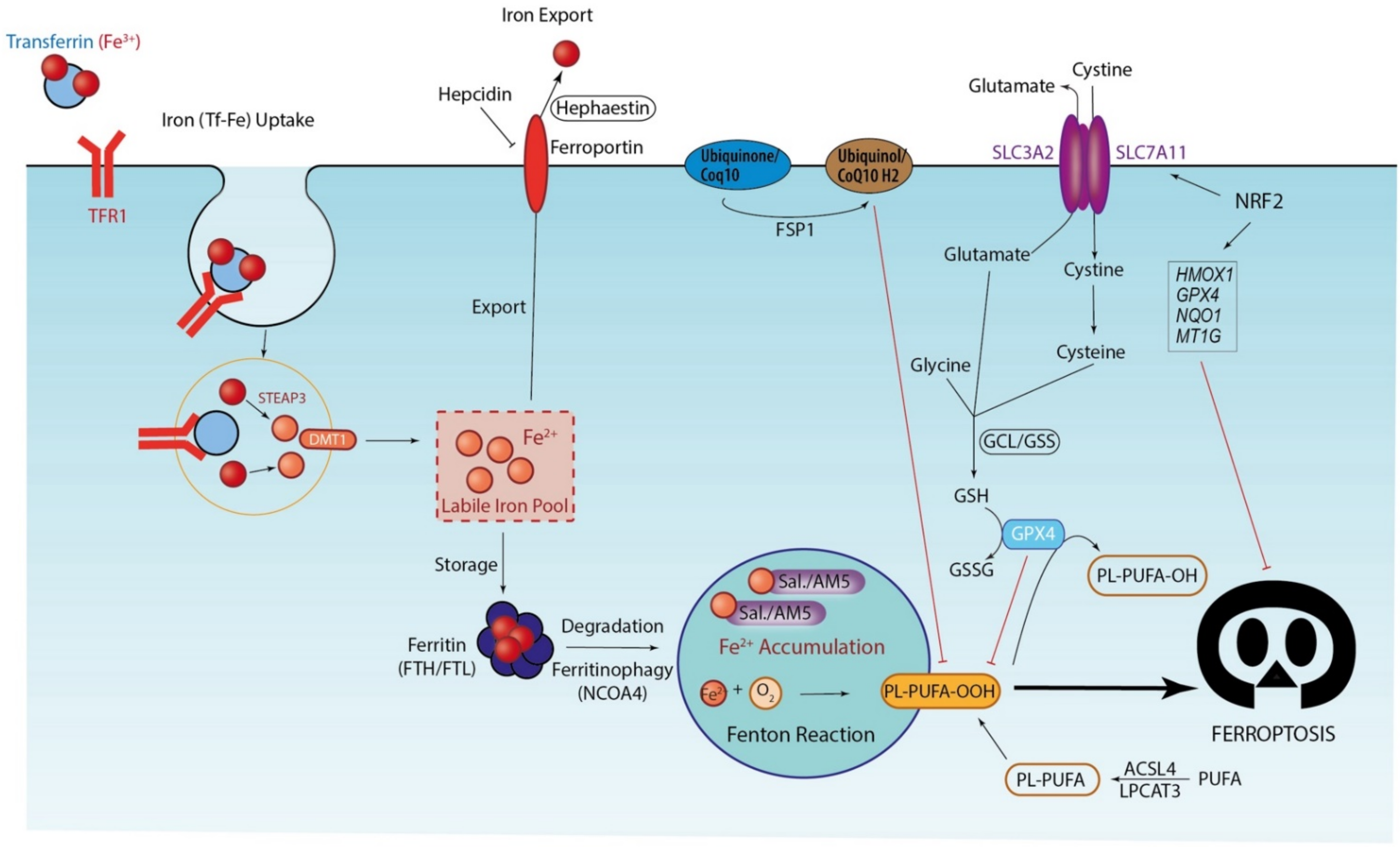
3. Ferroptosis, Iron-Driven Cell Death
3.1. Iron Accumulation and Lipid Peroxidation: Drivers of Ferroptosis Execution
3.2. Antioxidant Systems: The Last Defense before Ferroptosis Execution
4. Autophagy and Ferroptosis Regulation
4.1. Ferritinophagy: Drivers of Ferroptosis Initiation
4.2. Autophagy: A Dual Role in Ferroptosis Execution
5. Therapeutic Strategies to Target Ferroptosis in CSCs/Targeting Ferroptosis in CSCs
5.1. Through Manipulating Tumor-Suppressor p53
5.2. Through the Use of Ferroptosis Inducers Now Available
6. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Torti, S.V.; Torti, F.M. Iron: The cancer connection. Mol. Asp. Med. 2020, 75, 100860. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, F. Iron homeostasis and tumorigenesis: Molecular mechanisms and therapeutic opportunities. Protein Cell 2015, 6, 88–100. [Google Scholar] [CrossRef] [Green Version]
- Muñoz, M.; Gómez-Ramírez, S.; Martín-Montañez, E.; Auerbach, M. Perioperative anemia management in colorectal cancer patients: A pragmatic approach. World J. Gastroenterol. 2014, 20, 1972–1985. [Google Scholar] [CrossRef]
- Pantopoulos, K.; Porwal, S.K.; Tartakoff, A.; Devireddy, L. Mechanisms of mammalian iron homeostasis. Biochemistry 2012, 51, 5705–5724. [Google Scholar] [CrossRef]
- Ganz, T.; Nemeth, E. Hepcidin and iron homeostasis. Biochim. Biophys. Acta 2012, 1823, 1434–1443. [Google Scholar] [CrossRef] [Green Version]
- Manz, D.H.; Blanchette, N.L.; Paul, B.T.; Torti, F.M.; Torti, S.V. Iron and cancer: Recent insights. Ann. N. Y. Acad. Sci. 2016, 1368, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A Red Carpet for Iron Metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef] [Green Version]
- Walcher, L.; Kistenmacher, A.-K.; Suo, H.; Kitte, R.; Dluczek, S.; Strauß, A.; Blaudszun, A.-R.; Yevsa, T.; Fricke, S.; Kossatz-Boehlert, U. Cancer Stem Cells—Origins and Biomarkers: Perspectives for Targeted Personalized Therapies. Front. Immunol. 2020, 11, 1280. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, S.; Solanki, M.; Spitschak, A.; Vera, J.; Pützer, B.M. Emerging functional markers for cancer stem cell-based therapies: Understanding signaling networks for targeting metastasis. Semin. Cancer Biol. 2018, 53, 90–109. [Google Scholar] [CrossRef]
- Müller, S.; Sindikubwabo, F.; Cañeque, T.; Lafon, A.; Versini, A.; Lombard, B.; Loew, D.; Wu, T.-D.; Ginestier, C.; Charafe-Jauffret, E.; et al. CD44 regulates epigenetic plasticity by mediating iron endocytosis. Nat. Chem. 2020, 12, 929–938. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomita, H.; Tanaka, K.; Tanaka, T.; Hara, A. Aldehyde dehydrogenase 1A1 in stem cells and cancer. Oncotarget 2016, 7, 11018–11032. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.; Gasparetto, M.; Humphries, K.; Pollyea, D.A.; Vasiliou, V.; Jordan, C.T. Aldehyde dehydrogenases in acute myeloid leukemia: AML and ALDHs. Ann. N. Y. Acad. Sci. 2014, 1310, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Blume, R.; Rempel, E.; Manta, L.; Saeed, B.R.; Wang, W.; Raffel, S.; Ermakova, O.; Eckstein, V.; Benes, V.; Trumpp, A.; et al. The molecular signature of AML with increased ALDH activity suggests a stem cell origin. Leuk. Lymphoma. 2018, 59, 2201–2210. [Google Scholar] [CrossRef]
- Rasper, M.; Schafer, A.; Piontek, G.; Teufel, J.; Brockhoff, G.; Ringel, F.; Heindl, S.; Zimmer, C.; Schlegel, J. Aldehyde dehydrogenase 1 positive glioblastoma cells show brain tumor stem cell capacity. Neuro-Oncology 2010, 12, 1024–1033. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Dallaglio, K.; Chen, Y.; Robinson, W.A.; Robinson, S.E.; McCarter, M.D.; Wang, J.; Gonzalez, R.; Thompson, D.C.; Norris, D.A.; et al. ALDH1A Isozymes are Markers of Human Melanoma Stem Cells and Potential Therapeutic Targets: ALDH + Cancer Stem Cells in Human Melanoma. Stem Cells 2012, 30, 2100–2113. [Google Scholar] [CrossRef] [Green Version]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 Is a Marker of Normal and Malignant Human Mammary Stem Cells and a Predictor of Poor Clinical Outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef] [Green Version]
- Bourseau-Guilmain, E.; Griveau, A.; Benoit, J.-P.; Garcion, E. The importance of the stem cell marker prominin-1/CD133 in the uptake of transferrin and in iron metabolism in human colon cancer Caco-2 cells. PLoS ONE 2011, 6, e25515. [Google Scholar] [CrossRef] [Green Version]
- Glumac, P.M.; LeBeau, A.M. The role of CD133 in cancer: A concise review. Clin. Transl. Med. 2018, 7, e18. [Google Scholar] [CrossRef]
- Wang, Y.-C.; Yo, Y.-T.; Lee, H.-Y.; Liao, Y.-P.; Chao, T.-K.; Su, P.-H.; Lai, H.-C. ALDH1-bright epithelial ovarian cancer cells are associated with CD44 expression, drug resistance, and poor clinical outcome. Am. J. Pathol. 2012, 180, 1159–1169. [Google Scholar] [CrossRef]
- Collins, A.T.; Berry, P.A.; Hyde, C.; Stower, M.J.; Maitland, N.J. Prospective Identification of Tumorigenic Prostate Cancer Stem Cells. Cancer Res. 2005, 65, 10946–10951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schatton, T.; Murphy, G.F.; Frank, N.Y.; Yamaura, K.; Waaga-Gasser, A.M.; Gasser, M.; Zhan, Q.; Jordan, S.; Duncan, L.M.; Weishaupt, C.; et al. Identification of cells initiating human melanomas. Nature 2008, 451, 345–349. [Google Scholar] [CrossRef] [Green Version]
- Dalerba, P.; Dylla, S.J.; Park, I.-K.; Liu, R.; Wang, X.; Cho, R.W.; Hoey, T.; Gurney, A.; Huang, E.H.; Simeone, D.M.; et al. Phenotypic characterization of human colorectal cancer stem cells. Proc. Natl. Acad. Sci. USA 2007, 104, 10158–10163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prince, M.E.; Sivanandan, R.; Kaczorowski, A.; Wolf, G.T.; Kaplan, M.J.; Dalerba, P.; Weissman, I.L.; Clarke, M.F.; Ailles, L.E. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 2007, 104, 973–978. [Google Scholar] [CrossRef] [Green Version]
- Okano, M.; Konno, M.; Kano, Y.; Kim, H.; Kawamoto, K.; Ohkuma, M.; Haraguchi, N.; Yokobori, T.; Mimori, K.; Yamamoto, H.; et al. Human colorectal CD24+ cancer stem cells are susceptible to epithelial-mesenchymal transition. Int. J. Oncol. 2014, 45, 575–580. [Google Scholar] [CrossRef]
- Kemper, K.; Grandela, C.; Medema, J.P. Molecular identification and targeting of colorectal cancer stem cells. Oncotarget 2010, 1, 387–395. [Google Scholar] [CrossRef] [Green Version]
- Dana, H. CD166 as a Stem Cell Marker? A Potential Target for Therapy Colorectal Cancer? JSRT 2016, 1, 226–229. [Google Scholar] [CrossRef]
- Zakaria, N.; Yusoff, N.M.; Zakaria, Z.; Lim, M.N.; Baharuddin, P.J.N.; Fakiruddin, K.S.; Yahaya, B. Human non-small cell lung cancer expresses putative cancer stem cell markers and exhibits the transcriptomic profile of multipotent cells. BMC Cancer 2015, 15, 84. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Yu, D.; Zhang, H.; He, H.; Zhang, C.; Zhao, W.; Shao, R. CD133 + EpCAM + Phenotype Possesses More Characteristics of Tumor Initiating Cells in Hepatocellular Carcinoma Huh7 Cells. Int. J. Biol. Sci. 2012, 8, 992–1004. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of Pancreatic Cancer Stem Cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef] [Green Version]
- Saini, V.; Hose, C.D.; Monks, A.; Nagashima, K.; Han, B.; Newton, D.L.; Millione, A.; Shah, J.; Hollingshead, M.G.; Hite, K.M.; et al. Identification of CBX3 and ABCA5 as Putative Biomarkers for Tumor Stem Cells in Osteosarcoma. PLoS ONE 2012, 7, e41401. [Google Scholar] [CrossRef]
- Kemper, K.; Sprick, M.R.; de Bree, M.; Scopelliti, A.; Vermeulen, L.; Hoek, M.; Zeilstra, J.; Pals, S.T.; Mehmet, H.; Stassi, G.; et al. The AC133 Epitope, but not the CD133 Protein, Is Lost upon Cancer Stem Cell Differentiation. Cancer Res. 2010, 70, 719–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.F.; Ho, D.W.; Ng, M.N.; Lau, C.K.; Yu, W.C.; Ngai, P.; Chu, P.W.K.; Lam, C.T.; Poon, R.T.P.; Fan, S.T. Significance of CD90+ cancer stem cells in human liver cancer. Cancer Cell 2008, 13, 153–166. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- Kong, Y.; Yoshida, S.; Saito, Y.; Doi, T.; Nagatoshi, Y.; Fukata, M.; Saito, N.; Yang, S.M.; Iwamoto, C.; Okamura, J.; et al. CD34+CD38+CD19+ as well as CD34+CD38−CD19+ cells are leukemia-initiating cells with self-renewal capacity in human B-precursor ALL. Leukemia 2008, 22, 1207–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drakesmith, H.; Nemeth, E.; Ganz, T. Ironing out Ferroportin. Cell Metab. 2015, 22, 777–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vela, D.; Vela-Gaxha, Z. Differential regulation of hepcidin in cancer and non-cancer tissues and its clinical implications. Exp. Mol. Med. 2018, 50, e436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schonberg, D.L.; Miller, T.E.; Wu, Q.; Flavahan, W.A.; Das, N.K.; Hale, J.S.; Hubert, C.G.; Mack, S.C.; Jarrar, A.M.; Karl, R.T.; et al. Preferential Iron Trafficking Characterizes Glioblastoma Stem-like Cells. Cancer Cell 2015, 28, 441–455. [Google Scholar] [CrossRef] [Green Version]
- Mai, T.T.; Hamaï, A.; Hienzsch, A.; Cañeque, T.; Müller, S.; Wicinski, J.; Cabaud, O.; Leroy, C.; David, A.; Acevedo, V.; et al. Salinomycin kills cancer stem cells by sequestering iron in lysosomes. Nat. Chem. 2017, 9, 1025–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamaï, A.; Cañeque, T.; Müller, S.; Mai, T.T.; Hienzsch, A.; Ginestier, C.; Charafe-Jauffret, E.; Codogno, P.; Mehrpour, M.; Rodriguez, R. An iron hand over cancer stem cells. Autophagy 2017, 13, 1465–1466. [Google Scholar] [CrossRef]
- Chanvorachote, P.; Luanpitpong, S. Iron induces cancer stem cells and aggressive phenotypes in human lung cancer cells. Am. J. Physiol. Cell Physiol. 2016, 310, C728–C739. [Google Scholar] [CrossRef] [Green Version]
- Rychtarcikova, Z.; Lettlova, S.; Tomkova, V.; Korenkova, V.; Langerova, L.; Simonova, E.; Zjablovskaja, P.; Alberich-Jorda, M.; Neuzil, J.; Truksa, J. Tumor-initiating cells of breast and prostate origin show alterations in the expression of genes related to iron metabolism. Oncotarget 2017, 8, 6376–6398. [Google Scholar] [CrossRef] [Green Version]
- Raggi, C.; Gammella, E.; Correnti, M.; Buratti, P.; Forti, E.; Andersen, J.B.; Alpini, G.; Glaser, S.; Alvaro, D.; Invernizzi, P.; et al. Dysregulation of Iron Metabolism in Cholangiocarcinoma Stem-like Cells. Sci. Rep. 2017, 7, 17667. [Google Scholar] [CrossRef] [Green Version]
- Blanchette-Farra, N.; Kita, D.; Konstorum, A.; Tesfay, L.; Lemler, D.; Hegde, P.; Claffey, K.P.; Torti, F.M.; Torti, S.V. Contribution of three-dimensional architecture and tumor-associated fibroblasts to hepcidin regulation in breast cancer. Oncogene 2018, 37, 4013–4032. [Google Scholar] [CrossRef] [PubMed]
- Bootcov, M.R.; Bauskin, A.R.; Valenzuela, S.M.; Moore, A.G.; Bansal, M.; He, X.Y.; Zhang, H.P.; Donnellan, M.; Mahler, S.; Pryor, K.; et al. MIC-1, a novel macrophage inhibitory cytokine, is a divergent member of the TGF-beta superfamily. Proc. Natl. Acad. Sci. USA 1997, 94, 11514–11519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.J.; Lee, H.; Lee, J.-H. Macrophage inhibitory cytokine-1 transactivates ErbB family receptors via the activation of Src in SK-BR-3 human breast cancer cells. BMB Rep. 2010, 43, 91–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasahara, A.; Tominaga, K.; Nishimura, T.; Yano, M.; Kiyokawa, E.; Noguchi, M.; Noguchi, M.; Kanauchi, H.; Ogawa, T.; Minato, H.; et al. An autocrine/paracrine circuit of growth differentiation factor (GDF) 15 has a role for maintenance of breast cancer stem-like cells. Oncotarget 2017, 8, 24869–24881. [Google Scholar] [CrossRef]
- Basuli, D.; Tesfay, L.; Deng, Z.; Paul, B.; Yamamoto, Y.; Ning, G.; Xian, W.; McKeon, F.; Lynch, M.; Crum, C.P.; et al. Iron addiction: A novel therapeutic target in ovarian cancer. Oncogene 2017, 36, 4089–4099. [Google Scholar] [CrossRef] [Green Version]
- Song, S.; Christova, T.; Perusini, S.; Alizadeh, S.; Bao, R.-Y.; Miller, B.W.; Hurren, R.; Jitkova, Y.; Gronda, M.; Isaac, M.; et al. Wnt inhibitor screen reveals iron dependence of β-catenin signaling in cancers. Cancer Res. 2011, 71, 7628–7639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doyard, M.; Fatih, N.; Monnier, A.; Island, M.L.; Aubry, M.; Leroyer, P.; Bouvet, R.; Chalès, G.; Mosser, J.; Loréal, O.; et al. Iron excess limits HHIPL-2 gene expression and decreases osteoblastic activity in human MG-63 cells. Osteoporos. Int. 2012, 23, 2435–2445. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct. Target. Ther. 2020, 5, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, N.R.; Murray, J.I.; Watson, P.H. Oncostatin-M promotes phenotypic changes associated with mesenchymal and stem cell-like differentiation in breast cancer. Oncogene 2014, 33, 1485–1494. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Yao, Q.; Liu, Y.; Du, S.; Liu, A.; Guo, Z.; Sun, A.; Ruan, J.; Chen, L.; Ye, C.; et al. IL-6-induced epithelial-mesenchymal transition promotes the generation of breast cancer stem-like cells analogous to mammosphere cultures. Int. J. Oncol. 2012, 40, 1171–1179. [Google Scholar] [CrossRef] [Green Version]
- Marotta, L.L.C.; Almendro, V.; Marusyk, A.; Shipitsin, M.; Schemme, J.; Walker, S.R.; Bloushtain-Qimron, N.; Kim, J.J.; Choudhury, S.A.; Maruyama, R.; et al. The JAK2/STAT3 signaling pathway is required for growth of CD44+CD24− stem cell-like breast cancer cells in human tumors. J. Clin. Investig. 2011, 121, 2723–2735. [Google Scholar] [CrossRef]
- Guo, L.; Chen, C.; Shi, M.; Wang, F.; Chen, X.; Diao, D.; Hu, M.; Yu, M.; Qian, L.; Guo, N. Stat3-coordinated Lin-28-let-7-HMGA2 and miR-200-ZEB1 circuits initiate and maintain oncostatin M-driven epithelial-mesenchymal transition. Oncogene 2013, 32, 5272–5282. [Google Scholar] [CrossRef]
- Lobello, N.; Biamonte, F.; Pisanu, M.E.; Faniello, M.C.; Jakopin, Ž.; Chiarella, E.; Giovannone, E.D.; Mancini, R.; Ciliberto, G.; Cuda, G.; et al. Ferritin heavy chain is a negative regulator of ovarian cancer stem cell expansion and epithelial to mesenchymal transition. Oncotarget 2016, 7, 62019–62033. [Google Scholar] [CrossRef] [Green Version]
- Samimi, A.; Khodayar, M.J.; Alidadi, H.; Khodadi, E. The Dual Role of ROS in Hematological Malignancies: Stem Cell Protection and Cancer Cell Metastasis. Stem. Cell Rev. Rep. 2020, 16, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, C.I.; Suda, T. Regulation of reactive oxygen species in stem cells and cancer stem cells. J. Cell Physiol. 2012, 227, 421–430. [Google Scholar] [CrossRef]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef] [PubMed]
- Arnold, C.R.; Mangesius, J.; Skvortsova, I.-I.; Ganswindt, U. The Role of Cancer Stem Cells in Radiation Resistance. Front. Oncol. 2020, 10, 164. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.S.; Kang, M.J.; Cho, Y.M. Low production of reactive oxygen species and high DNA repair: Mechanism of radioresistance of prostate cancer stem cells. Anticancer Res. 2013, 33, 4469–4474. [Google Scholar]
- Scaramuzzino, L.; Lucchino, V.; Scalise, S.; Lo Conte, M.; Zannino, C.; Sacco, A.; Biamonte, F.; Parrotta, E.I.; Costanzo, F.S.; Cuda, G. Uncovering the Metabolic and Stress Responses of Human Embryonic Stem Cells to FTH1 Gene Silencing. Cells 2021, 10, 2431. [Google Scholar] [CrossRef]
- Zolea, F.; Battaglia, A.M.; Chiarella, E.; Malanga, D.; Marco, C.D.; Bond, H.M.; Morrone, G.; Costanzo, F.; Biamonte, F. Ferritin Heavy Subunit Silencing Blocks the Erythroid Commitment of K562 Cells via miR-150 up-Regulation and GATA-1 Repression. Int. J. Mol. Sci. 2017, 18, 2167. [Google Scholar] [CrossRef] [Green Version]
- Salatino, A.; Aversa, I.; Battaglia, A.M.; Sacco, A.; Di Vito, A.; Santamaria, G.; Chirillo, R.; Veltri, P.; Tradigo, G.; Di Cello, A.; et al. H-Ferritin Affects Cisplatin-Induced Cytotoxicity in Ovarian Cancer Cells through the Modulation of ROS. Oxidative Med. Cell. Longev. 2019, 2019, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.Ng.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aversa, I.; Zolea, F.; Ieranò, C.; Bulotta, S.; Trotta, A.M.; Faniello, M.C.; De Marco, C.; Malanga, D.; Biamonte, F.; Viglietto, G.; et al. Epithelial-to-mesenchymal transition in FHC-silenced cells: The role of CXCR4/CXCL12 axis. J. Exp. Clin. Cancer Res. 2017, 36, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Luo, C.; Mines, M.; Zhang, J.; Fan, G.-H. Chemokine CXCL12 Induces Binding of Ferritin Heavy Chain to the Chemokine Receptor CXCR4, Alters CXCR4 Signaling, and Induces Phosphorylation and Nuclear Translocation of Ferritin Heavy Chain. J. Biol. Chem. 2006, 281, 37616–37627. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.-H.; Tian, H.-Y.; Gao, X.; Lei, W.-W.; Hu, Y.; Wang, D.-M.; Pan, X.-C.; Yu, M.-L.; Xu, G.-J.; Zhao, F.-K.; et al. Ferritin Heavy Chain–Mediated Iron Homeostasis and Subsequent Increased Reactive Oxygen Species Production Are Essential for Epithelial-Mesenchymal Transition. Cancer Res. 2009, 69, 5340–5348. [Google Scholar] [CrossRef] [Green Version]
- Chirillo, R.; Aversa, I.; Di Vito, A.; Salatino, A.; Battaglia, A.M.; Sacco, A.; Di Sanzo, M.A.; Faniello, M.C.; Quaresima, B.; Palmieri, C.; et al. FtH-Mediated ROS Dysregulation Promotes CXCL12/CXCR4 Axis Activation and EMT-Like Trans-Differentiation in Erythroleukemia K562 Cells. Front. Oncol. 2020, 10, 698. [Google Scholar] [CrossRef]
- Gong, A.-H.; Wei, P.; Zhang, S.; Yao, J.; Yuan, Y.; Zhou, A.-D.; Lang, F.F.; Heimberger, A.B.; Rao, G.; Huang, S. FoxM1 Drives a Feed-Forward STAT3-Activation Signaling Loop That Promotes the Self-Renewal and Tumorigenicity of Glioblastoma Stem-like Cells. Cancer Res. 2015, 75, 2337–2348. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Zhao, B.S.; Zhou, A.; Lin, K.; Zheng, S.; Lu, Z.; Chen, Y.; Sulman, E.P.; Xie, K.; Bögler, O.; et al. m6A Demethylase ALKBH5 Maintains Tumorigenicity of Glioblastoma Stem-like Cells by Sustaining FOXM1 Expression and Cell Proliferation Program. Cancer Cell 2017, 31, 591–606.E6. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Yuan, Q.; Xie, L. The AlkB Family of Fe (II)/Alpha-Ketoglutarate-Dependent Dioxygenases Modulates Embryogenesis through Epigenetic Regulation. Curr. Stem Cell Res. Ther. 2018, 13, 136–143. [Google Scholar] [CrossRef]
- Zhang, C.; Zhi, W.I.; Lu, H.; Samanta, D.; Chen, I.; Gabrielson, E.; Semenza, G.L. Hypoxia-inducible factors regulate pluripotency factor expression by ZNF217- and ALKBH5-mediated modulation of RNA methylation in breast cancer cells. Oncotarget 2016, 7, 64527–64542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Zhang, C.; Qi, S.; Guo, S.; Chen, Y.; Du, E.; Zhang, H.; Wang, X.; Liu, R.; Qiao, B.; et al. Elevated expression of ZNF217 promotes prostate cancer growth by restraining ferroportin-conducted iron egress. Oncotarget 2016, 7, 84893–84906. [Google Scholar] [CrossRef] [Green Version]
- Giovannetti, E.; Zucali, P.A.; Assaraf, Y.G.; Funel, N.; Gemelli, M.; Stark, M.; Thunnissen, E.; Hou, Z.; Muller, I.B.; Struys, E.A.; et al. Role of proton-coupled folate transporter in pemetrexed resistance of mesothelioma: Clinical evidence and new pharmacological tools. Ann. Oncol. 2017, 28, 2725–2732. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.-H.; Li, C.; Xu, X.; Zheng, Y.; Xiao, C.; Zerfas, P.; Cooperman, S.; Eckhaus, M.; Rouault, T.; Mishra, L.; et al. A role of SMAD4 in iron metabolism through the positive regulation of hepcidin expression. Cell Metab. 2005, 2, 399–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Di, X.; Torti, S.V.; Torti, F.M. Ferritin H induction by histone deacetylase inhibitors. Biochem. Pharmacol. 2010, 80, 316–324. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-F.; Zhang, J.; Su, Y.; Shen, Y.-Y.; Jiang, D.-X.; Hou, Y.-Y.; Geng, M.-Y.; Ding, J.; Chen, Y. G9a regulates breast cancer growth by modulating iron homeostasis through the repression of ferroxidase hephaestin. Nat. Commun. 2017, 8, 274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roatsch, M.; Hoffmann, I.; Abboud, M.I.; Hancock, R.L.; Tarhonskaya, H.; Hsu, K.-F.; Wilkins, S.E.; Yeh, T.-L.; Lippl, K.; Serrer, K.; et al. The Clinically Used Iron Chelator Deferasirox Is an Inhibitor of Epigenetic JumonjiC Domain-Containing Histone Demethylases. ACS Chem. Biol. 2019, 14, 1737–1750. [Google Scholar] [CrossRef]
- Sarno, F.; Papulino, C.; Franci, G.; Andersen, J.H.; Cautain, B.; Melardo, C.; Altucci, L.; Nebbioso, A. 3-Chloro-N’-(2-hydroxybenzylidene) benzohydrazide: An LSD1-Selective Inhibitor and Iron-Chelating Agent for Anticancer Therapy. Front. Pharmacol. 2018, 9, 1006. [Google Scholar] [CrossRef]
- Cao, L.-L.; Liu, H.; Yue, Z.; Liu, L.; Pei, L.; Gu, J.; Wang, H.; Jia, M. Iron chelation inhibits cancer cell growth and modulates global histone methylation status in colorectal cancer. Biometals 2018, 31, 797–805. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Ohlsson, R.; Henikoff, S. The epigenetic progenitor origin of human cancer. Nat. Rev. Genet. 2006, 7, 21–33. [Google Scholar] [CrossRef]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Roesch, A.; Fukunaga-Kalabis, M.; Schmidt, E.C.; Zabierowski, S.E.; Brafford, P.A.; Vultur, A.; Basu, D.; Gimotty, P.; Vogt, T.; Herlyn, M. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell 2010, 141, 583–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Yagoda, N.; von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 864–868. [Google Scholar] [CrossRef] [Green Version]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [Green Version]
- Ray, P.D.; Huang, B.-W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [Green Version]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.F.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.Y.; Dixon, S.J. Mechanisms of ferroptosis. Cell. Mol. Life Sci. 2016, 73, 2195–2209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Herde, K.; Krysko, D.V. Ferroptosis: Oxidized PEs trigger death. Nat. Chem. Biol. 2017, 13, 4–5. [Google Scholar] [CrossRef] [PubMed]
- Louandre, C.; Ezzoukhry, Z.; Godin, C.; Barbare, J.-C.; Mazière, J.-C.; Chauffert, B.; Galmiche, A. Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. Int. J. Cancer 2013, 133, 1732–1742. [Google Scholar] [CrossRef]
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol. Cell. 2015, 59, 298–308. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; Stockwell, B.R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef] [Green Version]
- Angeli, J.P.F.; Shah, R.; Pratt, D.A.; Conrad, M. Ferroptosis Inhibition: Mechanisms and Opportunities. Trends Pharmacol. Sci. 2017, 38, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Yant, L.J.; Ran, Q.; Rao, L.; Van Remmen, H.; Shibatani, T.; Belter, J.G.; Motta, L.; Richardson, A.; Prolla, T.A. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic. Biol. Med. 2003, 34, 496–502. [Google Scholar] [CrossRef]
- Linkermann, A.; Skouta, R.; Himmerkus, N.; Mulay, S.R.; Dewitz, C.; De Zen, F.; Prokai, A.; Zuchtriegel, G.; Krombach, F.; Welz, P.-S.; et al. Synchronized renal tubular cell death involves ferroptosis. Proc. Natl. Acad. Sci. USA 2014, 111, 16836–16841. [Google Scholar] [CrossRef] [Green Version]
- Skouta, R.; Dixon, S.J.; Wang, J.; Dunn, D.E.; Orman, M.; Shimada, K.; Rosenberg, P.A.; Lo, D.C.; Weinberg, J.M.; Linkermann, A.; et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J. Am. Chem. Soc. 2014, 136, 4551–4556. [Google Scholar] [CrossRef] [PubMed]
- Hao, S.; Liang, B.; Huang, Q.; Dong, S.; Wu, Z.; He, W.; Shi, M. Metabolic networks in ferroptosis. Oncol. Lett. 2018, 15, 5405–5411. [Google Scholar] [CrossRef] [PubMed]
- Lewerenz, J.; Hewett, S.J.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; et al. The cystine/glutamate antiporter system xc− in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal. 2013, 18, 522–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latunde-Dada, G.O. Ferroptosis: Role of lipid peroxidation, iron and ferritinophagy. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 1893–1900. [Google Scholar] [CrossRef] [Green Version]
- Hayano, M.; Yang, W.S.; Corn, C.K.; Pagano, N.C.; Stockwell, B.R. Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ. 2016, 23, 270–278. [Google Scholar] [CrossRef] [Green Version]
- Shimada, K.; Skouta, R.; Kaplan, A.; Yang, W.S.; Hayano, M.; Dixon, S.J.; Brown, L.M.; Valenzuela, C.A.; Wolpaw, A.J.; Stockwell, B.R. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol. 2016, 12, 497–503. [Google Scholar] [CrossRef] [Green Version]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef]
- Krishan, S.; Jansson, P.J.; Gutierrez, E.; Lane, D.J.R.; Richardson, D.E.S.; Sahni, S. Iron metabolism and autophagy: A poorly explored relationship that has important consequences for health and disease. Nagoya J. Med. Sci. 2015, 77, 1–6. [Google Scholar]
- Mancias, J.D.; Pontano Vaites, L.; Nissim, S.; Biancur, D.E.; Kim, A.J.; Wang, X.; Liu, Y.; Goessling, W.; Kimmelman, A.C.; Harper, J.W. Ferritinophagy via NCOA4 is required for erythropoiesis and is regulated by iron dependent HERC2-mediated proteolysis. eLife 2015, 4, e10308. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Monian, P.; Pan, Q.; Zhang, W.; Xiang, J.; Jiang, X. Ferroptosis is an autophagic cell death process. Cell. Res. 2016, 26, 1021–1032. [Google Scholar] [CrossRef] [Green Version]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef]
- Du, J.; Wang, T.; Li, Y.; Zhou, Y.; Wang, X.; Yu, X.; Ren, X.; An, Y.; Wu, Y.; Sun, W.; et al. DHA inhibits proliferation and induces ferroptosis of leukemia cells through autophagy dependent degradation of ferritin. Free Radic. Biol. Med. 2019, 131, 356–369. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Zhang, Z.; Chen, L.; Zhou, Y.; Zou, P.; Feng, C.; Wang, L.; Liang, G. Dihydroartemisinin (DHA) induces ferroptosis and causes cell cycle arrest in head and neck carcinoma cells. Cancer Lett. 2016, 381, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Zhang, Q.; Sun, X.; Zeh, H.J.; Lotze, M.T.; Kang, R.; Tang, D. HSPA5 Regulates Ferroptotic Cell Death in Cancer Cells. Cancer Res. 2017, 77, 2064–2077. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Geng, Y.; Lu, X.; Shi, Y.; Wu, G.; Zhang, M.; Shan, B.; Pan, H.; Yuan, J. Chaperone-mediated autophagy is involved in the execution of ferroptosis. Proc. Natl. Acad. Sci. USA 2019, 116, 2996–3005. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Czaja, M.J. Regulation of lipid stores and metabolism by lipophagy. Cell Death Differ. 2013, 20, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Yuan, Y.; Meng, Y.; Rong, Y.; Bai, H.; Chen, L. Autophagy upregulation ameliorates cell injury in Sequestosome 1 knockout podocytes in vitro. Biochem. Biophys. Res. Commun. 2017, 490, 98–103. [Google Scholar] [CrossRef]
- Schroeder, B.; Schulze, R.J.; Weller, S.G.; Sletten, A.C.; Casey, C.A.; McNiven, M.A. The small GTPase Rab7 as a central regulator of hepatocellular lipophagy. Hepatology 2015, 61, 1896–1907. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Chen, P.; Liu, J.; Zhu, S.; Kroemer, G.; Klionsky, D.J.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. Clockophagy is a novel selective autophagy process favoring ferroptosis. Sci Adv. 2019, 5, eaaw2238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, X.; Zhu, S.; Chen, P.; Hou, W.; Wen, Q.; Liu, J.; Xie, Y.; Liu, J.; Klionsky, D.J.; Kroemer, G.; et al. AMPK-Mediated BECN1 Phosphorylation Promotes Ferroptosis by Directly Blocking System Xc− Activity. Curr. Biol. 2018, 28, 2388–2399.e5. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Yao, Z.; Wang, L.; Ding, H.; Shao, J.; Chen, A.; Zhang, F.; Zheng, S. Activation of ferritinophagy is required for the RNA-binding protein ELAVL1/HuR to regulate ferroptosis in hepatic stellate cells. Autophagy 2018, 14, 2083–2103. [Google Scholar] [CrossRef] [Green Version]
- Ye, F.; Chai, W.; Xie, M.; Yang, M.; Yu, Y.; Cao, L.; Yang, L. HMGB1 regulates erastin-induced ferroptosis via RAS-JNK/p38 signaling in HL-60/NRASQ61L cells. Am. J. Cancer Res. 2019, 9, 730–739. [Google Scholar] [PubMed]
- Tang, D.; Kang, R.; Livesey, K.M.; Cheh, C.-W.; Farkas, A.; Loughran, P.; Hoppe, G.; Bianchi, M.E.; Tracey, K.J.; Zeh, H.J.; et al. Endogenous HMGB1 regulates autophagy. J. Cell. Biol. 2010, 190, 881–892. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Cheh, C.-W.; Livesey, K.M.; Liang, X.; Schapiro, N.E.; Benschop, R.; Sparvero, L.J.; Amoscato, A.A.; Tracey, K.J.; et al. HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene 2010, 29, 5299–5310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Liu, J.; Xu, Y.; Wu, R.; Chen, X.; Song, X.; Zeh, H.; Kang, R.; Klionsky, D.J.; Wang, X.; et al. Tumor heterogeneity in autophagy-dependent ferroptosis. Autophagy 2021, 17, 1–14. [Google Scholar] [CrossRef]
- Zhang, J.-F.; Yan, X.-M.; Lan, B.; Lei, Y.-R.; Li, X.-H.; Gao, S.; Guo, Y.-F.; Guo, F. Molecular mechanisms of synergistic induction of apoptosis by the combination therapy with hyperthermia and cisplatin in prostate cancer cells. Biochem. Biophys. Res. Commun. 2016, 479, 159–165. [Google Scholar] [CrossRef]
- Wang, Y.-Q.; Chang, S.-Y.; Wu, Q.; Gou, Y.-J.; Jia, L.; Cui, Y.-M.; Yu, P.; Shi, Z.-H.; Wu, W.-S.; Gao, G.; et al. The Protective Role of Mitochondrial Ferritin on Erastin-Induced Ferroptosis. Front. Aging Neurosci. 2016, 8, 308. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef]
- Wu, K.C.; Cui, J.Y.; Klaassen, C.D. Beneficial role of Nrf2 in regulating NADPH generation and consumption. Toxicol. Sci. 2011, 123, 590–600. [Google Scholar] [CrossRef] [Green Version]
- Adedoyin, O.; Boddu, R.; Traylor, A.; Lever, J.M.; Bolisetty, S.; George, J.F.; Agarwal, A. Heme oxygenase-1 mitigates ferroptosis in renal proximal tubule cells. Am. J. Physiol. Renal. Physiol. 2018, 314, F702–F714. [Google Scholar] [CrossRef] [Green Version]
- Shin, D.; Kim, E.H.; Lee, J.; Roh, J.-L. Nrf2 inhibition reverses resistance to GPX4 inhibitor-induced ferroptosis in head and neck cancer. Free Radic. Biol. Med. 2018, 129, 454–462. [Google Scholar] [CrossRef]
- Chen, D.; Tavana, O.; Chu, B.; Erber, L.; Chen, Y.; Baer, R.; Gu, W. NRF2 Is a Major Target of ARF in p53-Independent Tumor Suppression. Mol. Cell 2017, 68, 224–232.e4. [Google Scholar] [CrossRef] [Green Version]
- Garg, A.D.; Dudek, A.M.; Ferreira, G.B.; Verfaillie, T.; Vandenabeele, P.; Krysko, D.V.; Mathieu, C.; Agostinis, P. ROS-induced autophagy in cancer cells assists in evasion from determinants of immunogenic cell death. Autophagy 2013, 9, 1292–1307. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, Y.; Liu, J.; Kang, R.; Tang, D. Interplay between MTOR and GPX4 signaling modulates autophagy-dependent ferroptotic cancer cell death. Cancer Gene Ther. 2021, 28, 55–63. [Google Scholar] [CrossRef]
- Zhang, Y.; Swanda, R.V.; Nie, L.; Liu, X.; Wang, C.; Lee, H.; Lei, G.; Mao, C.; Koppula, P.; Cheng, W.; et al. mTORC1 couples cyst(e)ine availability with GPX4 protein synthesis and ferroptosis regulation. Nat. Commun. 2021, 12, 1589. [Google Scholar] [CrossRef]
- Lu, B.; Chen, X.B.; Ying, M.D.; He, Q.J.; Cao, J.; Yang, B. The Role of Ferroptosis in Cancer Development and Treatment Response. Front. Pharmacol. 2017, 8, 992. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Kroemer, G.; Tang, D. The tumor suppressor protein p53 and the ferroptosis network. Free Radic. Biol. Med. 2019, 133, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, C.; Wang, J.; Hu, W.; Feng, Z. The Regulation of Ferroptosis by Tumor Suppressor p53 and its Pathway. Int. J. Mol. Sci. 2020, 21, 8387. [Google Scholar] [CrossRef]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.-J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Ou, Y.; Wang, S.-J.; Li, D.; Chu, B.; Gu, W. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc. Natl. Acad. Sci. USA 2016, 113, E6806–E6812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, W.; Zhang, C.; Wu, R.; Sun, Y.; Levine, A.; Feng, Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc. Natl. Acad. Sci. USA 2010, 107, 7455–7460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Zhu, S.; Song, X.; Sun, X.; Fan, Y.; Liu, J.; Zhong, M.; Yuan, H.; Zhang, L.; Billiar, T.R.; et al. The Tumor Suppressor p53 Limits Ferroptosis by Blocking DPP4 Activity. Cell. Rep. 2017, 20, 1692–1704. [Google Scholar] [CrossRef] [Green Version]
- Lam, C.S.-C.; Cheung, A.H.-K.; Wong, S.K.-M.; Wan, T.M.-H.; Ng, L.; Chow, A.K.-M.; Cheng, N.S.-M.; Pak, R.C.-H.; Li, H.-S.; Man, J.H.-W.; et al. Prognostic significance of CD26 in patients with colorectal cancer. PLoS ONE 2014, 9, e98582. [Google Scholar] [CrossRef] [Green Version]
- Pang, R.; Law, W.L.; Chu, A.C.Y.; Poon, J.T.; Lam, C.S.C.; Chow, A.K.M.; Ng, L.; Cheung, L.W.H.; Lan, X.R.; Lan, H.Y.; et al. A subpopulation of CD26+ cancer stem cells with metastatic capacity in human colorectal cancer. Cell Stem Cell 2010, 6, 603–615. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Chu, B.; Yang, X.; Liu, Z.; Jin, Y.; Kon, N.; Rabadan, R.; Jiang, X.; Stockwell, B.R.; Gu, W. iPLA2β-mediated lipid detoxification controls p53-driven ferroptosis independent of GPX4. Nat. Commun. 2021, 12, 3644. [Google Scholar] [CrossRef]
- Yi, M.; Li, J.; Chen, S.; Cai, J.; Ban, Y.; Peng, Q.; Zhou, Y.; Zeng, Z.; Peng, S.; Li, X.; et al. Emerging role of lipid metabolism alterations in Cancer stem cells. J. Exp. Clin. Cancer Res. 2018, 37, 118. [Google Scholar] [CrossRef]
- Fujihara, K.M.; Zhang, B.Z.; Clemons, N.J. Opportunities for Ferroptosis in Cancer Therapy. Antioxidants 2021, 10, 986. [Google Scholar] [CrossRef] [PubMed]
- Birsen, R.; Larrue, C.; Decroocq, J.; Johnson, N.; Guiraud, N.; Gotanegre, M.; Cantero-Aguilar, L.; Grignano, E.; Huynh, T.; Fontenay, M.; et al. APR-246 induces early cell death by ferroptosis in acute myeloid leukemia. Haematologica 2021. [Google Scholar] [CrossRef]
- Liu, D.S.; Duong, C.P.; Haupt, S.; Montgomery, K.G.; House, C.M.; Azar, W.J.; Pearson, H.B.; Fisher, O.M.; Read, M.; Guerra, G.R.; et al. Inhibiting the system xC-/glutathione axis selectively targets cancers with mutant-p53 accumulation. Nat. Commun. 2017, 8, 14844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, C.L.; Stevens, B.M.; D’Alessandro, A.; Culp-Hill, R.; Reisz, J.A.; Pei, S.; Gustafson, A.; Khan, N.; DeGregori, J.; Pollyea, D.A.; et al. Cysteine depletion targets leukemia stem cells through inhibition of electron transport complex II. Blood 2019, 134, 389–394. [Google Scholar] [CrossRef]
- Ishimoto, T.; Nagano, O.; Yae, T.; Tamada, M.; Motohara, T.; Oshima, H.; Oshima, M.; Ikeda, T.; Asaba, R.; Yagi, H.; et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc− and thereby promotes tumor growth. Cancer Cell 2011, 19, 387–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagano, O. Cancer therapy targeted to CD44v-xCT-mediated antioxidant system. Ann. Oncol. 2016, 27, vii40. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, M.; Tsuchihashi, K.; Ishimoto, T.; Yae, T.; Motohara, T.; Sugihara, E.; Onishi, N.; Masuko, T.; Yoshizawa, K.; Kawashiri, S.; et al. xCT inhibition depletes CD44v-expressing tumor cells that are resistant to EGFR-targeted therapy in head and neck squamous cell carcinoma. Cancer Res. 2013, 73, 1855–1866. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Ou, Z.; Xie, M.; Kang, R.; Fan, Y.; Niu, X.; Wang, H.; Cao, L.; Tang, D. HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene 2015, 34, 5617–5625. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Kang, R.; Livesey, K.M.; Kroemer, G.; Billiar, T.R.; Van Houten, B.; Zeh, H.J.; Lotze, M.T. High-mobility group box 1 is essential for mitochondrial quality control. Cell Metab. 2011, 13, 701–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, L.; Qi, Z.; Zhu, Y.; Song, X.; Xuan, C.; Ben, P.; Lan, L.; Luo, L.; Yin, Z. Phosphorylated heat shock protein 27 promotes lipid clearance in hepatic cells through interacting with STAT3 and activating autophagy. Cell. Signal. 2016, 28, 1086–1098. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-L.; Uthaya Kumar, D.B.; Punj, V.; Xu, J.; Sher, L.; Tahara, S.M.; Hess, S.; Machida, K. NANOG Metabolically Reprograms Tumor-Initiating Stem-like Cells through Tumorigenic Changes in Oxidative Phosphorylation and Fatty Acid Metabolism. Cell Metab. 2016, 23, 206–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marzenell, P.; Hagen, H.; Sellner, L.; Zenz, T.; Grinyte, R.; Pavlov, V.; Daum, S.; Mokhir, A. Aminoferrocene-based prodrugs and their effects on human normal and cancer cells as well as bacterial cells. J. Med. Chem. 2013, 56, 6935–6944. [Google Scholar] [CrossRef]
- Hagen, H.; Marzenell, P.; Jentzsch, E.; Wenz, F.; Veldwijk, M.R.; Mokhir, A. Aminoferrocene-based prodrugs activated by reactive oxygen species. J. Med. Chem. 2012, 55, 924–934. [Google Scholar] [CrossRef]
- Trujillo-Alonso, V.; Pratt, E.C.; Zong, H.; Lara-Martinez, A.; Kaittanis, C.; Rabie, M.O.; Longo, V.; Becker, M.W.; Roboz, G.J.; Grimm, J.; et al. FDA-approved ferumoxytol displays anti-leukaemia efficacy against cells with low ferroportin levels. Nat. Nanotechnol. 2019, 14, 616–622. [Google Scholar] [CrossRef]
- Sagasser, J.; Ma, B.N.; Baecker, D.; Salcher, S.; Hermann, M.; Lamprecht, J.; Angerer, S.; Obexer, P.; Kircher, B.; Gust, R. A New Approach in Cancer Treatment: Discovery of Chlorido[N,N’-disalicylidene-1,2-phenylenediamine]iron(III) Complexes as Ferroptosis Inducers. J. Med. Chem. 2019, 62, 8053–8061. [Google Scholar] [CrossRef]
- Gao, J.; Luo, T.; Wang, J. Gene interfered-ferroptosis therapy for cancers. Nat. Commun. 2021, 12, 5311. [Google Scholar] [CrossRef]
- Yu, H.; Yang, C.; Jian, L.; Guo, S.; Chen, R.; Li, K.; Qu, F.; Tao, K.; Fu, Y.; Luo, F.; et al. Sulfasalazine-induced ferroptosis in breast cancer cells is reduced by the inhibitory effect of estrogen receptor on the transferrin receptor. Oncol. Rep. 2019, 42, 826–838. [Google Scholar] [CrossRef]
- Okazaki, S.; Shintani, S.; Hirata, Y.; Suina, K.; Semba, T.; Yamasaki, J.; Umene, K.; Ishikawa, M.; Saya, H.; Nagano, O. Synthetic lethality of the ALDH3A1 inhibitor dyclonine and xCT inhibitors in glutathione deficiency-resistant cancer cells. Oncotarget 2018, 9, 33832–33843. [Google Scholar] [CrossRef] [Green Version]
- Gout, P.W.; Buckley, A.R.; Simms, C.R.; Bruchovsky, N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the xc− cystine transporter: A new action for an old drug. Leukemia 2001, 15, 1633–1640. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, Y.; Kasukabe, T.; Kumakura, S. Piperlongumine rapidly induces the death of human pancreatic cancer cells mainly through the induction of ferroptosis. Int. J. Oncol. 2018, 52, 1011–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, S.M.; Buckingham, S.C.; Campbell, S.L.; Robel, S.; Holt, K.T.; Ogunrinu-Babarinde, T.; Warren, P.P.; White, D.M.; Reid, M.A.; Eschbacher, J.M.; et al. SLC7A11 expression is associated with seizures and predicts poor survival in patients with malignant glioma. Sci. Transl. Med. 2015, 7, 289ra86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robe, P.A.; Martin, D.H.; Nguyen-Khac, M.T.; Artesi, M.; Deprez, M.; Albert, A.; Vanbelle, S.; Califice, S.; Bredel, M.; Bours, V. Early termination of ISRCTN45828668, a phase 1/2 prospective, randomized study of sulfasalazine for the treatment of progressing malignant gliomas in adults. BMC Cancer 2009, 9, 372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lachaier, E.; Louandre, C.; Godin, C.; Saidak, Z.; Baert, M.; Diouf, M.; Chauffert, B.; Galmiche, A. Sorafenib induces ferroptosis in human cancer cell lines originating from different solid tumors. Anticancer Res. 2014, 34, 6417–6422. [Google Scholar]
- Shaw, A.T.; Winslow, M.M.; Magendantz, M.; Ouyang, C.; Dowdle, J.; Subramanian, A.; Lewis, T.A.; Maglathin, R.L.; Tolliday, N.; Jacks, T. Selective killing of K-ras mutant cancer cells by small molecule inducers of oxidative stress. Proc. Natl. Acad. Sci. USA 2011, 108, 8773–8778. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Xie, Y.; Cao, L.; Yang, L.; Yang, M.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. The ferroptosis inducer erastin enhances sensitivity of acute myeloid leukemia cells to chemotherapeutic agents. Mol. Cell. Oncol. 2015, 2, e1054549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [Green Version]
- Malfa, G.A.; Tomasello, B.; Acquaviva, R.; Genovese, C.; La Mantia, A.; Cammarata, F.P.; Ragusa, M.; Renis, M.; Di Giacomo, C. Betula etnensis Raf. (Betulaceae) Extract Induced HO-1 Expression and Ferroptosis Cell Death in Human Colon Cancer Cells. Int. J. Mol. Sci. 2019, 20, 2723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaschler, M.M.; Andia, A.A.; Liu, H.; Csuka, J.M.; Hurlocker, B.; Vaiana, C.A.; Heindel, D.W.; Zuckerman, D.S.; Bos, P.H.; Reznik, E.; et al. FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat. Chem. Biol. 2018, 14, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Abrams, R.P.; Carroll, W.L.; Woerpel, K.A. Five-Membered Ring Peroxide Selectively Initiates Ferroptosis in Cancer Cells. ACS Chem. Biol. 2016, 11, 1305–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eling, N.; Reuter, L.; Hazin, J.; Hamacher-Brady, A.; Brady, N.R. Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience 2015, 2, 517–532. [Google Scholar] [CrossRef] [Green Version]
- Yang, N.-D.; Tan, S.-H.; Ng, S.; Shi, Y.; Zhou, J.; Tan, K.S.W.; Wong, W.-S.F.; Shen, H.-M. Artesunate induces cell death in human cancer cells via enhancing lysosomal function and lysosomal degradation of ferritin. J. Biol. Chem. 2014, 289, 33425–33441. [Google Scholar] [CrossRef] [Green Version]
- Ooko, E.; Saeed, M.E.M.; Kadioglu, O.; Sarvi, S.; Colak, M.; Elmasaoudi, K.; Janah, R.; Greten, H.J.; Efferth, T. Artemisinin derivatives induce iron-dependent cell death (ferroptosis) in tumor cells. Phytomedicine 2015, 22, 1045–1054. [Google Scholar] [CrossRef]
- Wang, N.; Zeng, G.-Z.; Yin, J.-L.; Bian, Z.-X. Artesunate activates the ATF4-CHOP-CHAC1 pathway and affects ferroptosis in Burkitt’s Lymphoma. Biochem. Biophys. Res. Commun. 2019, 519, 533–539. [Google Scholar] [CrossRef]
- Chen, Y.; Mi, Y.; Zhang, X.; Ma, Q.; Song, Y.; Zhang, L.; Wang, D.; Xing, J.; Hou, B.; Li, H.; et al. Dihydroartemisinin-induced unfolded protein response feedback attenuates ferroptosis via PERK/ATF4/HSPA5 pathway in glioma cells. J. Exp. Clin. Cancer Res. 2019, 38, 402. [Google Scholar] [CrossRef]
- Bebber, C.M.; Thomas, E.S.; Stroh, J.; Chen, Z.; Androulidaki, A.; Schmitt, A.; Höhne, M.N.; Stüker, L.; de Pádua Alves, C.; Khonsari, A.; et al. Ferroptosis response segregates small cell lung cancer (SCLC) neuroendocrine subtypes. Nat. Commun. 2021, 12, 2048. [Google Scholar] [CrossRef]
- Ma, S.; Henson, E.S.; Chen, Y.; Gibson, S.B. Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis. 2016, 7, e2307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, S.; Dielschneider, R.F.; Henson, E.S.; Xiao, W.; Choquette, T.R.; Blankstein, A.R.; Chen, Y.; Gibson, S.B. Ferroptosis and autophagy induced cell death occur independently after siramesine and lapatinib treatment in breast cancer cells. PLoS ONE 2017, 12, e0182921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentric, G.; Kieffer, Y.; Mieulet, V.; Goundiam, O.; Bonneau, C.; Nemati, F.; Hurbain, I.; Raposo, G.; Popova, T.; Stern, M.-H.; et al. PML-Regulated Mitochondrial Metabolism Enhances Chemosensitivity in Human Ovarian Cancers. Cell Metab. 2019, 29, 156–173.e10. [Google Scholar] [CrossRef] [Green Version]
- Badgley, M.A.; Kremer, D.M.; Maurer, H.C.; DelGiorno, K.E.; Lee, H.-J.; Purohit, V.; Sagalovskiy, I.R.; Ma, A.; Kapilian, J.; Firl, C.E.M.; et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 2020, 368, 85–89. [Google Scholar] [CrossRef]
- Cramer, S.L.; Saha, A.; Liu, J.; Tadi, S.; Tiziani, S.; Yan, W.; Triplett, K.; Lamb, C.; Alters, S.E.; Rowlinson, S.; et al. Systemic depletion of L-cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth. Nat. Med. 2017, 23, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Green, M.; Choi, J.E.; Gijón, M.; Kennedy, P.D.; Johnson, J.K.; Liao, P.; Lang, X.; Kryczek, I.; Sell, A.; et al. CD8+ T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 2019, 569, 270–274. [Google Scholar] [CrossRef]
- Bjarnadottir, O.; Romero, Q.; Bendahl, P.-O.; Jirström, K.; Rydén, L.; Loman, N.; Uhlén, M.; Johannesson, H.; Rose, C.; Grabau, D.; et al. Targeting HMG-CoA reductase with statins in a window-of-opportunity breast cancer trial. Breast Cancer Res. Treat. 2013, 138, 499–508. [Google Scholar] [CrossRef]
- Graf, H.; Jüngst, C.; Straub, G.; Dogan, S.; Hoffmann, R.-T.; Jakobs, T.; Reiser, M.; Waggershauser, T.; Helmberger, T.; Walter, A.; et al. Chemoembolization combined with pravastatin improves survival in patients with hepatocellular carcinoma. Digestion 2008, 78, 34–38. [Google Scholar] [CrossRef] [Green Version]
- Kornblau, S.M.; Banker, D.E.; Stirewalt, D.; Shen, D.; Lemker, E.; Verstovsek, S.; Estrov, Z.; Faderl, S.; Cortes, J.; Beran, M.; et al. Blockade of adaptive defensive changes in cholesterol uptake and synthesis in AML by the addition of pravastatin to idarubicin + high-dose Ara-C: A phase 1 study. Blood 2007, 109, 2999–3006. [Google Scholar] [CrossRef]
- Sondergaard, T.E.; Pedersen, P.T.; Andersen, T.L.; Søe, K.; Lund, T.; Ostergaard, B.; Garnero, P.; Delaisse, J.-M.; Plesner, T. A phase II clinical trial does not show that high dose simvastatin has beneficial effect on markers of bone turnover in multiple myeloma. Hematol. Oncol. 2009, 27, 17–22. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Markers | Cancer | Ref |
|---|---|---|
| CD44+CD24- | Breast | [10,11] |
| ALDH1+ | Colon, Brain, Acute Myeloid Leukemia, Breast, Stomach, Melanoma | [12,13,14,15,16,17] |
| CD133+ | Brain, Colon, Pancreas, Lung, Ovarian, Prostate, Stomach | [18,19] |
| CD44+ALDH1+ | Ovary | [10,20] |
| CD44+a2b1highCD133+ | Prostate | [10,18,21] |
| ABCB5 | Melanoma | [22] |
| CD44+ | Colon, Head and Neck | [10,23,24] |
| CD24+ | Colon | [25] |
| CD166+ | Colon, non-small cell lung cancer | [26,27,28] |
| CD133+EpCAM+ | Liver | [18,29] |
| CD44+EpCAM+ | Colon | [10,23] |
| ESA+CD44+CD24+ | Pancreas | [30] |
| CBX3+ABCA5+ | Osteosarcoma | [31] |
| LGR5+ | Colon | [32] |
| CD90+ | Liver | [33] |
| CD34+CD38- | Acute Myeloid Leukemia | [34] |
| CD34+CD38+CD19+/CD34+CD38-CD19+ | Leukemia | [35] |
| Name | Class/Action | Cancer | Ref |
|---|---|---|---|
| Erastin and its derivatives (better stability): Aldehyde erastin Piperazine Morpholine erastin II | Targets the mitochondrial voltage-dependent anion channel 2/3 (VDAC2/3); Class I inducer through the binding of SLC7A5, a subunit of system Xc−/induces of Beclin1-SCL7A11 complex formation/inhibits cystine uptake leading to GSH depletion/induces also the GPX4 protein degradation | Kras-mutant tumor cells, lung, leukemia, CRC | [73,163,118] |
| Sulfasalazine (SAS) | Class I inducer/Induces Beclin1-SCL7A11 complex formation/inhibits cystine uptake leading to GSH depletion | Lymphoma, SCLC, prostate cancer, breast cancer, glioblastoma, combined with dyclonine, targets ALDH3A1+ tumors cells in head and neck squamous cell carcinoma and in gastric tumors, leukemia, pancreatic cancer | [85,163,164,165,166,167,168] |
| Sorafenib | Class I inducer/inhibits the activity of system Xc− | Liver, kidney, lung or pancreatic derived cell lines, AML, HCC, neuroblastoma, NSCLC, RCC | [95,169] |
| Tat-beclin1 | Enhances erastin anti-cancer activity/direct inhibitor of the activity of system Xc−/leads to lipid peroxidation | Colon, pancreas, lung (NSCLC), cervical | [120] |
| Lanperisone (FDA-approved drug) | Class I inducer/inhibits cystine uptake leading to GSH depletion | Kras-mutant tumor cells | [170] |
| RSL3/5 | Class 2 inducer/binds GPX4 to inhibit its enzymatic activity/induces also the GPX4 protein degradation | Kras-mutant tumor cells; AML cells, Head and neck cancer, Colorectal cancer | [97,106,113,171,172,173] |
| FIN(ferroptosis inducing)56 | Class2 inducer, downregulates GPX4 expression at mRNA level/targets GPX4 degradation/causes depletion of mevalonate-derived coenzyme Q10 (CoQ10) | Osteosarcoma, lung adenocarcinoma, fibrosarcoma | [106] |
| FINO2 (endoperoxide-containing 1,2-dioxolane) | Class 2 inducer/represses indirectly the enzymatic function of GPX4 leading widespread lipid peroxidation/is able to oxidize ferrous iron directly/can also oxidize lipids, providing another source of lipid peroxides | NIH60, a range of cancer cell lines from different tissues, engineered cancer cells such as RCC cells and fibrosarcoma cells | [174,175] |
| Artesunate (artemisinin derivative, anti-malaria drug) | Targets iron/induces ferritin degradation leading to the lysosomal iron release and Fenton reaction with ROS | mutationaly-active Kras pancreatic ductal adenocarcinoma cell lines, lymphoma | [176,177,178,179] |
| Dihydroartemisinin (DHA, semi-synthetic artemisinin derivative) | Targets iron/inducs the autophagy-dependent degradation of ferritin by regulating the activity of the AMPK/mTOR/p70S6K pathway | Leukemia, glioma, head and neck cancer; | [112,180] |
| BSO (Buthionine sulfoximine) | Targets GCLC; prevents GSH synthesis; | HCC, Kras-mutant tumor cells SCLC cancer | [128,172,181] |
| Siramesine and lapatinib | Targets iron, decreases the expression of FPN and Ferritin and increases iron uptake through upregulation of TRFC; | Breast cancer cells | [182,183] |
| Salinomycine and its synthetic derivate Ironomycin | Sequesters lysosomal iron leading to cytoplasmic iron depletion/increases iron uptake through the up-expression of IRP2 and TFRC, along with the accelerated lysosomal degradation of ferritin | Breast cancer stem cells, ovarian cancer | [39,40,184] |
| Cyst(e)inase | Targets enzymatic degradation of cystine/cysteine, restricting its availability to cancer cells and triggering ferroptosis | Breast cancer, chronic lymphocytic leukemia, melanoma, pancreatic cancer, prostate cancer | [185,186,187] |
| Statins (Fluvastatin, Pravastin, lovastatin and simvastatin), inhibitors of HMGCR (HMG-CoA reductase), a rate-limiting enzyme in the mevalonate pathway | Are able to inhibit the biosynthesis of selenoproteins including GPX4 and CoQ10 | Breast cancer, HCC, AML, MM; | [188,189,190,191] |
| Eprenetapopt (APR-017/PRIMA-1, APR-246/PRIMA-1MET) | Mutant-p53 reactivators, has also the ability to conjugate free cysteine leading to GSH depletion and interferes with iron-sulfur cluster biogenesis | AML | [148] |
| Ferumoxytol (Feraheme, FDA-approved iron oxide nanoparticle) | Targets iron, fuels Fenton reaction leading to harmful production of ROS; | Leukemia cells | [160] |
| Iron salophen complexes (chemicaly-engineered transition-metal complexes) | Generate lipid ROS leading to ferroptosis | Leukemia, neuroblastoma cell lines | [161] |
| Fenugreek (trigonelline) | Inhibits NRF2 leading the blockage of MT-1G expression, and GSH depletion | HCC cells, head and neck cancer cells | [128] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cosialls, E.; El Hage, R.; Dos Santos, L.; Gong, C.; Mehrpour, M.; Hamaï, A. Ferroptosis: Cancer Stem Cells Rely on Iron until “to Die for” It. Cells 2021, 10, 2981. https://doi.org/10.3390/cells10112981
Cosialls E, El Hage R, Dos Santos L, Gong C, Mehrpour M, Hamaï A. Ferroptosis: Cancer Stem Cells Rely on Iron until “to Die for” It. Cells. 2021; 10(11):2981. https://doi.org/10.3390/cells10112981
Chicago/Turabian StyleCosialls, Emma, Rima El Hage, Leïla Dos Santos, Chang Gong, Maryam Mehrpour, and Ahmed Hamaï. 2021. "Ferroptosis: Cancer Stem Cells Rely on Iron until “to Die for” It" Cells 10, no. 11: 2981. https://doi.org/10.3390/cells10112981
APA StyleCosialls, E., El Hage, R., Dos Santos, L., Gong, C., Mehrpour, M., & Hamaï, A. (2021). Ferroptosis: Cancer Stem Cells Rely on Iron until “to Die for” It. Cells, 10(11), 2981. https://doi.org/10.3390/cells10112981