
Decreased Trabecular Bone Mass in Col22a1-Deficient Mice

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ex Vivo Cultures
2.2. Expression Analysis
2.3. Animal Husbandry and Experiments
2.4. Radiological Assessment
2.5. Skeletal Histology
2.6. Biomechanical Testing
2.7. Biochemical Assays
2.8. Statistical Analysis
3. Results
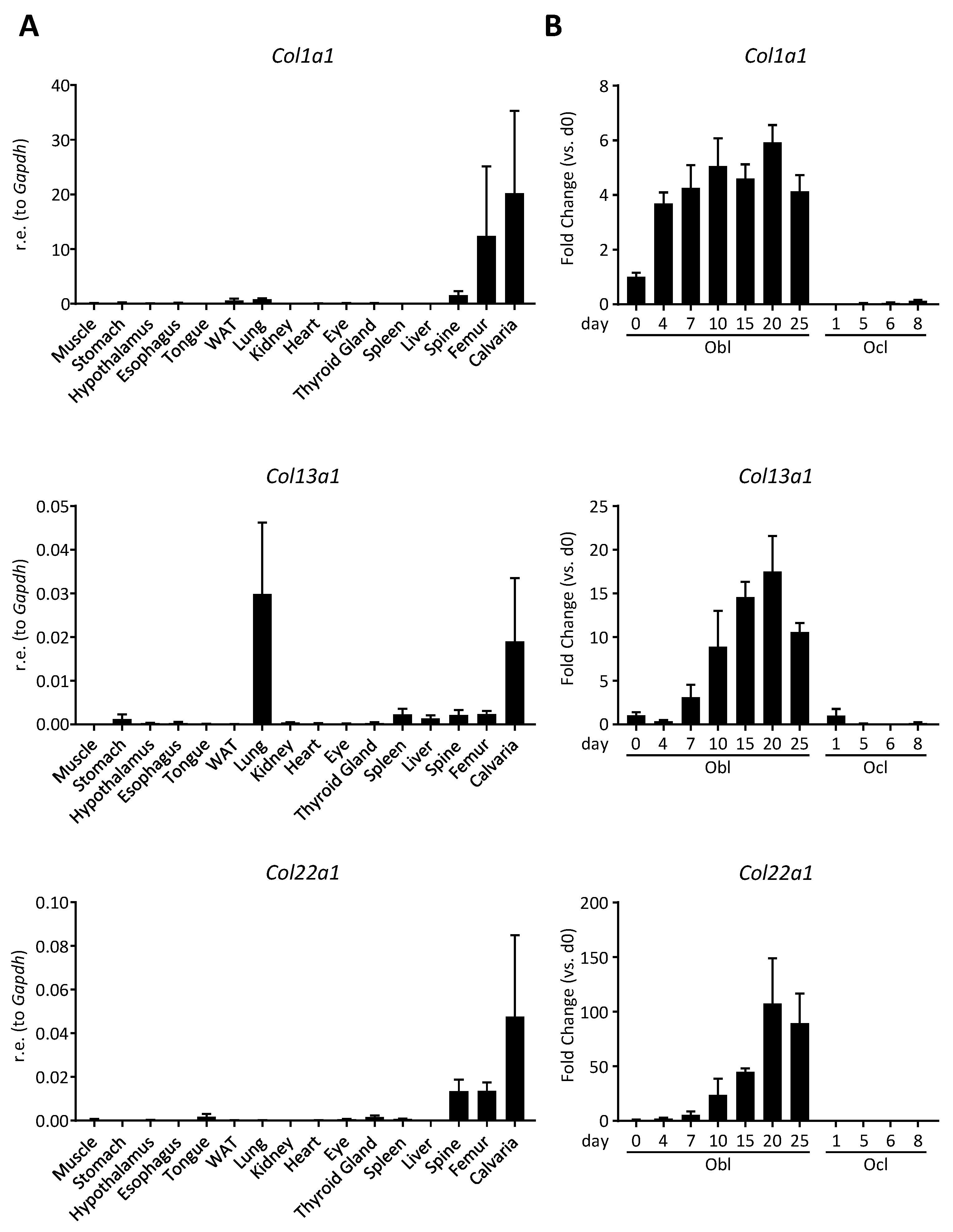
3.1. Col22a1 Is Predominantly Expressed in Bone-Forming Osteoblasts
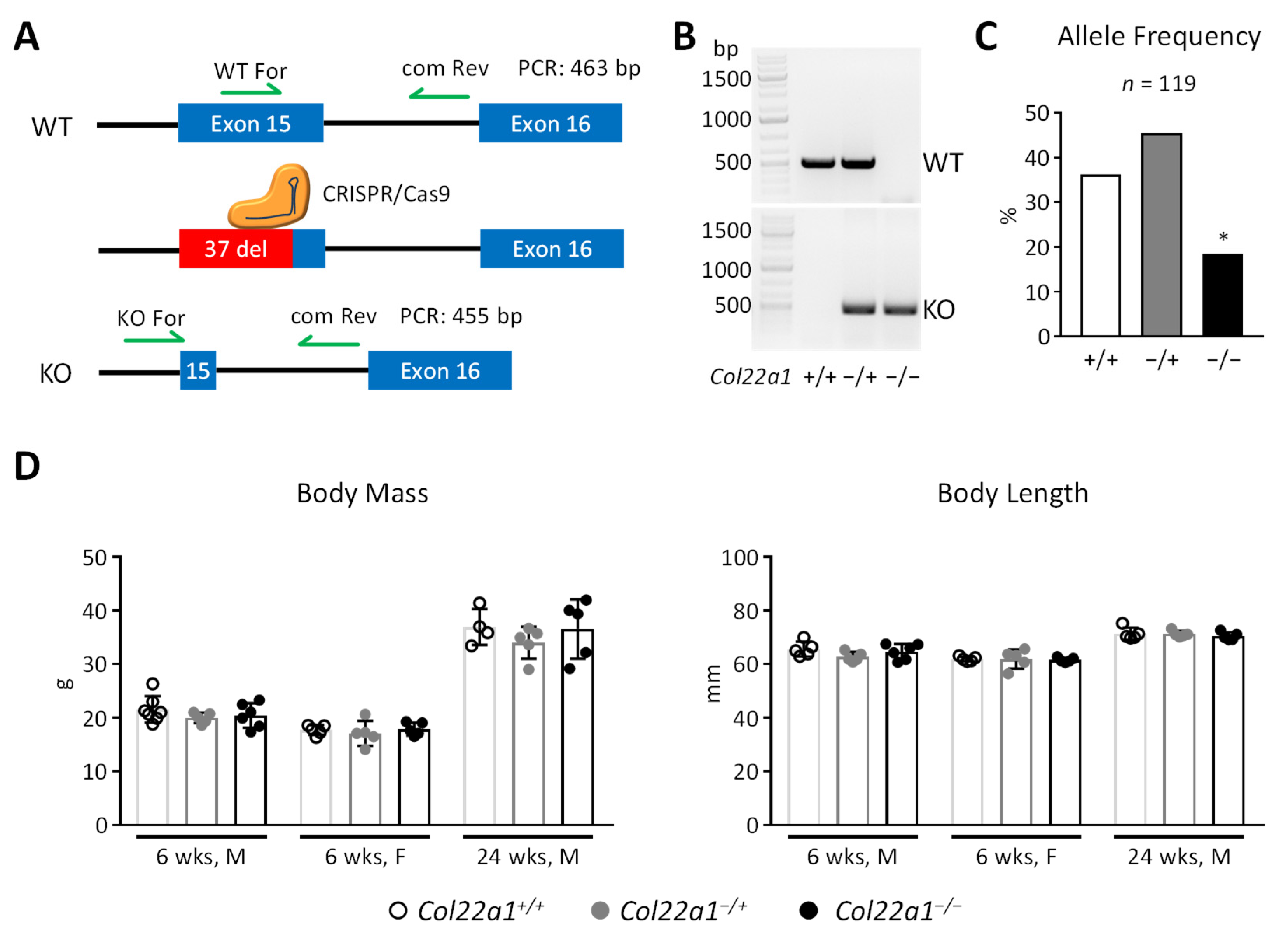
3.2. Generation of Col22a1-Deficient Mice
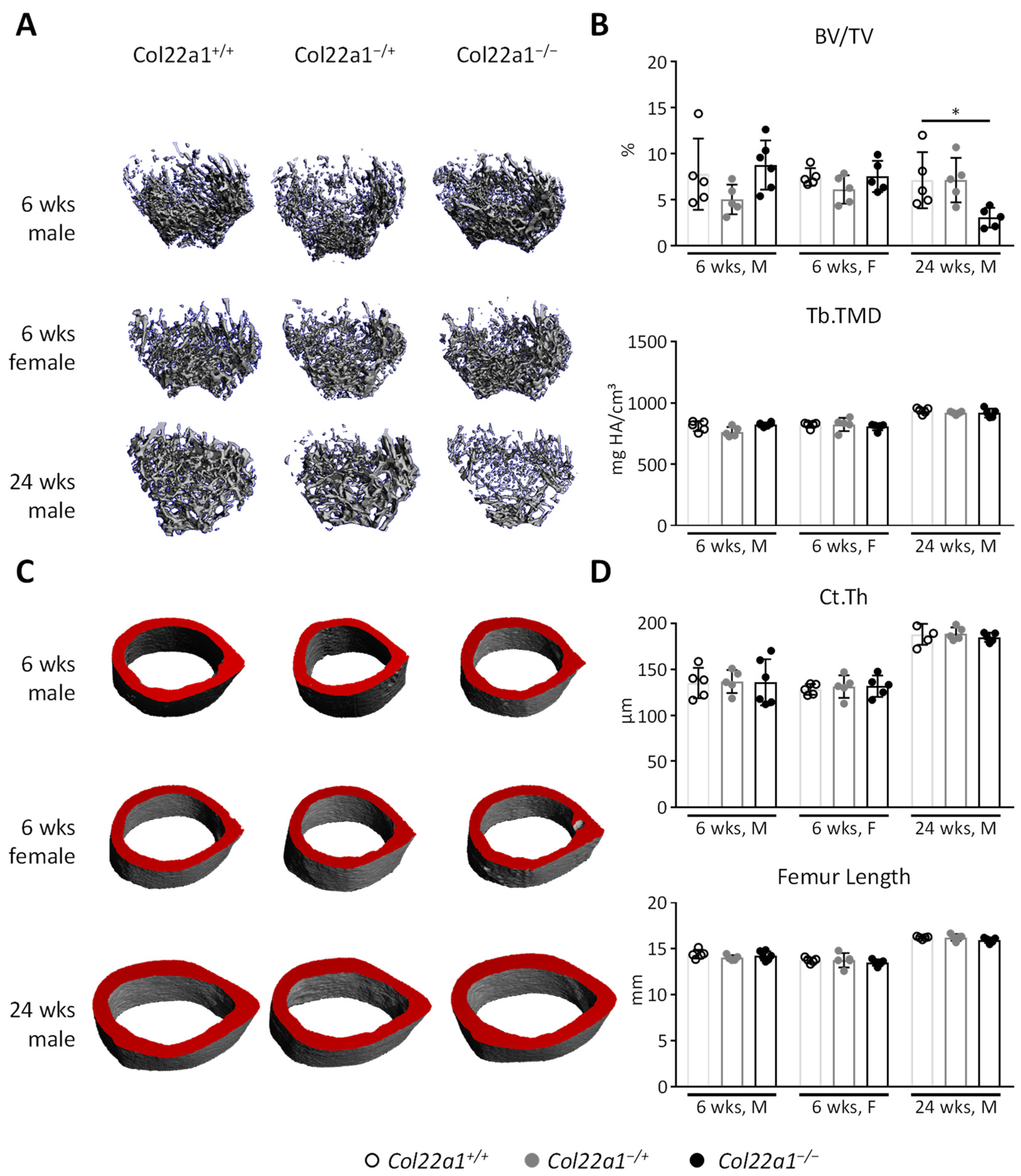
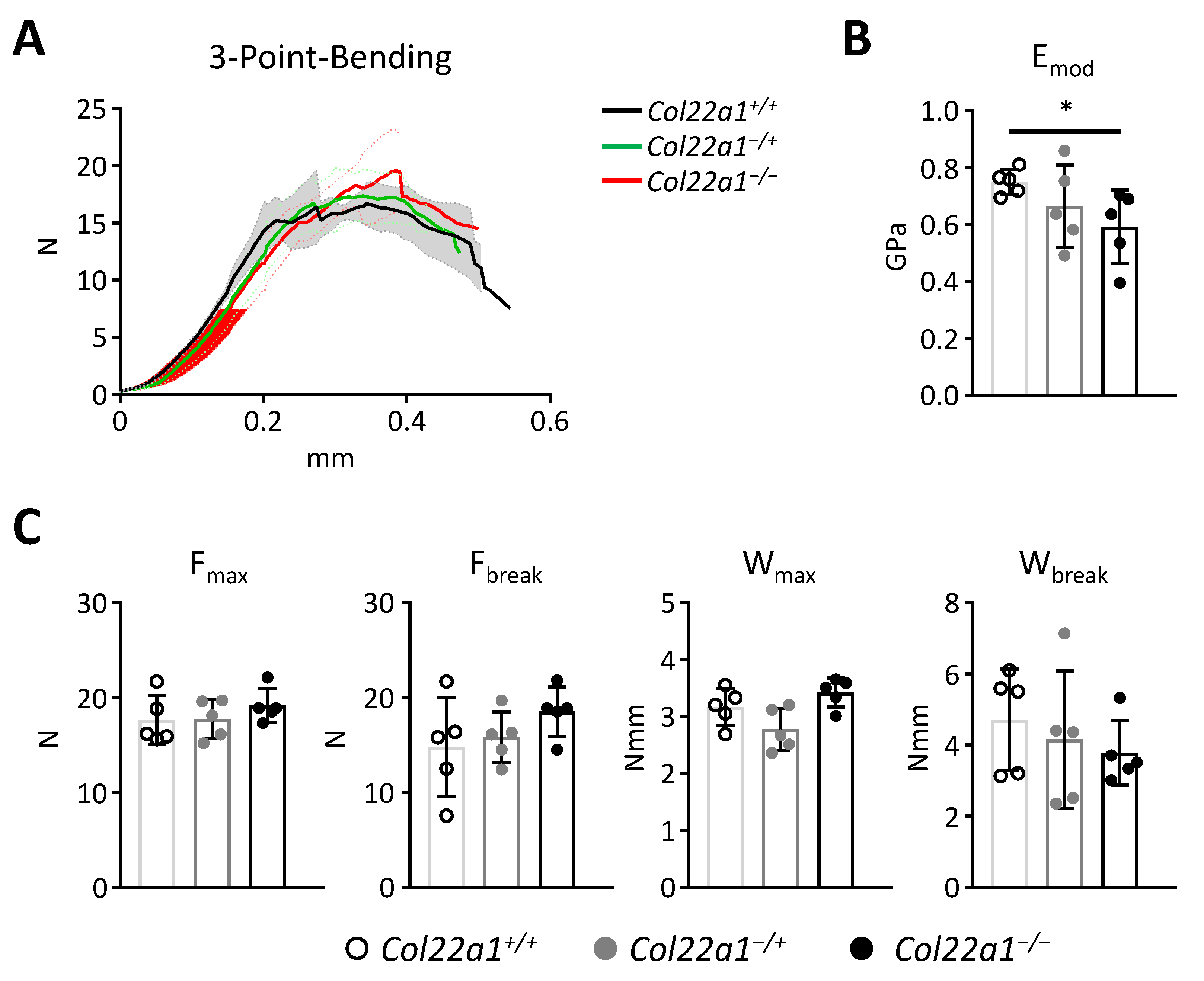
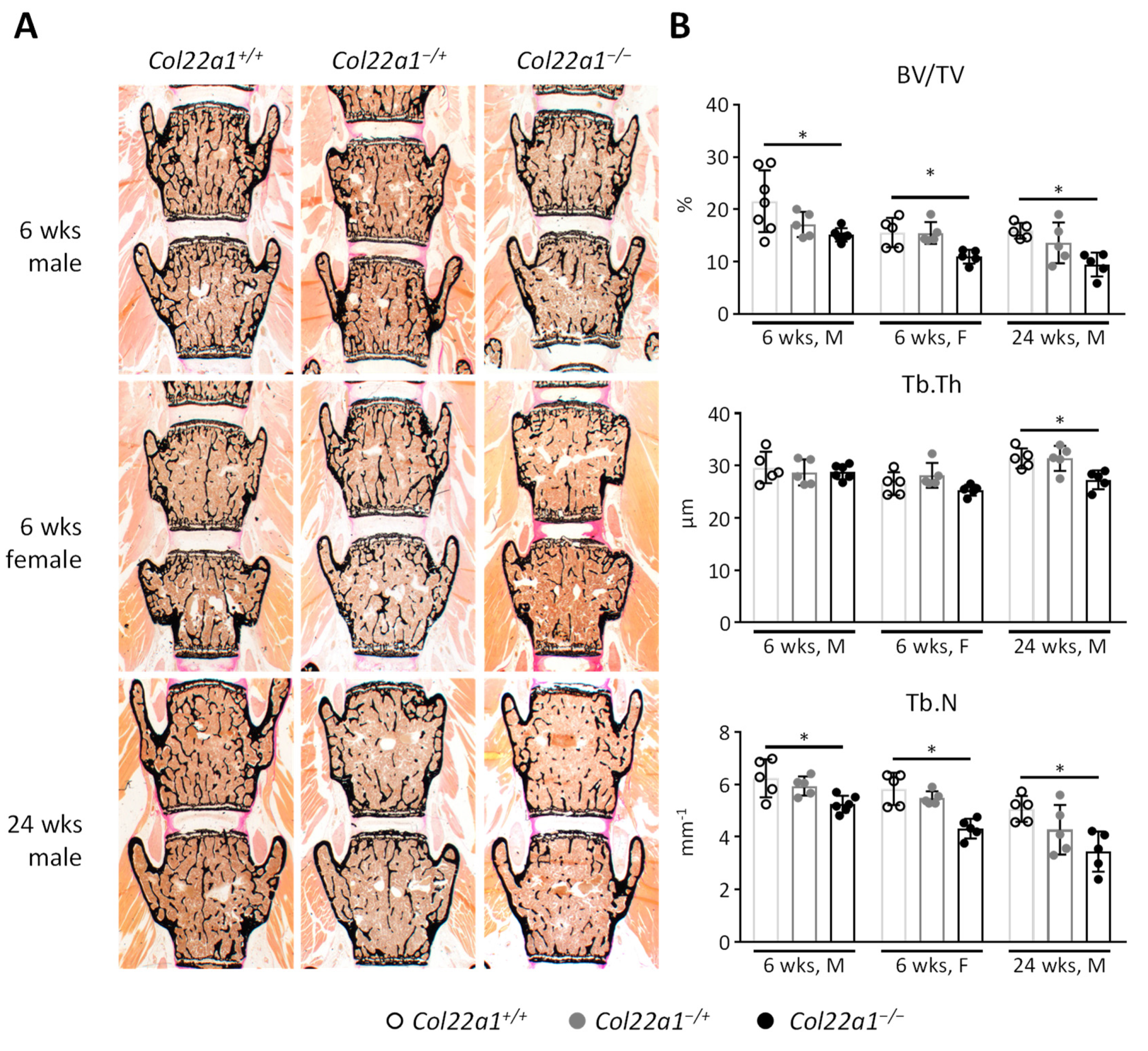
3.3. Moderate Trabecular Osteopenia, but Normal Cortical Bone Mass in Col22a1-Deficient Mice
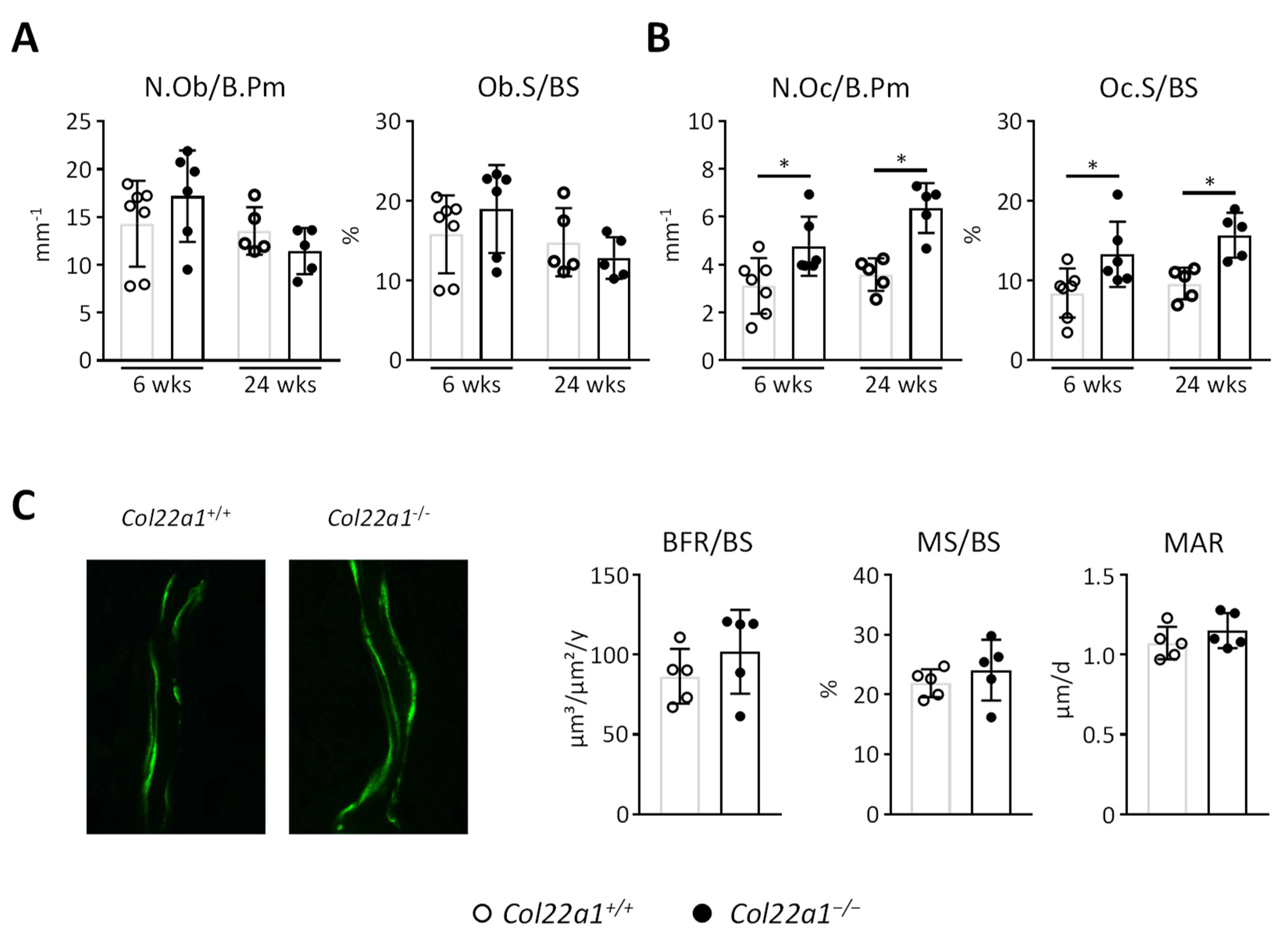
3.4. Increased Osteoclast Numbers at Trabecular Bone Surfaces of Col22a1-Deficient Mice
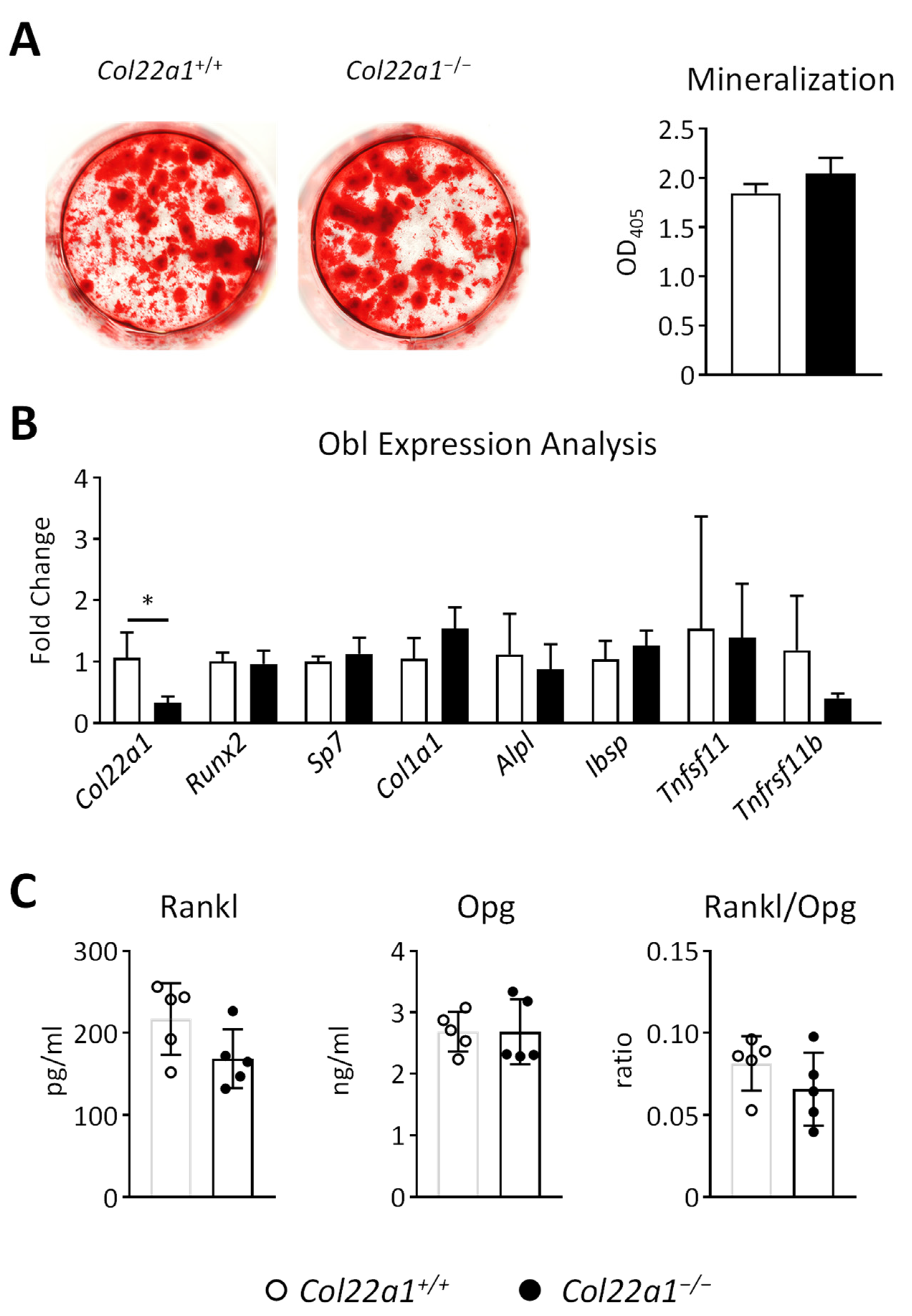
3.5. Primary Osteoblasts from Col22a1-Deficient Mice Do Not Display a Cell-Autonomous Osteogenesis Defect
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Katsimbri, P. The biology of normal bone remodelling. Eur. J. Cancer Care 2017, 26, e12740. [Google Scholar] [CrossRef]
- Zaidi, M. Skeletal remodeling in health and disease. Nat. Med. 2007, 13, 791–801. [Google Scholar] [CrossRef]
- Teitelbaum, S.L. Bone resorption by osteoclasts. Science 2000, 289, 1504–1508. [Google Scholar] [CrossRef] [PubMed]
- Capulli, M.; Paone, R.; Rucci, N. Osteoblast and osteocyte: Games without frontiers. Arch. Biochem. Biophys. 2014, 561, 3–12. [Google Scholar] [CrossRef]
- Henriksen, K.; Karsdal, M.A. Chapter 1—Type I collagen. In Biochemistry of Collagens, Laminins and Elastin, 2nd ed.; Karsdal, M.A., Ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 1–12. [Google Scholar] [CrossRef]
- Forlino, A.; Marini, J.C. Osteogenesis imperfecta. Lancet 2016, 387, 1657–1671. [Google Scholar] [CrossRef]
- Marom, R.; Rabenhorst, B.M.; Morello, R. Osteogenesis imperfecta: An update on clinical features and therapies. Eur. J. Endocrinol. 2020, 183, R95–R106. [Google Scholar] [CrossRef]
- Lin, X.; Patil, S.; Gao, Y.G.; Qian, A. The Bone Extracellular Matrix in Bone Formation and Regeneration. Front. Pharmacol. 2020, 11, 757. [Google Scholar] [CrossRef]
- Young, M.F. Bone matrix proteins: Their function, regulation, and relationship to osteoporosis. Osteoporos. Int. 2003, 14 (Suppl. 3), S35–S42. [Google Scholar] [CrossRef] [PubMed]
- Karsenty, G. Update on the Biology of Osteocalcin. Endocr. Pract. 2017, 23, 1270–1274. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Mizutani, S.; Muto, T.; Yoneya, T.; Hino, R.; Takeda, S.; Takeuchi, Y.; Fujita, T.; Fukumoto, S.; Yamashita, T. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc. Natl. Acad. Sci. USA 2001, 98, 6500–6505. [Google Scholar] [CrossRef] [Green Version]
- Ling, Y.; Rios, H.F.; Myers, E.R.; Lu, Y.; Feng, J.Q.; Boskey, A.L. DMP1 depletion decreases bone mineralization in vivo: An FTIR imaging analysis. J. Bone Miner. Res. 2005, 20, 2169–2177. [Google Scholar] [CrossRef]
- Feng, J.Q.; Ward, L.M.; Liu, S.; Lu, Y.; Xie, Y.; Yuan, B.; Yu, X.; Rauch, F.; Davis, S.I.; Zhang, S.; et al. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat. Genet. 2006, 38, 1310–1315. [Google Scholar] [CrossRef]
- Liu, S.; Zhou, J.; Tang, W.; Jiang, X.; Rowe, D.W.; Quarles, L.D. Pathogenic role of Fgf23 in Hyp mice. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E38–E49. [Google Scholar] [CrossRef]
- Bitzan, M.; Goodyer, P.R. Hypophosphatemic Rickets. Pediatr. Clin. N. Am. 2019, 66, 179–207. [Google Scholar] [CrossRef]
- Yorgan, T.A.; Peters, S.; Amling, M.; Schinke, T. Osteoblast-specific expression of Panx3 is dispensable for postnatal bone remodeling. Bone 2019, 127, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Koch, M.; Schulze, J.; Hansen, U.; Ashwodt, T.; Keene, D.R.; Brunken, W.J.; Burgeson, R.E.; Bruckner, P.; Bruckner-Tuderman, L. A novel marker of tissue junctions, collagen XXII. J. Biol. Chem. 2004, 279, 22514–22521. [Google Scholar] [CrossRef] [Green Version]
- Ricard-Blum, S.; Ruggiero, F. The collagen superfamily: From the extracellular matrix to the cell membrane. Pathol. Biol. 2005, 53, 430–442. [Google Scholar] [CrossRef] [PubMed]
- Schulze, J.; Bickert, T.; Beil, F.T.; Zaiss, M.M.; Albers, J.; Wintges, K.; Streichert, T.; Klaetschke, K.; Keller, J.; Hissnauer, T.N.; et al. Interleukin-33 is expressed in differentiated osteoblasts and blocks osteoclast formation from bone marrow precursor cells. J. Bone Miner. Res. 2011, 26, 704–717. [Google Scholar] [CrossRef]
- Vollersen, N.; Hermans-Borgmeyer, I.; Cornils, K.; Fehse, B.; Rolvien, T.; Triviai, I.; Jeschke, A.; Oheim, R.; Amling, M.; Schinke, T.; et al. High Bone Turnover in Mice Carrying a Pathogenic Notch2 Mutation Causing Hajdu-Cheney Syndrome. J. Bone Miner. Res. 2018, 33, 70–83. [Google Scholar] [CrossRef] [Green Version]
- Yorgan, T.A.; Peters, S.; Jeschke, A.; Benisch, P.; Jakob, F.; Amling, M.; Schinke, T. The Anti-Osteoanabolic Function of Sclerostin Is Blunted in Mice Carrying a High Bone Mass Mutation of Lrp5. J. Bone Miner. Res. 2015, 30, 1175–1183. [Google Scholar] [CrossRef]
- Bouxsein, M.L.; Boyd, S.K.; Christiansen, B.A.; Guldberg, R.E.; Jepsen, K.J.; Muller, R. Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. J. Bone Miner. Res. 2010, 25, 1468–1486. [Google Scholar] [CrossRef]
- Keller, J.; Catala-Lehnen, P.; Huebner, A.K.; Jeschke, A.; Heckt, T.; Lueth, A.; Krause, M.; Koehne, T.; Albers, J.; Schulze, J.; et al. Calcitonin controls bone formation by inhibiting the release of sphingosine 1-phosphate from osteoclasts. Nat. Commun. 2014, 5, 5215. [Google Scholar] [CrossRef]
- Schinke, T.; Schilling, A.F.; Baranowsky, A.; Seitz, S.; Marshall, R.P.; Linn, T.; Blaeker, M.; Huebner, A.K.; Schulz, A.; Simon, R.; et al. Impaired gastric acidification negatively affects calcium homeostasis and bone mass. Nat. Med. 2009, 15, 674–681. [Google Scholar] [CrossRef]
- Dempster, D.W.; Compston, J.E.; Drezner, M.K.; Glorieux, F.H.; Kanis, J.A.; Malluche, H.; Meunier, P.J.; Ott, S.M.; Recker, R.R.; Parfitt, A.M. Standardized nomenclature, symbols, and units for bone histomorphometry: A 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J. Bone Miner. Res. 2013, 28, 2–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faul, F.; Erdfelder, E.; Buchner, A.; Lang, A.G. Statistical power analyses using G*Power 3.1: Tests for correlation and regression analyses. Behav. Res. Methods 2009, 41, 1149–1160. [Google Scholar] [CrossRef] [Green Version]
- Charvet, B.; Guiraud, A.; Malbouyres, M.; Zwolanek, D.; Guillon, E.; Bretaud, S.; Monnot, C.; Schulze, J.; Bader, H.L.; Allard, B.; et al. Knockdown of col22a1 gene in zebrafish induces a muscular dystrophy by disruption of the myotendinous junction. Development 2013, 140, 4602–4613. [Google Scholar] [CrossRef] [Green Version]
- Ton, Q.V.; Leino, D.; Mowery, S.A.; Bredemeier, N.O.; Lafontant, P.J.; Lubert, A.; Gurung, S.; Farlow, J.L.; Foroud, T.M.; Broderick, J.; et al. Collagen COL22A1 maintains vascular stability and mutations in COL22A1 are potentially associated with intracranial aneurysms. Dis. Model. Mech. 2018, 11, dmm033654. [Google Scholar] [CrossRef] [Green Version]
- Farlow, J.L.; Lin, H.; Sauerbeck, L.; Lai, D.; Koller, D.L.; Pugh, E.; Hetrick, K.; Ling, H.; Kleinloog, R.; van der Vlies, P.; et al. Lessons learned from whole exome sequencing in multiplex families affected by a complex genetic disorder, intracranial aneurysm. PLoS ONE 2015, 10, e0121104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forlino, A.; Porter, F.D.; Lee, E.J.; Westphal, H.; Marini, J.C. Use of the Cre/lox recombination system to develop a non-lethal knock-in murine model for osteogenesis imperfecta with an alpha1(I) G349C substitution. Variability in phenotype in BrtlIV mice. J. Biol. Chem. 1999, 274, 37923–37931. [Google Scholar] [CrossRef] [Green Version]
- Chipman, S.D.; Sweet, H.O.; McBride, D.J., Jr.; Davisson, M.T.; Marks, S.C., Jr.; Shuldiner, A.R.; Wenstrup, R.J.; Rowe, D.W.; Shapiro, J.R. Defective pro alpha 2(I) collagen synthesis in a recessive mutation in mice: A model of human osteogenesis imperfecta. Proc. Natl. Acad. Sci. USA 1993, 90, 1701–1705. [Google Scholar] [CrossRef] [Green Version]
- Zwolanek, D.; Veit, G.; Eble, J.A.; Gullberg, D.; Ruggiero, F.; Heino, J.; Meier, M.; Stetefeld, J.; Koch, M. Collagen XXII binds to collagen-binding integrins via the novel motifs GLQGER and GFKGER. Biochem. J. 2014, 459, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Ricard-Blum, S. The collagen family. Cold Spring Harb. Perspect. Biol. 2011, 3, a004978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyce, B.F.; Xing, L. Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch. Biochem. Biophys. 2008, 473, 139–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, C.; Chan, W.C.W.; Lam, Y.; Wang, X.; Chen, P.; Niu, B.; Ng, V.C.W.; Yeo, J.C.; Stricker, S.; Cheah, K.S.E.; et al. Lgr5 and Col22a1 Mark Progenitor Cells in the Lineage toward Juvenile Articular Chondrocytes. Stem Cell Rep. 2019, 13, 713–729. [Google Scholar] [CrossRef] [PubMed]
- Duong, L.T.; Lakkakorpi, P.; Nakamura, I.; Rodan, G.A. Integrins and signaling in osteoclast function. Matrix Biol. 2000, 19, 97–105. [Google Scholar] [CrossRef]
- Faccio, R.; Takeshita, S.; Zallone, A.; Ross, F.P.; Teitelbaum, S.L. c-Fms and the alphavbeta3 integrin collaborate during osteoclast differentiation. J. Clin. Investig. 2003, 111, 749–758. [Google Scholar] [CrossRef] [Green Version]
- Teitelbaum, S.L. Osteoclasts and integrins. Ann. N. Y. Acad. Sci. 2006, 1068, 95–99. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, W.; Wiedemann, P.; Wölfel, E.M.; Neven, M.; Peters, S.; Imhof, T.; Koch, M.; Busse, B.; Amling, M.; Schinke, T.; et al. Decreased Trabecular Bone Mass in Col22a1-Deficient Mice. Cells 2021, 10, 3020. https://doi.org/10.3390/cells10113020
Zhao W, Wiedemann P, Wölfel EM, Neven M, Peters S, Imhof T, Koch M, Busse B, Amling M, Schinke T, et al. Decreased Trabecular Bone Mass in Col22a1-Deficient Mice. Cells. 2021; 10(11):3020. https://doi.org/10.3390/cells10113020
Chicago/Turabian StyleZhao, Wenbo, Philip Wiedemann, Eva Maria Wölfel, Mona Neven, Stephanie Peters, Thomas Imhof, Manuel Koch, Björn Busse, Michael Amling, Thorsten Schinke, and et al. 2021. "Decreased Trabecular Bone Mass in Col22a1-Deficient Mice" Cells 10, no. 11: 3020. https://doi.org/10.3390/cells10113020
APA StyleZhao, W., Wiedemann, P., Wölfel, E. M., Neven, M., Peters, S., Imhof, T., Koch, M., Busse, B., Amling, M., Schinke, T., & Yorgan, T. A. (2021). Decreased Trabecular Bone Mass in Col22a1-Deficient Mice. Cells, 10(11), 3020. https://doi.org/10.3390/cells10113020