
Roles for ADAM17 in TNF-R1 Mediated Cell Death and Survival in Human U937 and Jurkat Cells

 ,
,  , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Cell Culture
2.3. CRISPR/Cas9 Mediated Knock-Out
2.4. SDS-PAGE/Western Blot: IκB and Cell Death Assay, Whole Cell Lysate
2.5. Guava MUSE Cell Death Assay
2.6. TNF-R1 Surface Expression, Internalization and ADAM17 Activity Assay
3. Results
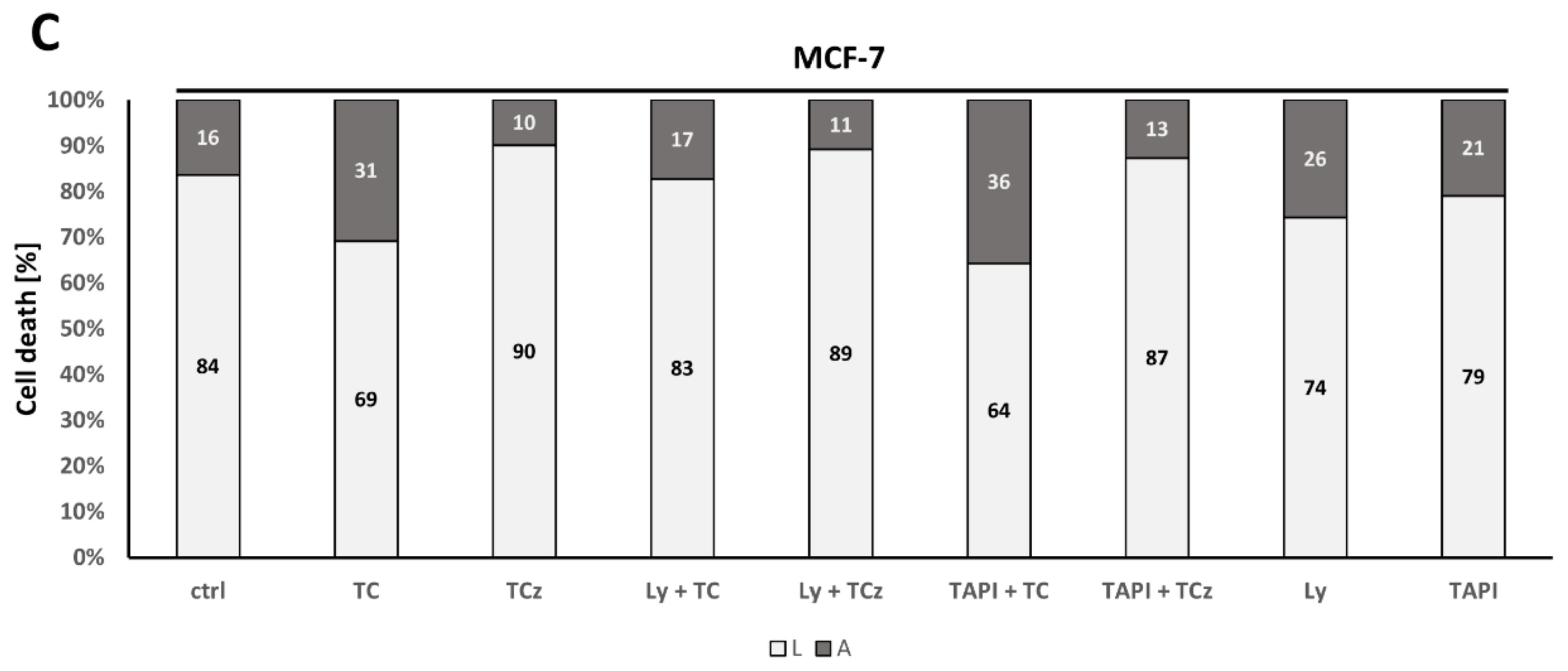
3.1. Analysis of Cell Death Induction upon Pharmacological ADAM17 Inhibition
3.2. Knock-Out of ADAM17 Does Not Alter TNF-R1 Surface Expression and Internalization
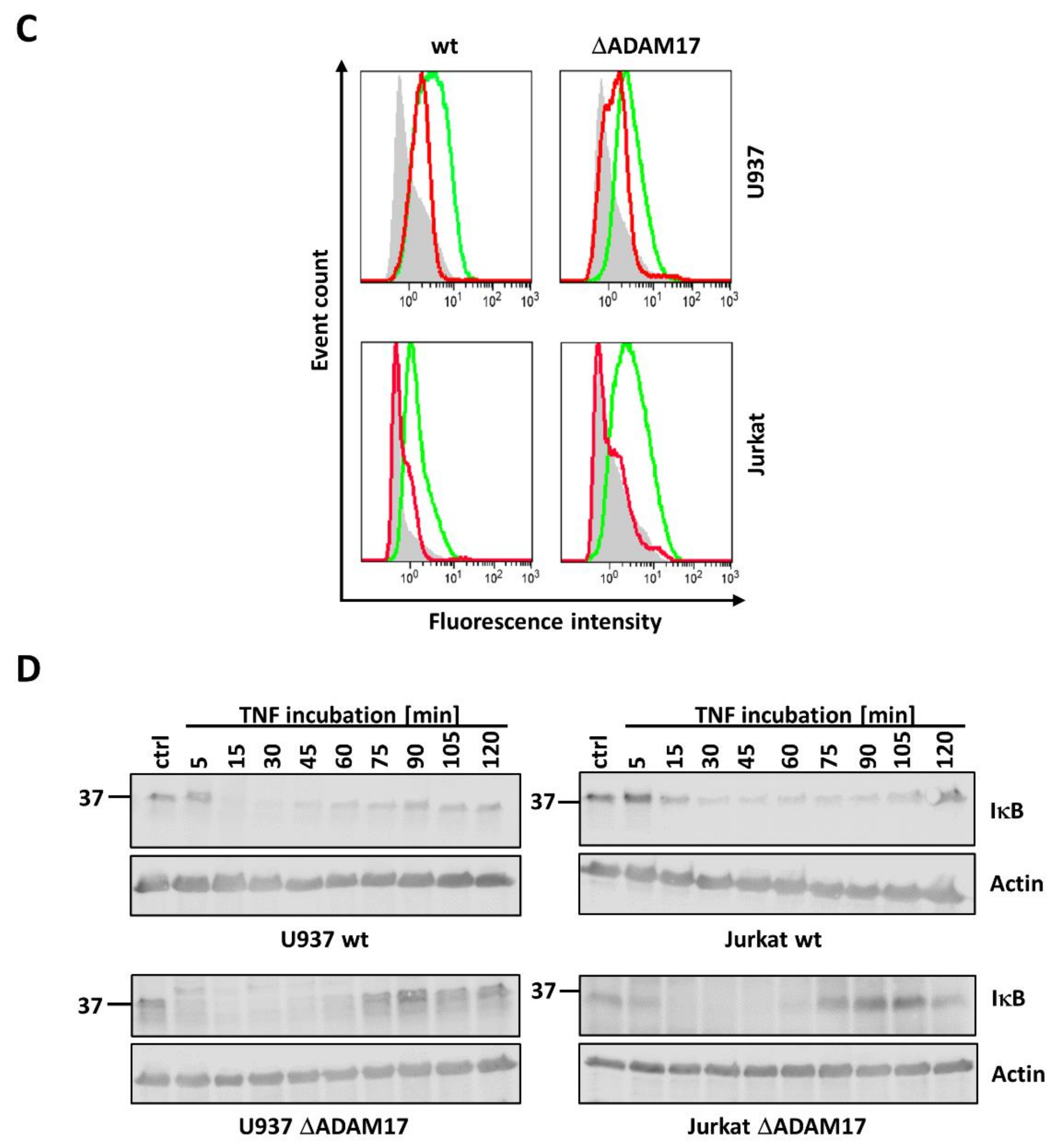
3.3. ADAM17 Knock-Out Does Not Affect TNF Induced NFκB Activation
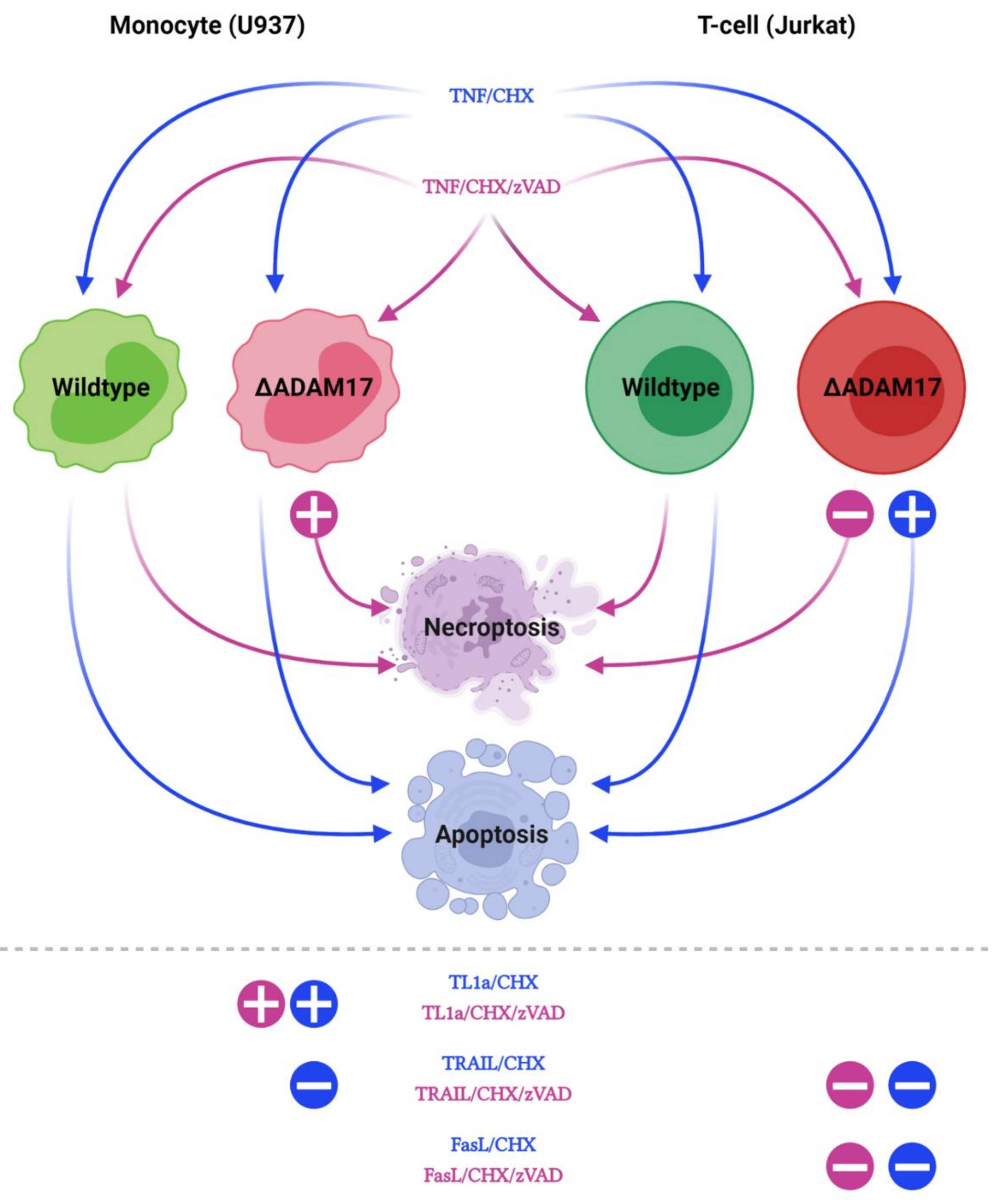
3.4. Genetic ADAM17 Deficiency Has Differential Effects on TNF Induced Cell Death in U937, Jurkat and mEF Cells
3.5. ADAM17 in the Regulation of DR3 Signal Transduction
3.6. Pharmacological Inhibition of γ-Secretase Does Not Alter TNF-R1 Mediated Cell Death Induction
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dostert, C.; Grusdat, M.; Letellier, E.; Brenner, D. The TNF Family of Ligands and Receptors: Communication Modules in the Immune System and Beyond. Physiol. Rev. 2019, 99, 115–160. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, J.; Särchen, V.; Schneider-Brachert, W. Regulation of Death Receptor Signaling by S-Palmitoylation and Detergent-Resistant Membrane Micro Domains-Greasing the Gears of Extrinsic Cell Death Induction, Survival, and Inflammation. Cancers 2021, 13, 2513. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; Fritsch, J.; Zigdon, H.; Pewzner-Jung, Y.; Schütze, S.; Futerman, A.H. Altering the sphingolipid acyl chain composition prevents LPS/GLN-mediated hepatic failure in mice by disrupting TNFR1 internalization. Cell Death Dis. 2013, 4, e929. [Google Scholar] [CrossRef]
- Edelmann, B.; Bertsch, U.; Tchikov, V.; Winoto-Morbach, S.; Perrotta, C.; Jakob, M.; Adam-Klages, S.; Kabelitz, D.; Schütze, S. Caspase-8 and caspase-7 sequentially mediate proteolytic activation of acid sphingomyelinase in TNF-R1 receptosomes. EMBO J. 2011, 30, 379–394. [Google Scholar] [CrossRef]
- Fritsch, J.; Stephan, M.; Tchikov, V.; Winoto-Morbach, S.; Gubkina, S.; Kabelitz, D.; Schütze, S. Cell Fate Decisions Regulated by K63 Ubiquitination of Tumor Necrosis Factor Receptor 1. Mol. Cell. Biol. 2014, 34, 3214–3228. [Google Scholar] [CrossRef] [Green Version]
- Schneider-Brachert, W.; Tchikov, V.; Neumeyer, J.; Jakob, M.; Winoto-Morbach, S.; Held-Feindt, J.; Heinrich, M.; Merkel, O.; Ehrenschwender, M.; Adam, D.; et al. Compartmentalization of TNF receptor 1 signaling: Internalized TNF receptosomes as death signaling vesicles. Immunity 2004, 21, 415–428. [Google Scholar] [CrossRef] [Green Version]
- Chhibber-Goel, J.; Coleman-Vaughan, C.; Agrawal, V.; Sawhney, N.; Hickey, E.; Powell, J.C.; McCarthy, J.V. γ-Secretase Activity Is Required for Regulated Intramembrane Proteolysis of Tumor Necrosis Factor (TNF) Receptor 1 and TNF-mediated Pro-apoptotic Signaling. J. Biol. Chem. 2016, 291, 5971–5985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schumacher, N.; Rose-John, S.; Schmidt-Arras, D. ADAM-Mediated Signalling Pathways in Gastrointestinal Cancer Formation. Int. J. Mol. Sci. 2020, 21, 5133. [Google Scholar] [CrossRef]
- Sommer, A.; Duppe, M.; Baumecker, L.; Kordowski, F.; Buch, J.; Chico, J.F.; Fritsch, J.; Schutze, S.; Adam, D.; Sperrhacke, M.; et al. Extracellular sphingomyelinase activity impairs TNF-alpha-induced endothelial cell death via ADAM17 activation and TNF receptor 1 shedding. Oncotarget 2017, 8, 72584–72596. [Google Scholar] [CrossRef] [Green Version]
- Fuchslocher Chico, J.; Falk-Paulsen, M.; Luzius, A.; Saggau, C.; Ruder, B.; Bolik, J.; Schmidt-Arras, D.; Linkermann, A.; Becker, C.; Rosenstiel, P.; et al. The enhanced susceptibility of ADAM-17 hypomorphic mice to DSS-induced colitis is not ameliorated by loss of RIPK3, revealing an unexpected function of ADAM-17 in necroptosis. Oncotarget 2018, 9, 12941–12958. [Google Scholar] [CrossRef] [Green Version]
- Bolik, J.; Krause, F.; Stevanovic, M.; Gandraß, M.; Thomsen, I.; Schacht, S.; Rieser, E.; Müller, M.; Schumacher, N.; Fritsch, J.; et al. Inhibition of ADAM17 impairs endothelial cell necroptosis and blocks metastasis. J. Exp. Med. 2021. [Google Scholar] [CrossRef]
- Stephan, M.; Edelmann, B.; Winoto-Morbach, S.; Janssen, O.; Bertsch, U.; Perrotta, C.; Schütze, S.; Fritsch, J. Role of caspases in CD95-induced biphasic activation of acid sphingomyelinase. Oncotarget 2017, 8, 20067–20085. [Google Scholar] [CrossRef] [Green Version]
- Chalaris, A.; Adam, N.; Sina, C.; Rosenstiel, P.; Lehmann-Koch, J.; Schirmacher, P.; Hartmann, D.; Cichy, J.; Gavrilova, O.; Schreiber, S.; et al. Critical role of the disintegrin metalloprotease ADAM17 for intestinal inflammation and regeneration in mice. J. Exp. Med. 2010, 207, 1617–1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambrecht, B.N.; Vanderkerken, M.; Hammad, H. The emerging role of ADAM metalloproteinases in immunity. Nat. Rev. Immunol. 2018, 18, 745–758. [Google Scholar] [CrossRef]
- Doedens, J.R.; Black, R.A. Stimulation-induced down-regulation of tumor necrosis factor-alpha converting enzyme. J. Biol. Chem. 2000, 275, 14598–14607. [Google Scholar] [CrossRef] [Green Version]
- Bittner, S.; Ehrenschwender, M. Multifaceted death receptor 3 signaling-promoting survival and triggering death. FEBS Lett. 2017, 591, 2543–2555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriwaki, K.; Bertin, J.; Gough, P.J.; Orlowski, G.M.; Chan, F.K. Differential roles of RIPK1 and RIPK3 in TNF-induced necroptosis and chemotherapeutic agent-induced cell death. Cell Death Dis. 2015, 6, e1636. [Google Scholar] [CrossRef] [Green Version]
- Fritsch, J.; Zingler, P.; Särchen, V.; Heck, A.L.; Schütze, S. Role of ubiquitination and proteolysis in the regulation of pro- and anti-apoptotic TNF-R1 signaling. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2138–2146. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Fujita, K.-I.; Xiao, Q.; Tchikov, V.; Yang, W.; Gunsor, M.; Garfield, S.; Goldsmith, P.; El-Deiry, W.S.; Schütze, S.; et al. Response: CARP1 regulates induction of NF-κB by TNFα. Curr. Biol. 2009, 19, R17–R19. [Google Scholar] [CrossRef] [Green Version]
- Liao, W.; Xiao, Q.; Tchikov, V.; Fujita, K.; Yang, W.; Wincovitch, S.; Garfield, S.; Conze, D.; El-Deiry, W.S.; Schütze, S.; et al. CARP-2 is an endosome-associated ubiquitin ligase for RIP and regulates TNF-induced NF-kappaB activation. Curr. Biol. 2008, 18, 641–649. [Google Scholar] [CrossRef] [Green Version]
- Yazdanpanah, B.; Wiegmann, K.; Tchikov, V.; Krut, O.; Pongratz, C.; Schramm, M.; Kleinridders, A.; Wunderlich, T.; Kashkar, H.; Utermohlen, O.; et al. Riboflavin kinase couples TNF receptor 1 to NADPH oxidase. Nature 2009, 460, 1159–1163. [Google Scholar] [CrossRef] [PubMed]
- Holdbrooks, A.T.; Britain, C.M.; Bellis, S.L. ST6Gal-I sialyltransferase promotes tumor necrosis factor (TNF)-mediated cancer cell survival via sialylation of the TNF receptor 1 (TNFR1) death receptor. J. Biol. Chem. 2018, 293, 1610–1622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Swindall, A.F.; Kesterson, R.A.; Schoeb, T.R.; Bullard, D.C.; Bellis, S.L. ST6Gal-I regulates macrophage apoptosis via alpha2-6 sialylation of the TNFR1 death receptor. J. Biol. Chem. 2011, 286, 39654–39662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.H.; Chang, L.S. Suppression of Akt/Foxp3-mediated miR-183 expression blocks Sp1-mediated ADAM17 expression and TNFalpha-mediated NFκB activation in piceatannol-treated human leukemia U937 cells. Biochem. Pharmacol. 2012, 84, 670–680. [Google Scholar] [CrossRef] [PubMed]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fritsch, J.; Frankenheim, J.; Marischen, L.; Vadasz, T.; Troeger, A.; Rose-John, S.; Schmidt-Arras, D.; Schneider-Brachert, W. Roles for ADAM17 in TNF-R1 Mediated Cell Death and Survival in Human U937 and Jurkat Cells. Cells 2021, 10, 3100. https://doi.org/10.3390/cells10113100
Fritsch J, Frankenheim J, Marischen L, Vadasz T, Troeger A, Rose-John S, Schmidt-Arras D, Schneider-Brachert W. Roles for ADAM17 in TNF-R1 Mediated Cell Death and Survival in Human U937 and Jurkat Cells. Cells. 2021; 10(11):3100. https://doi.org/10.3390/cells10113100
Chicago/Turabian StyleFritsch, Jürgen, Julia Frankenheim, Lothar Marischen, Timea Vadasz, Anja Troeger, Stefan Rose-John, Dirk Schmidt-Arras, and Wulf Schneider-Brachert. 2021. "Roles for ADAM17 in TNF-R1 Mediated Cell Death and Survival in Human U937 and Jurkat Cells" Cells 10, no. 11: 3100. https://doi.org/10.3390/cells10113100
APA StyleFritsch, J., Frankenheim, J., Marischen, L., Vadasz, T., Troeger, A., Rose-John, S., Schmidt-Arras, D., & Schneider-Brachert, W. (2021). Roles for ADAM17 in TNF-R1 Mediated Cell Death and Survival in Human U937 and Jurkat Cells. Cells, 10(11), 3100. https://doi.org/10.3390/cells10113100