
Heat Shock-Related Protein Responses and Inflammatory Protein Changes Are Associated with Mild Prolonged Hypoglycemia


 ,
,
Abstract
:
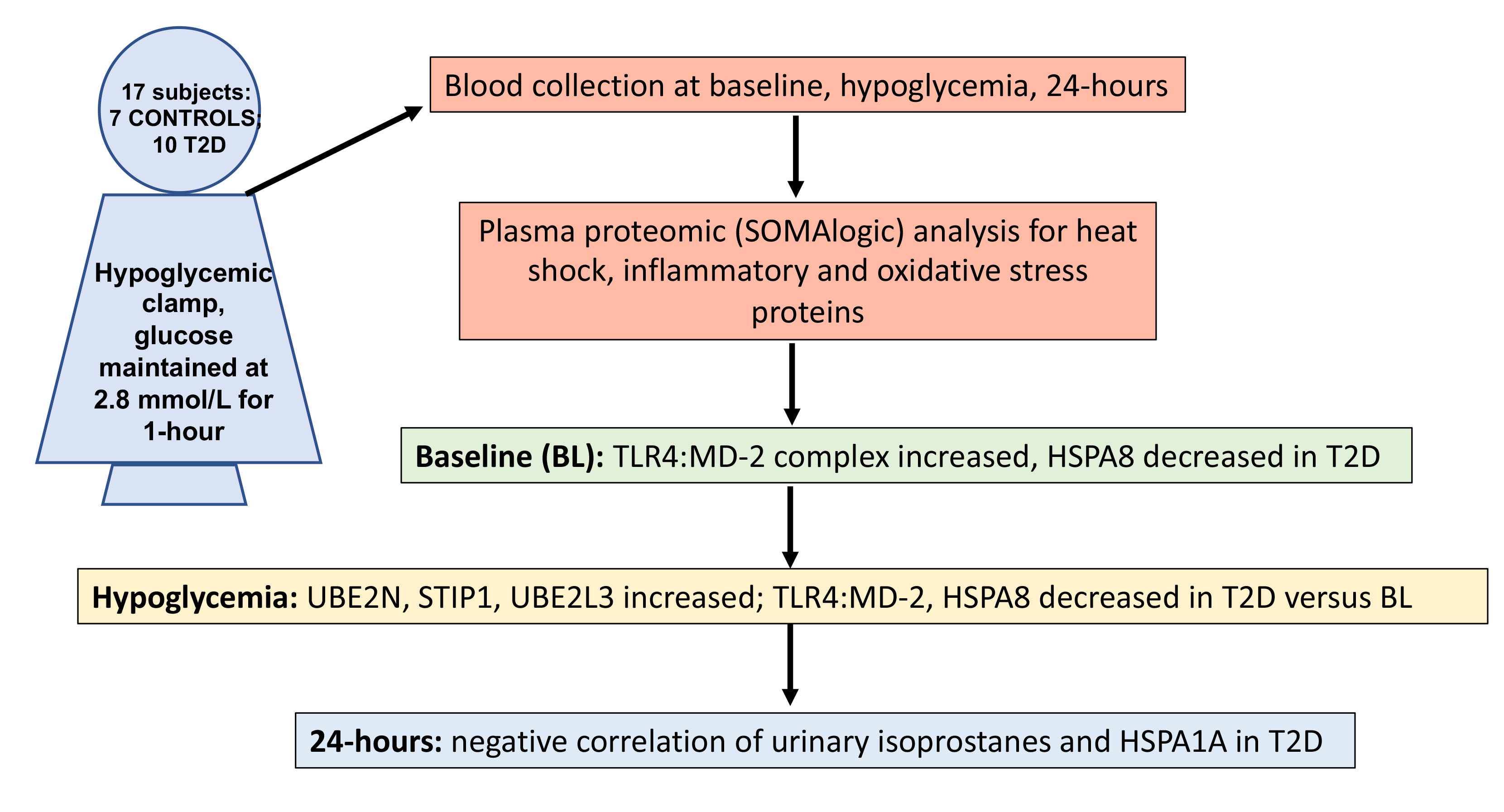
1. Background
2. Methods
2.1. Biochemical Markers
2.2. SOMA-Scan Assay and Statistical Analysis
2.3. Data Processing and Analysis
2.4. Protein–Protein Interaction Tools
3. Results
3.1. Study Participants
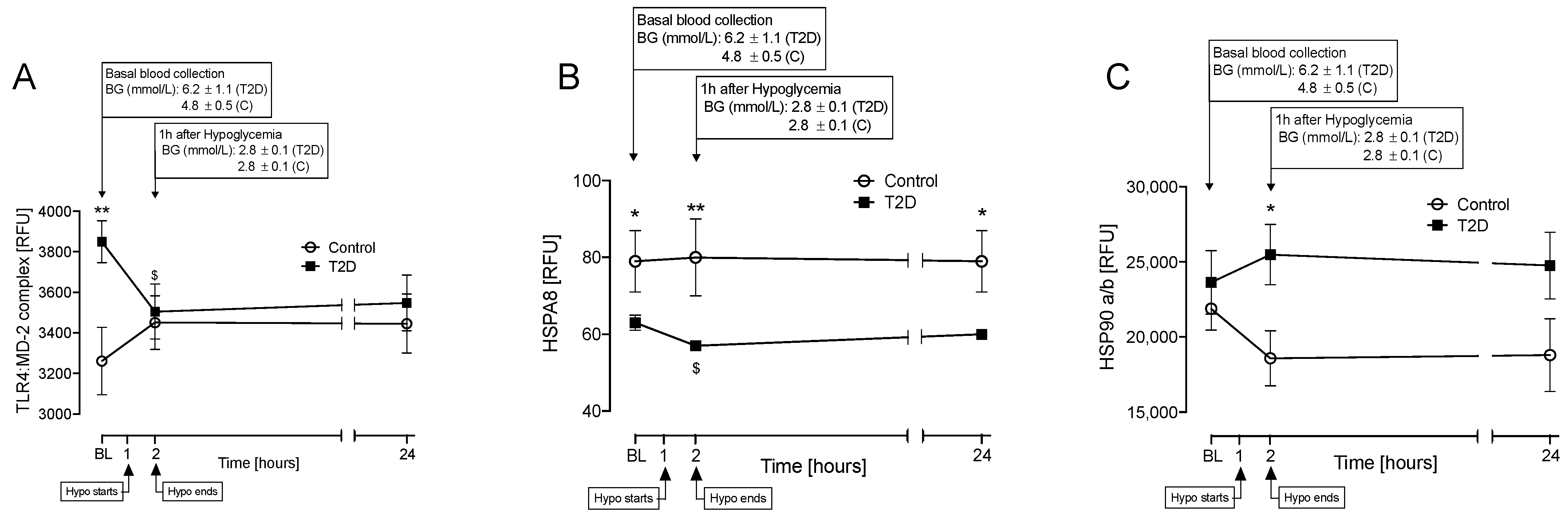
3.2. Proteins That Differed between T2D and Control at Baseline and at Hypoglycemia
3.3. Correlations between Inflammatory and Oxidative Stress Markers for HSPs That Differed Significantly between T2D and Control at Baseline (TL4:MD-2 Complex and HSPA8)
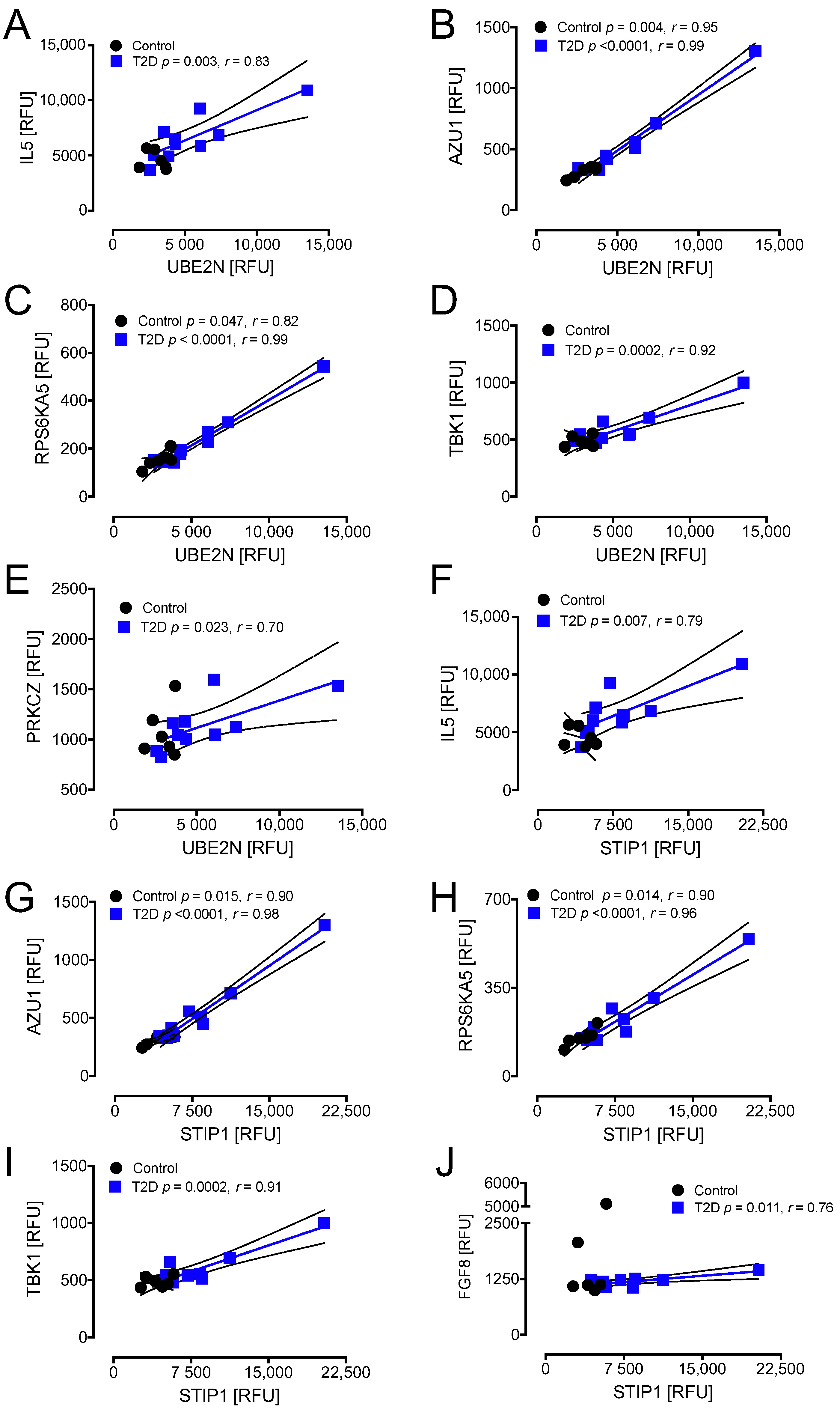
3.4. Correlations between Inflammatory and Oxidative Stress Markers for HSPs (UBE2N and STIP1) and UBE2L3 That Differed Significantly between T2D and Control at Hypoglycemia
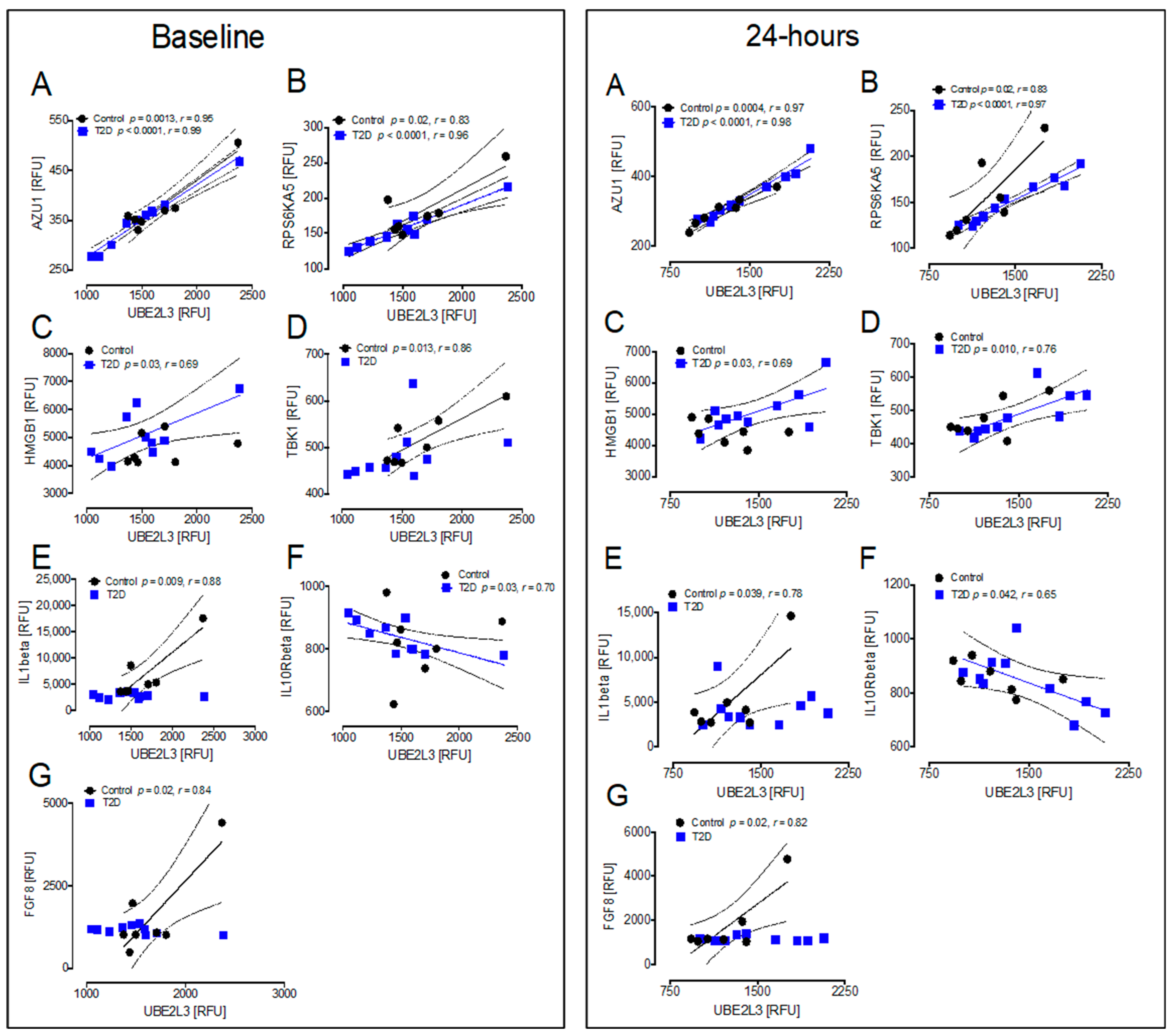
3.5. Correlations between Inflammatory and Oxidative Stress Markers for the HSP UBE2L3 That Differed Significantly between T2D and Controls at Baseline and at 24 h after Hypoglycemia
3.6. Correlations between Urinary Isoprostane 8-Iso PGF2α and HSPs
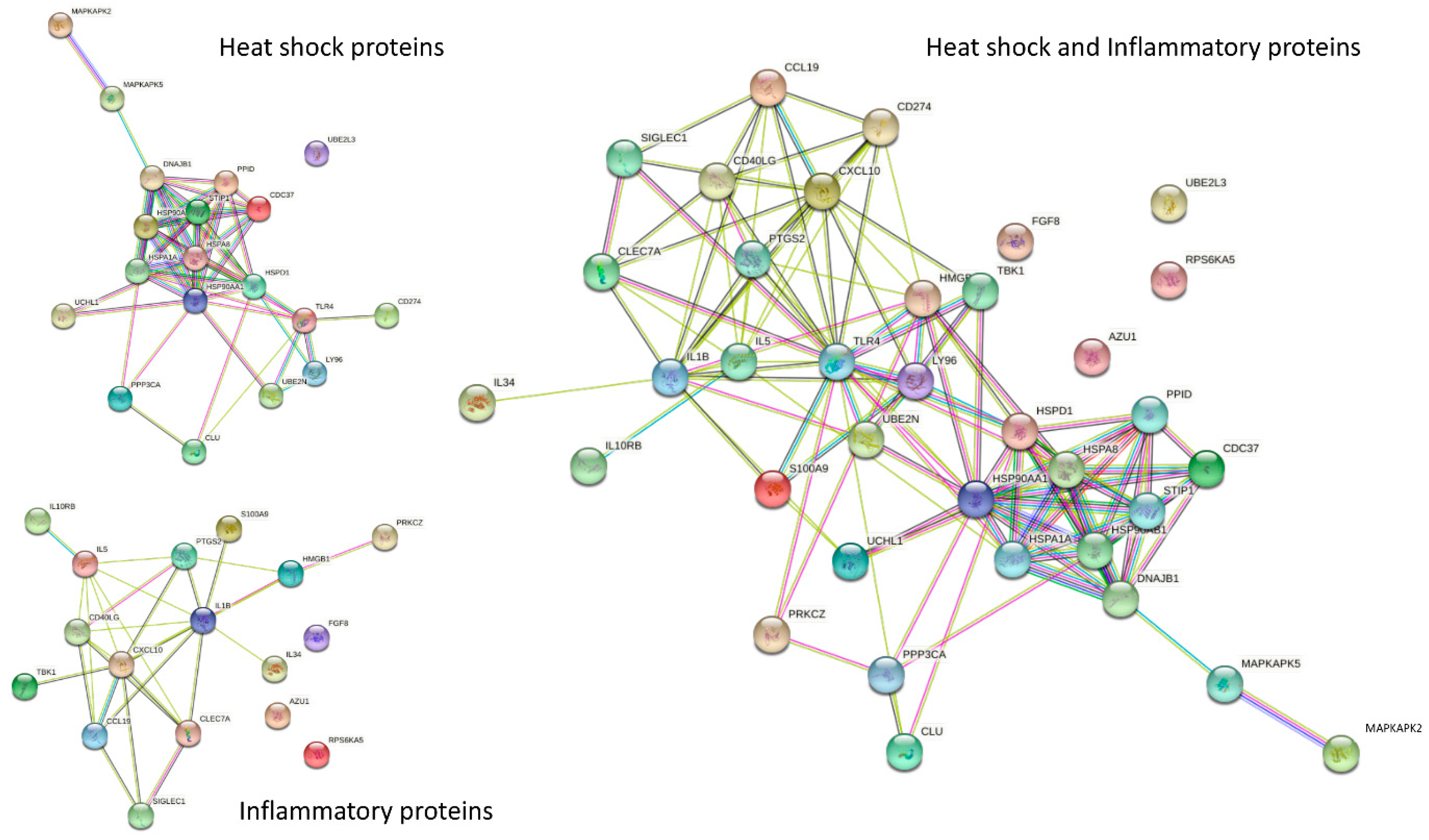
3.7. Interaction of HSPs by STRING Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| HSPA1A | Heat shock 70 kDa protein 1A |
| HSPA8 | Heat shock cognate 71 kDa protein |
| HSPD1 | Heat shock protein family D (Hsp60) member 1 |
| CDC37 | Hsp90 co-chaperone Cdc37 |
| CLU | Clusterin |
| DNAJB1 | DnaJ homolog subfamily B member 1 |
| MAPKAPK2 | MAP kinase-activated protein kinase 2 |
| MAPKAPK5 | MAP kinase-activated protein kinase 5 |
| PPID | Peptidyl-prolyl cis-trans isomerase D |
| STIP1 | Stress-induced-phosphoprotein 1 |
| TLR4:MD-2 complex | Toll-like receptor 4 in complex with MD-2 |
| HSP90a/b | HSP90 dimer |
| CD274 | Programmed cell death 1 ligand 1 |
| UBE2L3 | Ubiquitin-conjugating enzyme E2L 3 |
| UBE2N | Ubiquitin-conjugating enzyme E2 N |
| UCHL1 | Ubiquitin carboxyl-terminal hydrolase isozyme L1 |
| AZU1 | Azurocidin 1 |
| CD40 ligand | Cluster of differentiation 40 ligand |
| IL34 | Interleukin 34 |
| IL5 | Interleukin 5 |
| RPS6KA5 | Ribosomal protein S6 kinase 5 |
| TBK1 | TANK-binding kinase 1 |
| PRKCZ | Protein kinase C zeta type |
| FGF8 | Fibroblast growth factor 8 |
| HMGB1 | High mobility group box 1 |
| IL10Rbeta | Interleukin 10 receptor beta |
References
- Donnelly, L.A.; Morris, A.D.; Frier, B.M.; Ellis, J.D.; Donnan, P.T.; Durrant, R.; Band, M.M.; Reekie, G.; Leese, G.P. Frequency and predictors of hypoglycaemia in Type 1 and insulin-treated Type 2 diabetes: A population-based study. Diabet. Med. 2005, 22, 749–755. [Google Scholar] [CrossRef]
- Chin, S.O.; Rhee, S.Y.; Chon, S.; Baik, S.H.; Park, Y.; Nam, M.S.; Lee, K.W.; Chun, K.H.; Woo, J.-T.; Kim, Y.S. Hypoglycemia is associated with dementia in elderly patients with type 2 diabetes mellitus: An analysis based on the Korea National Diabetes Program Cohort. Diabetes Res. Clin. Pract. 2016, 122, 54–61. [Google Scholar] [CrossRef]
- Mukherjee, A.; Morales-Scheihing, D.; Butler, P.; Soto, C. Type 2 diabetes as a protein misfolding disease. Trends Mol. Med. 2015, 21, 439–449. [Google Scholar] [CrossRef] [Green Version]
- Costes, S.; Huang, C.-J.; Gurlo, T.; Daval, M.; Matveyenko, A.V.; Rizza, R.A.; Butler, A.E.; Butler, P.C. β-cell dysfunctional ERAD/ubiquitin/proteasome system in type 2 diabetes mediated by islet amyloid polypeptide-induced UCH-L1 deficiency. Diabetes 2011, 60, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Kampinga, H.H.; Hageman, J.; Vos, M.; Kubota, H.; Tanguay, R.M.; Bruford, E.; Cheetham, M.; Chen, B.; Hightower, L.E. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones 2009, 14, 105–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nitika; Porter, C.M.; Truman, A.W.; Truttmann, M.C. Post-translational modifications of Hsp70 family proteins: Expanding the chaperone code. J. Biol. Chem. 2020, 295, 10689–10708. [Google Scholar] [CrossRef]
- Backe, S.J.; Sager, R.A.; Woodford, M.R.; Makedon, A.M.; Mollapour, M. Post-translational modifications of Hsp90 and translating the chaperone code. J. Biol. Chem. 2020, 295, 11099–11117. [Google Scholar] [CrossRef]
- Takakuwa, J.E.; Nitika; Knighton, L.E.; Truman, A.W. Oligomerization of Hsp70: Current Perspectives on Regulation and Function. Front. Mol. Biosci. 2019, 6, 81. [Google Scholar] [CrossRef] [Green Version]
- Vigouroux, S.; Briand, M.; Briand, Y. Linkage between the proteasome pathway and neurodegenerative diseases and aging. Mol. Neurobiol. 2004, 30, 201–221. [Google Scholar] [CrossRef]
- Hooper, P.L.; Hooper, P.L. Inflammation, heat shock proteins, and type 2 diabetes. Cell Stress Chaperones 2009, 14, 113–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krause, M.; Keane, K.N.; Rodrigues-Krause, J.; Crognale, D.; Egan, B.; De Vito, G.; Murphy, C.; Newsholme, P. Elevated levels of extracellular heat-shock protein 72 (eHSP72) are positively correlated with insulin resistance in vivo and cause pancreatic β-cell dysfunction and death in vitro. Clin. Sci. 2014, 126, 739–752. [Google Scholar] [CrossRef]
- Jakhotia, S.; Sivaprasad, M.; Shalini, T.; Reddy, P.Y.; Viswanath, K.; Jakhotia, K.; Sahay, R.; Sahay, M.; Reddy, G.B. Circulating levels of Hsp27 in microvascular complications of diabetes: Prospects as a biomarker of diabetic nephropathy. J. Diabetes Complicat. 2018, 32, 221–225. [Google Scholar] [CrossRef]
- Gruden, G.; Bruno, G.; Chaturvedi, N.; Burt, D.; Pinach, S.; Schalkwijk, C.; Stehouwer, C.D.; Witte, D.; Fuller, J.H.; Cavallo-Perin, P.; et al. ANTI-HSP60 and ANTI-HSP70 antibody levels and micro/macrovascular complications in type 1 diabetes: The EURODIAB Study. J. Intern. Med. 2009, 266, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Yaribeygi, H.; Farrokhi, F.R.; Rezaee, R.; Sahebkar, A. Oxidative stress induces renal failure: A review of possible molecular pathways. J. Cell. Biochem. 2017, 119, 2990–2998. [Google Scholar] [CrossRef] [PubMed]
- Yaribeygi, H.; Sathyapalan, T.; Atkin, S.L.; Sahebkar, A. Molecular Mechanisms Linking Oxidative Stress and Diabetes Mellitus. Oxidative Med. Cell. Longev. 2020, 2020, 8609213. [Google Scholar] [CrossRef] [Green Version]
- Yaribeygi, H.; Atkin, S.L.; Sahebkar, A. A review of the molecular mechanisms of hyperglycemia-induced free radical generation leading to oxidative stress. J. Cell. Physiol. 2019, 234, 1300–1312. [Google Scholar] [CrossRef]
- Siti, H.N.; Kamisah, Y.; Kamsiah, J. The role of oxidative stress, antioxidants and vascular inflammation in cardiovascular disease (a review). Vasc. Pharmacol. 2015, 71, 40–56. [Google Scholar] [CrossRef] [PubMed]
- Sahebkar, A.; Panahi, Y.; Yaribeygi, H.; Javadi, J. Oxidative Stress in Neurodegenerative Diseases: A Review; CNS & Neurological Disorders Drug Targets; Bentham Science Publishers: Sharjah, United Arab Emirates, 2018. [Google Scholar]
- Touyz, R.M. Molecular and cellular mechanisms in vascular injury in hypertension: Role of angiotensin II. Curr. Opin. Nephrol. Hypertens. 2005, 14, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, T. Molecular hydrogen: New antioxidant and anti-inflammatory therapy for rheumatoid arthritis and related diseases. Curr. Pharm. Des. 2013, 19, 6375–6381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahal, H.; Halama, A.; Aburima, A.; Bhagwat, A.M.; Butler, A.E.; Graumann, J.; Suhre, K.; Sathyapalan, T.; Atkin, S.L. Effect of induced hypoglycemia on inflammation and oxidative stress in type 2 diabetes and control subjects. Sci. Rep. 2020, 10, 4750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahal, H.; Aburima, A.; Spurgeon, B.; Wraith, K.; Rigby, A.; Sathyapalan, T.; Kilpatrick, E.; Naseem, K.; Atkin, S. Platelet function following induced hypoglycaemia in type 2 diabetes. Diabetes Metab. 2018, 44, 431–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halama, A.; Kahal, H.; Bhagwat, A.M.; Zierer, J.; Sathyapalan, T.; Graumann, J.; Suhre, K.; Atkin, S.L.; Grauman, J. Metabolic and proteomic signatures of hypoglycaemia in type 2 diabetes. Diabetes Obes. Metab. 2019, 21, 909–919. [Google Scholar] [CrossRef] [PubMed]
- Atkin, A.S.; Moin, A.S.M.; Al-Qaissi, A.; Sathyapalan, T.; Atkin, S.L.; Butler, A.E. Plasma heat shock protein response to euglycemia in type 2 diabetes. BMJ Open Diabetes Res. Care 2021, 9, e002057. [Google Scholar] [CrossRef] [PubMed]
- Atkin, A.S.; Moin, A.S.M.; Nandakumar, M.; Al-Qaissi, A.; Sathyapalan, T.; Atkin, S.L.; Butler, A.E. Impact of severe hypoglycemia on the heat shock and related protein response. Sci. Rep. 2021, 11, 17057. [Google Scholar] [CrossRef] [PubMed]
- Moin, A.S.M.; Al-Qaissi, A.; Sathyapalan, T.; Atkin, S.L.; Butler, A.E. Hypoglycaemia in type 2 diabetes exacerbates amyloid-related proteins associated with dementia. Diabetes Obes. Metab. 2020, 23, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, S.; Vaught, J.D.; Bock, C.; Gold, L.; Katilius, E.; Keeney, T.R.; Kim, N.; Saccomano, N.A.; Wilcox, S.K.; Zichi, D. From SOMAmer-based biomarker discovery to diagnostic and clinical applications: A SOMAmer-based, streamlined multiplex proteomic assay. PLoS ONE 2011, 6, e26332. [Google Scholar] [CrossRef] [Green Version]
- Suhre, K.; Arnold, M.; Bhagwat, A.M.; Cotton, R.J.; Engelke, R.; Raffler, J.; Sarwath, H.; Thareja, G.; Wahl, A.; DeLisle, R.K.; et al. Connecting genetic risk to disease end points through the human blood plasma proteome. Nat. Commun. 2017, 8, 14357. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Benjamini, Y.; Drai, D.; Elmer, G.; Kafkafi, N.; Golani, I. Controlling the false discovery rate in behavior genetics research. Behav. Brain Res. 2001, 125, 279–284. [Google Scholar] [CrossRef] [Green Version]
- Kavanagh, K.; Flynn, D.M.; Jenkins, K.A.; Zhang, L.; Wagner, J.D. Restoring HSP70 deficiencies improves glucose tolerance in diabetic monkeys. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E894–E901. [Google Scholar] [CrossRef] [Green Version]
- Gillies, A.T.; Taylor, R.; Gestwicki, J.E. Synthetic lethal interactions in yeast reveal functional roles of J protein co-chaperones. Mol. Biosyst. 2012, 8, 2901–2908. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, F.F.; Haines, D.; Dashti, A.A.; El-Shazly, S.; Al-Najjar, F. Correlation between heat shock proteins, adiponectin, and T lymphocyte cytokine expression in type 2 diabetics. Cell Stress Chaperones 2018, 23, 955–965. [Google Scholar] [CrossRef] [PubMed]
- Henstridge, D.C.; Whitham, M.; Febbraio, M.A. Chaperoning to the metabolic party: The emerging therapeutic role of heat-shock proteins in obesity and type 2 diabetes. Mol. Metab. 2014, 3, 781–793. [Google Scholar] [CrossRef] [PubMed]
- Trinchieri, G.; Sher, A. Cooperation of Toll-like receptor signals in innate immune defence. Nat. Rev. Immunol. 2007, 7, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Akira, S. Roles of Toll-like receptors in innate immune responses. Genes Cells 2001, 6, 733–742. [Google Scholar] [CrossRef]
- Abreu, M.T. Toll-like receptor signalling in the intestinal epithelium: How bacterial recognition shapes intestinal function. Nat. Rev. Immunol. 2010, 10, 131–144. [Google Scholar] [CrossRef]
- Sonnenburg, J.L.; Bäckhed, F. Diet–microbiota interactions as moderators of human metabolism. Nat. Cell Biol. 2016, 535, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Taha, I.M.; Allah, A.M.A.; El Gayed, E.M.A. Expression of toll-like receptor 4 and its connection with type 2 diabetes mellitus. Cell. Mol. Biol. 2018, 64, 15–20. [Google Scholar] [CrossRef]
- Dasu, M.R.; Devaraj, S.; Zhao, L.; Hwang, D.H.; Jialal, I. High Glucose Induces Toll-Like Receptor Expression in Human Monocytes: Mechanism of Activation. Diabetes 2008, 57, 3090–3098. [Google Scholar] [CrossRef] [Green Version]
- Popovic, D.; Vucic, D.; Dikic, I. Ubiquitination in disease pathogenesis and treatment. Nat. Med. 2014, 20, 1242–1253. [Google Scholar] [CrossRef] [PubMed]
- Ullah, K.; Chen, S.; Lu, J.; Wang, X.; Liu, Q.; Zhang, Y.; Long, Y.; Hu, Z.; Xu, G. The E3 ubiquitin ligase STUB1 attenuates cell senescence by promoting the ubiquitination and degradation of the core circadian regulator BMAL1. J. Biol. Chem. 2020, 295, 4696–4708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zininga, T.; Ramatsui, L.; Shonhai, A. Heat Shock Proteins as Immunomodulants. Molecules 2018, 23, 2846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.Y.; Lee, Z.-H.; Song, Y.W. CXCL10 and autoimmune diseases. Autoimmun. Rev. 2009, 8, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Schulthess, F.T.; Paroni, F.; Sauter, N.S.; Shu, L.; Ribaux, P.; Haataja, L.; Strieter, R.M.; Oberholzer, J.; King, C.C.; Maedler, K. CXCL10 impairs beta cell function and viability in diabetes through TLR4 signaling. Cell Metab. 2009, 9, 125–139. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Wu, Y.; Chen, G.-Y. Siglec-1 negatively regulates TLR4-mediated inflammatory response by uniquely controlling Src phosphorylation at Ser17. J. Immunol. 2019, 202 (Suppl. S1), 64.17. [Google Scholar]
- Lei, J.T.; Mazumdar, T.; Martinez-Moczygemba, M. Three Lysine Residues in the Common β Chain of the Interleukin-5 Receptor Are Required for Janus Kinase (JAK)-dependent receptor ubiquitination, endocytosis, and signaling. J. Biol. Chem. 2011, 286, 40091–40103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nozawa, T.; Sano, S.; Minowa-Nozawa, A.; Toh, H.; Nakajima, S.; Murase, K.; Aikawa, C.; Nakagawa, I. TBC1D9 regulates TBK1 activation through Ca2+ signaling in selective autophagy. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Della Peruta, M.; Giagulli, C.; Laudanna, C.; Scarpa, A.; Sorio, C. RHOA and PRKCZ control different aspects of cell motility in pancreatic cancer metastatic clones. Mol. Cancer 2010, 9, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowak, M.; Machate, A.; Yu, S.R.; Gupta, M.; Brand, M. Interpretation of the FGF8 morphogen gradient is regulated by endocytic trafficking. Nat. Cell Biol. 2011, 13, 153–158. [Google Scholar] [CrossRef]
- Eldridge, M.J.; Sanchez-Garrido, J.; Hoben, G.F.; Goddard, P.J.; Shenoy, A.R. The Atypical Ubiquitin E2 Conjugase UBE2L3 Is an Indirect Caspase-1 Target and Controls IL-1β Secretion by Inflammasomes. Cell Rep. 2017, 18, 1285–1297. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Controls (n = 7) | Type 2 Diabetes (n = 10) | |
| Age (years) | 47 ± 6 | 46 ± 6 |
| Sex (M/F) | 4M/3F | 7M/3F |
| BMI (kg/m2) | 29 ± 4 | 36 ± 7 |
| Systolic BP (mmHg) | 126 ± 15 | 127 ± 20 |
| Diastolic BP (mmHg) | 75 ± 13 | 75 ± 11 |
| Duration of diabetes (years) | N/A | 3.3 ± 2.3 |
| HbA1c (mmol/mol) | 33.6 ± 2.9 | 49 ± 12 |
| HbA1c (%) | 5.2 ± 0.3 | 6.6 ± 1.0 |
| Total cholesterol (mmol/L) | 5.1 ± 0.8 | 5.3 ± 0.7 |
| Triglyceride (mmol/L) | 1.2 ± 0.5 | 1.7 ± 0.8 |
| CRP (mg/L) | 0.8 ± 0.0 | 2.8 ± 1.8 |
| Urinary isoprostane 8-iso PGF2α (baseline) (ng/mL) | 53.7 ± 17.1 | 73.4 ± 9.6 |
| Urinary isoprostane 8-iso PGF2α (24 h) (ng/mL) | 85.0 ± 21.6 | 91.7 ± 5.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moin, A.S.M.; Nandakumar, M.; Kahal, H.; Sathyapalan, T.; Atkin, S.L.; Butler, A.E. Heat Shock-Related Protein Responses and Inflammatory Protein Changes Are Associated with Mild Prolonged Hypoglycemia. Cells 2021, 10, 3109. https://doi.org/10.3390/cells10113109
Moin ASM, Nandakumar M, Kahal H, Sathyapalan T, Atkin SL, Butler AE. Heat Shock-Related Protein Responses and Inflammatory Protein Changes Are Associated with Mild Prolonged Hypoglycemia. Cells. 2021; 10(11):3109. https://doi.org/10.3390/cells10113109
Chicago/Turabian StyleMoin, Abu Saleh Md, Manjula Nandakumar, Hassan Kahal, Thozhukat Sathyapalan, Stephen L. Atkin, and Alexandra E. Butler. 2021. "Heat Shock-Related Protein Responses and Inflammatory Protein Changes Are Associated with Mild Prolonged Hypoglycemia" Cells 10, no. 11: 3109. https://doi.org/10.3390/cells10113109
APA StyleMoin, A. S. M., Nandakumar, M., Kahal, H., Sathyapalan, T., Atkin, S. L., & Butler, A. E. (2021). Heat Shock-Related Protein Responses and Inflammatory Protein Changes Are Associated with Mild Prolonged Hypoglycemia. Cells, 10(11), 3109. https://doi.org/10.3390/cells10113109