
Lymphocyte Counts and Multiple Sclerosis Therapeutics: Between Mechanisms of Action and Treatment-Limiting Side Effects



Abstract
:1. Introduction
2. General Information
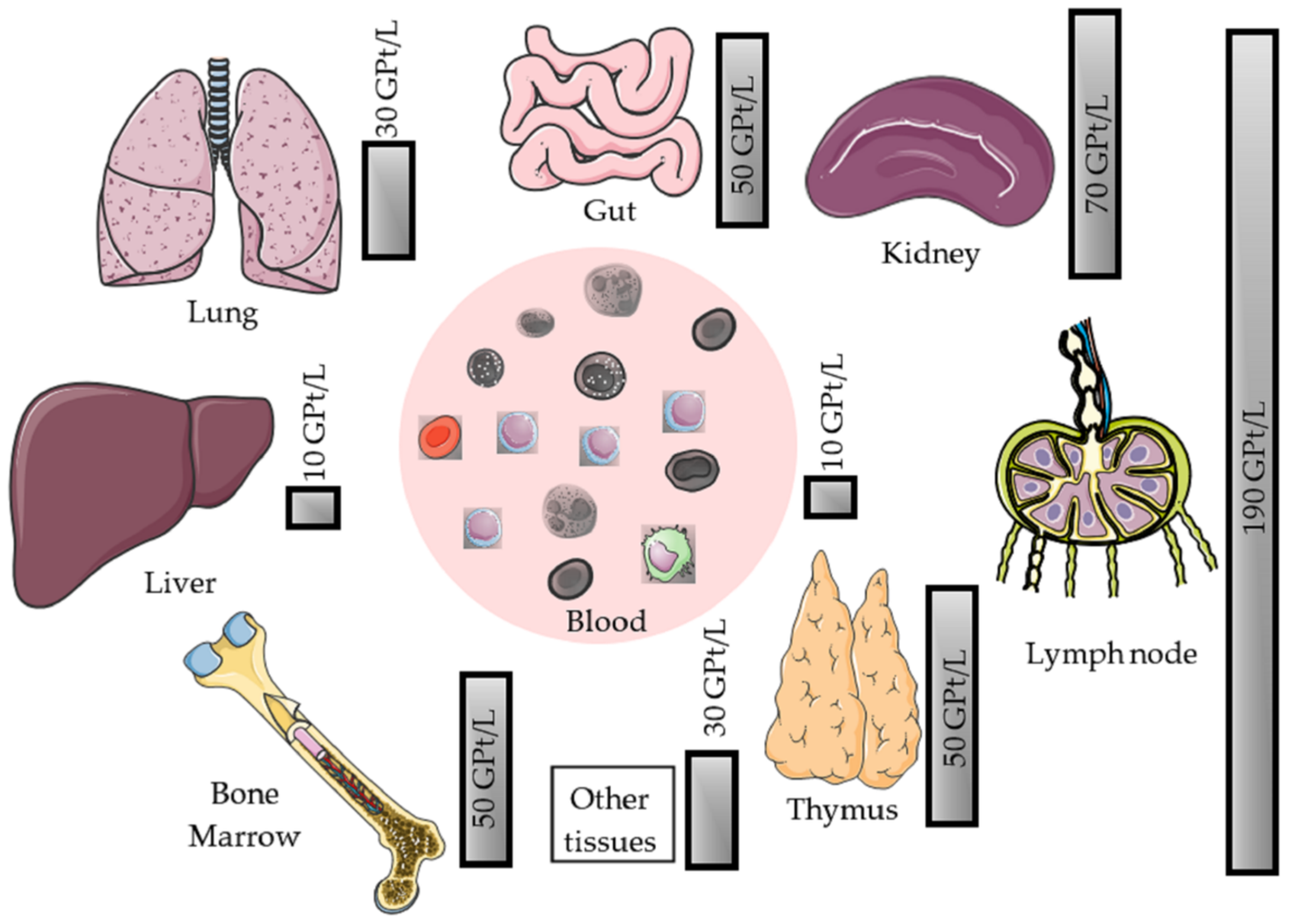
2.1. Physiology of Peripheral Blood Lymphocytes
2.2. Role of Lymphocytes in the Pathogenesis of MS
2.3. Effects of Disease-Modifying Therapy (DMT) on Lymphocyte Number and Function
2.4. Definition of Lymphocytopenia (Lymphopenia)
2.5. Potential Relevant Lymphopenia-Associated Complications
2.5.1. Opportunistic Infections
2.5.2. Immune Reconstitution Inflammatory Syndrome
2.5.3. Secondary Autoimmunity
2.6. Recommended Monitoring of Lymphocyte Count
3. Disease-Modifying Drugs and Their Effects on Lymphocyte Count
3.1. Mechanism of Action: Immunomodulation
3.1.1. Glatiramer Acetate
General Facts and Clinical Trial Data
Mechanism of Action and Impact on Lymphocyte Count
Recommended Monitoring
3.1.2. Interferons
General Facts and Clinical Trial Data
Mechanism of Action and Impact on Lymphocyte Count
- (a)
- IFN-β leads to a reduction of dendritic cells and down-regulates the antigen presentation by APCs in peripheral blood and in the CNS by microglia and monocytes.
- (b)
- The expression of toll-like receptor (TLR) 3, TLR7, and myeloid differentiation primary response 88 (MyD88) on dendritic cells increases, which leads to an altered immune response.
- (c)
- INF-β induces CD4+, CD8+, CD25+, FOXP3+, and FOXA1+ T cells (regulatory T cells). A reduced inflammatory T cell response is observed by inhibiting the stimulation and activation of T cells (e.g., by modulation of co-stimulating molecules on dendritic cells), inhibition of the expression of MHCII molecules, and co-stimulating factors like CD80 and CD28 on APC [53,54].
- (d)
- The secretion of cytokines and chemokines is altered during IFN-β treatment (interleukin (IL)-10 and IL-4 increased; IL-2 and TNFα decreased). The differentiation of CD4+ cells shift from Th1 to a Th2 phenotype; thereby, resulting in a less pro-inflammatory but more anti-inflammatory cytokine milieu [55].
- (e)
- (f)
Recommended Monitoring
3.1.3. Dimethyl Fumarate
General Facts and Clinical Trial Data
Mechanism of Action and Impact on Lymphocyte Count
- (a)
- (b)
- (c)
- (d)
- (e)
Recommended Monitoring
3.2. Mechanism of Action: Target Lymphocyte Proliferation
Teriflunomide
General Facts and Clinical Trial Data
Mechanism of Action and Impact on Lymphocyte Count
Recommended Monitoring
3.3. Mechanism of Action: Target Lymphocyte Migration
3.3.1. Sphingosine-1-Phosphate Receptor Modulation
General Facts and Clinical Trial Data
Mechanism of Action and Impact on Lymphocyte Counts
Recommended Monitoring
3.3.2. Natalizumab
General Facts and Clinical Trial Data
Mechanism of Action and Impact on Lymphocyte Counts
Recommended Monitoring
3.4. Lysis of Specific Lymphocytes Subsets
3.4.1. B Cell Depletion
General Facts and Clinical Trial Data
Mechanism of Action and Impact on Lymphocyte Count
Recommended Monitoring
3.4.2. Alemtuzumab
General Facts and Clinical Trial Data
Mechanism of Action and Impact on Lymphocyte Count
Recommended Monitoring
3.4.3. Cladribine
General Facts and Clinical Trial Data
Mechanism of Action and Impact on Lymphocyte Count
Recommended Monitoring
3.5. New Treatment Options under Investigation: Bruton’s Tyrosine Kinase Inhibitors—Non-Cell-Depleting Alternative to B Cell Modulation
4. Summary
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AIDS | Acquired Immune Deficiency Syndrome |
| ALC | Absolute lymphocyte count |
| APC | Antigen-presenting cells |
| ARR | Annualized relapse rate |
| BDNF | Brain-derived neurotrophic factor |
| BTKi | Bruton’s tyrosine kinase inhibitor |
| CCR7+ | C-C chemokine receptor type 7 |
| CI | Confidence interval |
| CIS | Clinically isolated syndrome |
| CNS | Central nervous system |
| COX-1 | Cyclooxygenase-1 |
| CSF | Cerebrospinal fluid |
| DCK | Deoxycytidine kinase |
| DHODH | Dihydroorotate dehydrogenase |
| DMF | Dimethyl fumarate |
| DMT | Disease-modifying therapy |
| DNA | Deoxyribonucleic acid |
| EAE | Experimental autoimmune encephalopathy |
| EBV | Epstein-Barr virus |
| EDSS | Expanded Disability Status Scale |
| e.g., | Exempli gratia |
| EU | European Union |
| FDA | Food and Drug Administration |
| FOXA1+ | Forkhead box protein A1 |
| FOXP3 | Forkhead box P3 |
| GA | Glatiramer acetate |
| GSH | Glutathione |
| HCA2 | Hydroxy-carboxylic acid receptor 2 |
| HIF-1α | Hypoxia-inducible factor -1α |
| HIV | Human immunodeficiency virus |
| HO-1 | Heme oxygenase-1 |
| HR | Hazard Ratio |
| HSV | Herpes simplex virus |
| i.e., | Id est |
| i.v. | Intravenous |
| IFN | Interferon |
| IFNAR | Interferon-alpha/beta receptor |
| Ig | Immunoglobulin |
| IgG | Immunoglobulin G |
| IL | Interleukin |
| IRIS | Immune reconstitution inflammatory Syndrome |
| ITP | Immune thrombocytopenic purpura |
| JAK/STAT | Janus kinases/signal transducer and activator of transcription proteins |
| JCV | John Cunningham virus |
| LLN | Lower limit of normal |
| LN | Lymph nodes |
| MBP | Myelin basic protein |
| MMF | Monomethylfumarate |
| MHC | Major Histocompatibility Complex |
| MMF | Monomethylfumarate |
| MOG | Myelin oligodendrocyte glycoprotein |
| MRI | Magnetic Resonance Imaging |
| MS | Multiple sclerosis |
| MX1 | Interferon-induced GTP-binding protein Mx1 |
| MyD88 | Myeloid differentiation primary response 88 |
| NCI- | National Cancer Institute Common |
| CTAE | Terminology Criteria for Adverse Events |
| NF-κB | Nuclear factor ‘kappa-light-chain- enhancer’ of activated B-cells |
| NK cells | Natural killer cells |
| Nrf2 | nuclear factor erythroid-derived 2-like 2 |
| PGE2 | Prostaglandin E2 |
| PLP | Proteolipidprotein |
| PML | Progressive multifocal leukoencephalopathy |
| PP | Peyers patches |
| PPMS | Primary progressive multiple sclerosis |
| RNA | Ribonucleic acid |
| RRMS | Relapsing remitting multiple sclerosis |
| S1P | Sphingosine-1-phosphate |
| SPMS | Secondary progressive multiple sclerosis |
| TCR | T-cell receptor |
| Th1/2 cells | T helper 1/2 cells |
| TLR | Toll-like receptor |
| TNF-α | Tumor necrosis factor-α |
| Tregs | Regulatory T cells |
| US | United States |
| VCAM | Vascular cell adhesion molecule |
| VLA | Very late antigen |
| vs. | Versus |
| VZV | Varicella-zoster virus |
References
- Klotz, L.; Berthele, A.; Brück, W.; Chan, A.; Flachenecker, P.; Gold, R.; Haghikia, A.; Hellwig, K.; Hemmer, B.; Hohlfeld, R.; et al. Monitoring von Blutparametern unter verlaufsmodifizierender MS-Therapie. Der Nervenarzt 2016, 87, 645–659. [Google Scholar] [CrossRef] [PubMed]
- Brück, W.; Gold, R.; Lund, B.T.; Oreja-Guevara, C.; Prat, A.; Spencer, C.M.; Steinman, L.; Tintoré, M.; Vollmer, T.L.; Weber, M.S.; et al. Therapeutic Decisions in Multiple Sclerosis: Moving Beyond Efficacy. JAMA Neurol. 2013, 70, 1315–1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiendl, H.; Kieseier, B. Multiple sclerosis: Reprogramming the immune repertoire with alemtuzumab in MS. Nat. Rev. Neurol. 2013, 9, 125. [Google Scholar] [CrossRef] [PubMed]
- Villar, L.M.; García-Sánchez, M.I.; Costa-Frossard, L.; Espiño, M.; Roldán, E.; Páramo, D.; Lucas, M.; Izquierdo, G.; Álvarez-Cermeño, J.C. Immunological Markers of Optimal Response to Natalizumab in Multiple Sclerosis. Arch. Neurol.-Chic. 2012, 69, 191–197. [Google Scholar] [CrossRef] [Green Version]
- Thomas, K.; Eisele, J.; Rodriguez-Leal, F.; Hainke, U.; Ziemssen, T. Acute effects of alemtuzumab infusion in patients with active relapsing-remitting MS. Neurol.-Neuroimmunol. Neuroinflamm. 2016, 3, e228. [Google Scholar] [CrossRef] [Green Version]
- Brass, D.; McKay, P.; Scott, F. Investigating an incidental finding of lymphopenia. BMJ 2014, 348, g1721. [Google Scholar] [CrossRef]
- Bradley, L.M.; Watson, S.R. Lymphocyte migration into tissue: The paradigm derived from CD4 subsets. Curr. Opin. Immunol. 1996, 8, 312–320. [Google Scholar] [CrossRef]
- Westermann, J.; Pabst, R. Lymphocyte subsets in the blood: A diagnostic window on the lymphoid system? Immunol. Today 1990, 11, 406–410. [Google Scholar] [CrossRef]
- Govender, S.; Otwombe, K.; Essien, T.; Panchia, R.; de Bruyn, G.; Mohapi, L.; Gray, G.; Martinson, N. CD4 Counts and Viral Loads of Newly Diagnosed HIV-Infected Individuals: Implications for Treatment as Prevention. PLoS ONE 2014, 9, e90754. [Google Scholar]
- Blum, K.S.; Pabst, R. Lymphocyte numbers and subsets in the human blood Do they mirror the situation in all organs? Immunol. Lett. 2007, 108, 45–51. [Google Scholar] [CrossRef]
- Ganusov, V.V.; Auerbach, J. Mathematical Modeling Reveals Kinetics of Lymphocyte Recirculation in the Whole Organism. PLoS Comput. Biol. 2014, 10, e1003586. [Google Scholar] [CrossRef] [Green Version]
- Dhabhar, F.S.; Miller, A.H.; Stein, M.; Mcewen, B.S.; Spencer, R.L. Diurnal and Acute Stress-Induced Changes in Distribution of Peripheral Blood Leukocyte Subpopulations. Brain Behav. Immun. 1994, 8, 66–79. [Google Scholar] [CrossRef] [Green Version]
- Thomas, K.; Proschmann, U.; Ziemssen, T. Fingolimod hydrochloride for the treatment of relapsing remitting multiple sclerosis. Expert Opin. Pharmacother. 2017, 18, 1649–1660. [Google Scholar] [CrossRef]
- Prat, A.; Biernacki, K.; Lavoie, J.-F.; Poirier, J.; Duquette, P.; Antel, J.P. Migration of Multiple Sclerosis Lymphocytes Through Brain Endothelium. Arch. Neurol.-Chic. 2002, 59, 391–397. [Google Scholar] [CrossRef]
- De Flon, P.; Söderström, L.; Laurell, K.; Dring, A.; Sundström, P.; Gunnarsson, M.; Svenningsson, A. Immunological profile in cerebrospinal fluid of patients with multiple sclerosis after treatment switch to rituximab and compared with healthy controls. PLoS ONE 2018, 13, e0192516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gastaldi, M.; Zardini, E.; Franciotta, D. An update on the use of cerebrospinal fluid analysis as a diagnostic tool in multiple sclerosis. Expert Rev. Mol. Diagn. 2016, 17, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Loleit, V.; Biberacher, V.; Hemmer, B. Current and future therapies targeting the immune system in multiple sclerosis. Curr. Pharm. Biotechnol. 2014, 15, 276–296. [Google Scholar] [CrossRef]
- Chen, H.; Assmann, J.C.; Krenz, A.; Rahman, M.; Grimm, M.; Karsten, C.M.; Köhl, J.; Offermanns, S.; Wettschureck, N.; Schwaninger, M. Hydroxycarboxylic acid receptor 2 mediates dimethyl fumarate’s protective effect in EAE. J. Clin. Investig. 2014, 124, 2188–2192. [Google Scholar] [CrossRef]
- Nakhaei-Nejad, M.; Barilla, D.; Lee, C.-H.; Blevins, G.; Giuliani, F. Characterization of lymphopenia in patients with MS treated with dimethyl fumarate and fingolimod. Neurol.-Neuroimmunol. Neuroinflamm. 2018, 5, e432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill-Cawthorne, G.A.; Button, T.; Tuohy, O.; Jones, J.L.; May, K.; Somerfield, J.; Green, A.; Giovannoni, G.; Compston, D.A.; Fahey M., T.; et al. Long term lymphocyte reconstitution after alemtuzumab treatment of multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 2012, 83, 298–304. [Google Scholar] [CrossRef]
- Kaufmann, M.; Haase, R.; Proschmann, U.; Ziemssen, T.; Akgün, K. Real World Lab Data: Patterns of Lymphocyte Counts in Fingolimod Treated Patients. Front. Immunol. 2018, 9, 2669. [Google Scholar] [CrossRef]
- Warny, M.; Helby, J.; Nordestgaard, B.G.; Birgens, H.; Bojesen, S.E. Lymphopenia and Risk of Infection and Infection-Related Death in 98,344 Individuals from a Prospective Danish Population-Based Study. PLoS Med. 2018, 15, e1002685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, Z.W.; Elwood, E.; Naveed, H.; Galea, I. Lymphopenia in treatment-naive relapsing multiple sclerosis. Neurol.-Neuroimmunol. Neuroinflamm. 2016, 3, e275. [Google Scholar] [CrossRef] [Green Version]
- Otero-Romero, S.; Sánchez-Montalvá, A.; Vidal-Jordana, A. Assessing and mitigating risk of infection in patients with multiple sclerosis on disease modifying treatment. Expert Rev. Clin. Immunol. 2021, 17, 1–16. [Google Scholar] [CrossRef]
- Castelo-Branco, A.; Chiesa, F.; Conte, S.; Bengtsson, C.; Lee, S.; Minton, N.; Niemcryk, S.; Lindholm, A.; Rosenlund, M.; Piehl, F.; et al. Infections in patients with multiple sclerosis: A national cohort study in Sweden. Mult. Scler. Relat. Dis. 2020, 45, 102420. [Google Scholar] [CrossRef] [PubMed]
- Khatri, B.O.; Man, S.; Giovannoni, G.; Koo, A.P.; Lee, J.-C.; Tucky, B.; Lynn, F.; Jurgensen, S.; Woodworth, J.; Goelz, S.; et al. Effect of plasma exchange in accelerating natalizumab clearance and restoring leukocyte function. Neurology 2020, 72, 402–409. [Google Scholar] [CrossRef] [Green Version]
- Wenning, W.; Haghikia, A.; Laubenberger, J.; Clifford, D.B.; Behrens, P.F.; Chan, A.; Gold, R. Treatment of progressive multifocal leukoencephalopathy associated with natalizumab. N. Engl. J. Med. 2009, 361, 1075–1080. [Google Scholar] [CrossRef]
- Miralles, P.; Berenguer, J.; Lacruz, C.; Cosín, J.; López, J.C.; Padilla, B.; Muñoz, L.; García-de-Viedma, D. Inflammatory reactions in progressive multifocal leukoencephalopathy after highly active antiretroviral therapy. AIDS 2001, 15, 1900–1902. [Google Scholar] [CrossRef] [PubMed]
- Vendrely, A.; Bienvenu, B.; Gasnault, J.; Thiebault, J.B.; Salmon, D.; Gray, F. Fulminant inflammatory leukoencephalopathy associated with HAART-induced immune restoration in AIDS-related progressive multifocal leukoencephalopathy. Acta Neuropathol. 2005, 109, 449–455. [Google Scholar] [CrossRef]
- Metz, I.; Radue, E.W.; Oterino, A.; Kümpfel, T.; Wiendl, H.; Schippling, S.; Kuhle, J.; Sahraian, M.A.; Gray, F.; Jakl, V.; et al. Pathology of immune reconstitution inflammatory syndrome in multiple sclerosis with natalizumab-associated progressive multifocal leukoencephalopathy. Acta Neuropathol. 2012, 123, 235–245. [Google Scholar] [CrossRef] [Green Version]
- Burt, R.K.; Muraro, P.A.; Farge, D.; Oliveira, M.C.; Snowden, J.A.; Saccardi, R.; Han, X.; Quigley, K.; Bueno, V.; Frasca, D.; et al. New autoimmune diseases after autologous hematopoietic stem cell transplantation for multiple sclerosis. Bone Marrow Transpl. 2021, 56, 1509–1517. [Google Scholar] [CrossRef] [PubMed]
- Sellner, J.; Rommer, P.S. Immunological consequences of immune reconstitution therapy” in multiple sclerosis: A systematic review. Autoimmun. Rev. 2020, 19, 102492. [Google Scholar] [CrossRef] [PubMed]
- Weetman, A. Immune reconstitution syndrome and the thyroid. Best Pract. Res. Clin. Endocrinol. Metab. 2009, 23, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Teitelbaum, D.; Arnon, R.; Sela, M. Copolymer 1: From basic research to clinical application. Cell. Mol. Life Sci. 1997, 53, 24–28. [Google Scholar] [CrossRef]
- Arnal-Garcia, C.; Amigo-Jorrin Mdel, C.; Lopez-Real, A.M.; Lema-Devesa, C.; Llopis, N.; Sanchez-de la Rosa, R.; XPERIENCIA-5 Study Group. Long-term effectiveness of glatiramer acetate in clinical practice conditions. J. Clin. Neurosci. 2014, 21, 2212–2218. [Google Scholar] [CrossRef] [PubMed]
- Comi, G.; Filippi, M.; Wolinsky, J.S. European/Canadian multicenter, double-blind, randomized, placebo-controlled study of the effects of glatiramer acetate on magnetic resonance imaging—Measured disease activity and burden in patients with relapsing multiple sclerosis. Ann. Neurol. 2001, 49, 290–297. [Google Scholar] [CrossRef]
- Dhib-Jalbut, S. Mechanisms of action of interferons and glatiramer acetate in multiple sclerosis. Neurology 2002, 58, 3–9. [Google Scholar] [CrossRef]
- Fridkis-Hareli, M.; Teitelbaum, D.; Gurevich, E.; Pecht, I.; Brautbar, C.; Kwon, O.J. Direct binding of myelin basic protein and synthetic copolymer 1 to class II major histocompatibility complex molecules on living antigen-presenting cells-specificity and promiscuity. Proc. Natl. Acad. Sci. USA 1994, 91, 4872–4876. [Google Scholar] [CrossRef] [Green Version]
- Ben-Nun, A.; Mendel, I.; Bakimer, R.; Fridkis-Hareli, M.; Teitelbaum, D.; Arnon, R. The autoimmunne reactivity to myelin oligodendrocyte glycoprotein (MOG) in multiple sclerosis is potentially pathogenic: Effect of copolymer 1 on MOG-induced disease. J. Neurol. 1996, 243, 14–22. [Google Scholar] [CrossRef]
- Teitelbaum, D.; Fridkis-Hareli, M.; Arnon, R.; Sela, M. Copolymer 1 inhibits chronic relapsing experimental allergic encephalomyelitis induced by proteolipid protein (PLP) peptides in mice and interferes with PLP-specific T cell responses. J. Neuroimmunol. 1996, 64, 209–217. [Google Scholar] [CrossRef]
- Neuhaus, O.; Farina, C.; Yassouridis, A.; Wiendl, H.; Then Bergh, F.; Dose, T. Multiple sclerosis: Comparison of copolymer-1-reactive T cell lines from treated and untreated subjects reveals cytokine shift from T helper 1 to T helper 2 cells. Proc. Natl. Acad. Sci. USA 2000, 97, 7452–7457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aharoni, R. Immunmodulation neuroprotection and remyelination—The fundamental therapeutic effects of glatiramer acetate: A critical review. J. Autoimmun. 2014, 54, 81–92. [Google Scholar] [CrossRef]
- Ziemssen, T.; Kümpfel, T.; Klinkert, W.E.; Neuhaus, O.; Hohlfeld, R. Glatiramer acetate-specific-T-helper 1- and 2-type cell lines produce BDNF: Implications for multiple sclerosis therapy. Brain-derived neurotrophic factor. Brain J. Neurol. 2002, 125, 2381–2391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, J.; Li, N.; Zhang, X.; Zheng, B.; Zhang, J.Z. Induction of CD4+CD25+ regulatory T cells by copolymer-1 through activation of transcription factor Foxp3. Proc. Natl. Acad. Sci. USA 2005, 102, 6449–6454. [Google Scholar] [CrossRef] [Green Version]
- Kuerten, S.; Jackson, L.J.; Kaye, J.; Vollmer, T.L. Impact of glatiramer acetate on B cell-mediated pathogenesis of multiple sclerosis. CNS Drugs 2018, 32, 1039–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkelmann, A.; Loebermann, M.; Reisinger, E.C.; Hartung, H.P.; Zettl, U.K. Disease-modifying therapies and infectious risks in multiple sclerosis. Nat. Rev. Neurol. 2016, 12, 217–233. [Google Scholar] [CrossRef]
- Winkelmann, A.; Loebermann, M.; Reisinger, E.C.; Zettl, U.K. Multiple sclerosis treatment and infectious issues: Update 2013. Clin. Exp. Immunol. 2014, 175, 425–438. [Google Scholar] [CrossRef]
- PRISMS (Prevention of Relapses and Disability by Interferon Beta-1a Subcutaneously in Multiple Sclerosis) Study Group. Randomised double-blind placebo-controlled study of interferon beta-1a in relapsing/remitting multiple sclerosis. Lancet 1998, 7, 352. [Google Scholar]
- Dobson, R.; Dassan, P.; Roberts, M.; Giovannoni, G.; Nelson-Piercy, C.; Brex, P.A. UK consensus on pregnancy in multiple sclerosis: ‘Association of British Neurologists’ guidelines. Pract. Neurol. 2019, 19, 106–114. [Google Scholar] [CrossRef] [Green Version]
- Varytė, G.; Zakarevičienė, J.; Ramašauskaitė, D.; Laužikienė, D.; Arlauskienė, A. Pregnancy and Multiple Sclerosis: An Update on the Disease Modifying Treatment Strategy and a Review of Pregnancy’s Impact on Disease Activity. Medicina (Kaunas) 2020, 56, 49. [Google Scholar] [CrossRef] [Green Version]
- Madsen, C. The innovative development in interferon beta treatments of relapsing-remitting multiple sclerosis. Brain Behav. 2017, 7, e00696. [Google Scholar] [CrossRef] [Green Version]
- De Andrea, M.; Ravera, R.; Gioia, D.; Gariglio, M.; Landolfo, S. The interferon system: An overview. Eur. Paediatr. Neurol. Soc. 2002, 6, A41–A46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markowitz, C.E. Interferon-beta: Mechanism of action and dosing issues. Neurology 2007, 68, 8–11. [Google Scholar] [CrossRef]
- Zhang, J.; Hutton, G.; Zhang, Y. A comparison of the mechanisms of action of interferon-beta and glatiramer acetate in the treatment of multiple sclerosis. Clin. Ther. 2002, 24, 1998–2021. [Google Scholar] [CrossRef]
- Wandinger, K.P.; Stürzebecher, C.S.; Bielekova, B.; Detore, G.; Rosenwald, A.; Staudt, L.M. Complex immunmodulatory effects of interferon-beta in multiple sclerosis include the upregulation of T helper1-associated marker genes. Ann. Neurol. 2001, 50, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Haji Abdolvahab, M.; Mofrad, M.R.K.; Schellekens, H. Interferon beta: From molecular level to therapeutic effects. Int. Rev. Cell Mol. Biol. 2016, 326, 343–372. [Google Scholar]
- Rommer, P.S.; Zettl, U.K. Managing the side effects of multiple sclerosis therapy: Pharmacotherapy options for patients. Expert Opin. Pharmacother. 2018, 19(5), 483–498. [Google Scholar] [CrossRef] [PubMed]
- Schreiner, B.; Mitsdoerffer, M.; Kieseier, B.C.; Chen, L.; Hartung, H.-P.; Weller, M. Interferon-beta enhances monocyte and dendritic cell expression of B7-H1 (PD-L1), a strong inhibitor of autologous T-cell activation: Relevance for the immune modulatory effect in multiple sclerosis. J. Neuroimmunol. 2004, 155, 172–182. [Google Scholar] [CrossRef]
- Dhib-Jalbut, S.; Marks, S. Interferon-beta mechanisms of action in multiple sclerosis. Neurology 2010, 74, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Hartrich, L.; Weinstock-Guttman, B.; Hall, D.; Badgett, D.; Baier, M.; Patrick, K.; Feichter, J.; Hong, J.; Ramanathan, M. Dynamics of immune cell trafficking in interferon-β treated multiple sclerosis patients. J. Neuroimmunol. 2003, 139, 84–92. [Google Scholar] [CrossRef]
- Moser, T.; Akgün, K.; Proschmann, U.; Sellner, J.; Ziemssen, T. The role of TH17 cells in multiple sclerosis: Therapeutic implications. Autoimmun Rev. 2020, 19, 102647. [Google Scholar] [CrossRef]
- Rieckmann, P.; O’Connor, P.; Franncis, G.S.; Wetherill, G.; Alteri, E. Haematological effects of interferon-beta 1a (Rebif) therapy in multiple sclerosis. Drug Saf. 2004, 27, 745–756. [Google Scholar] [CrossRef]
- Mehling, M.; Fritz, S.; Hafner, P. Preserved antigen-specific immune response in patients with multiple sclerosis responding to IFN-beta-therapy. PLoS ONE 2013, 8, e78532. [Google Scholar] [CrossRef]
- Schwind, S.R.; Decker, M.D.; Lopez-Bresnahan, M. Rebif-Influenza Vaccine Study Investigators. Immune response to influenza vaccine is maintained in patients with multile sclerosis receiving interferon beta-1a. Neurology 2005, 65, 1964–1966. [Google Scholar] [CrossRef] [PubMed]
- Longbrake, E.E.; Naismith, R.T.; Parks, B.J.; Wu, G.F.; Cross, A.H. Dimethyl fumarate-associated lymphopenia: Risk factors and clinical significance. Mult. Scler. J. Exp. Transl. Clin. 2015, 1, 2055217315596994. [Google Scholar] [CrossRef] [Green Version]
- Fox, R.J. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N. Engl. J. Med. 2012, 367, 1087–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gold, R. Placebo-controlled phase 3 study of oral BG-12 for relapsing-remitting multiple sclerosis. N. Engl. J. Med. 2012, 367, 1098–1107. [Google Scholar] [CrossRef] [Green Version]
- Wilms, H.; Sievers, J.; Rickert, U.; Rostami-Yazdi, M.; Mrowietz, U.; Lucius, R. Dimethylfumarate inhibits microglial and astrocytic inflammation by suppressing the synthesis of nitric oxide, IL-1beta, TNF-alpha and IL-6 in an in-vitro model of brain inflammation. J. Neuroinflamm. 2010, 19, 1742–2094. [Google Scholar]
- Helwa, I.; Choudhary, V.; Chen, X.; Kaddour-Djebbar, I.; Bollag, W.B. Anti-Psoriatic Drug Monomethylfumarate Increases Nuclear Factor Erythroid 2-Related Factor 2 Levels and Induces Aquaporin-3 mRNA and Protein Expression. J. Pharmacol. Exp. Ther. 2017, 362, 243–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghoreschi, K.; Brück, J.; Kellerer, C.; Deng, C.; Peng, H.; Rothfuss, O.; Hussain, R.Z.; Gocke, A.R.; Respa, A.; Glocova, I.; et al. Fumarates improve psoriasis and and multiple sclerosis by inducing type II dendritic cells. J. Exp. Med. 2011, 208, 2291–2303. [Google Scholar] [CrossRef]
- Dibbert, S.; Clement, B.; Skak-Nielsen, T.; Mrowietz, U.; Rostami-Yazdi, M. Detection of fumarate-glutathione adducts in the portal vein blood of rats: Evidence for rapid dimethylfumarate metabolism. Arch. Dermatol. Res. 2013, 305, 447–451. [Google Scholar] [CrossRef]
- Vandermeeren, M.; Janssens, S.; Wouters, H.; Borghmans, L.; Borgers, M.; Beyaert, R.; Geysen, J. Dimethylfumarate is an inhibitor of cytokine-induced nuclear translocation of NF-kappa B1, but not RelA in normal human dermal fibroblast cells. J. Investig. Dermatol. 2001, 116, 124–130. [Google Scholar] [PubMed] [Green Version]
- Litjens, N.H.; Rademaker, M.; Ravensbergen, B.; Rea, D.; van der Plas, M.J.; Thio, B.; Walding, A.; van Dissel, J.T.; Nibbering, P.H. Monomethylfumarate affects polarization of monocyte-derived dendritic cells resulting in down-regulated Th1 lymphocyte responses. Eur. J. Immunol. 2004, 34, 565–575. [Google Scholar] [CrossRef]
- Zhao, G.; Liu, Y.; Fang, J.; Chen, Y.; Li, H.; Gao, K. Dimethyl fumarate inhibits the expression and function of hypoxia-inducible factor-1α (HIF-1α). Biochem. Biophys. Res. Commun. 2014, 448, 303–307. [Google Scholar] [CrossRef]
- Li, Y.; Tang, J.; Hu, Y. Dimethyl fumarate protection against collagen II degradation. Biochem. Biophys. Res. Commun. 2014, 454, 257–261. [Google Scholar] [CrossRef]
- Tang, H.; Lu, J.Y.; Zheng, X.; Yang, Y.; Reagan, J.D. The psoriasis drug monomethylfumarate is a potent nicotinic acid receptor agonist. Biochem. Biophys. Res. Commun. 2008, 375, 562–565. [Google Scholar] [CrossRef] [PubMed]
- Hanson, J.; Gille, A.; Offermanns, S. Role of HCA2 (GPR109A) in nicotinic acid and fumaric acid ester-induced effects on the skin. Pharmacol. Ther. 2012, 136, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Khatri, B.O.; Garland, J.; Berger, J.; Kramer, J.; Sershon, L.; Olapo, T.; Sesing, J.; Dukic, M.; Rehn, E. The effect of dimethyl fumarate (Tecfidera™) on lymphocyte counts: A potential contributor to progressive multifocal leukoencephalopathy risk. Mult. Scler. Relat. Disord. 2015, 4, 377–379. [Google Scholar] [CrossRef]
- Spencer, C.M.; Crabtree-Hartman, E.C.; Lehmann-Horn, K.; Cree, B.A.; Zamvil, S.S. Reduction of CD8(+) T lymphocytes in multiple sclerosis patients treated with dimethyl fumarate. Neurol.-Neuroimmunol. Neuroinflamm. 2015, 12, e76. [Google Scholar] [CrossRef] [Green Version]
- Montes Diaz, G.; Fraussen, J.; Van Wijmeersch, B.; Hupperts, R.; Somers, V. Dimethyl fumarate induces a persistent change in the composition of the innate and adaptive immune system in multiple sclerosis patients. Sci. Rep. 2018, 8, 8194. [Google Scholar] [CrossRef]
- Mehta, D.; Miller, C.; Arnold, D.L.; Bame, E.; Bar-Or, A.; Gold, R.; Hanna, J. Effect of dimethyl fumarate on lymphocytes in RRMS: Implications for clinical practice. Neurology 2019, 92, e1724–e1738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, R.J.; Chan, A.; Gold, R. Characterizing absolute lymphocyte count profiles in dimethyl fumarate-treated patients with MS: Patient management considerations. Neurol. Clin. Pract. 2016, 6, 220–229. [Google Scholar] [CrossRef] [Green Version]
- Delgado-Alvarado, M.; Sedano, M.J.; Gonzalez-Quintanilla, V.; de Lucas, E.M.; Polo, J.M.; Berciano, J. Progressive multifocal leukoencephalopathy and idiopathic CD4 lymphocytopenia. J. Neurol. Sci. 2013, 327, 75–79. [Google Scholar] [CrossRef]
- Nieukamp, D.J.; Murk, J.L.; van Oosten, B.W.; Cremers, C.H.; Killestein, J.; Viveen, M.C.; Van Hecke, W.; Frijlink, D.W.; Wattjes, M.P. PML in a patient without severe lymphocytopenia receiving dimethyl fumarate. N. Engl. J. Med. 2015, 372, 14. [Google Scholar] [CrossRef] [PubMed]
- Aly, L.; Hemmer, B.; Korn, T. From leflunomide to teriflunomide: Drug development and immunosuppressive oral drugs in the treatment of multiple sclerosis. Curr. Neuropharmacol. 2017, 15, 874–891. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, P.W.; Li, D.; Freedman, M.S.; Bar-Or, A.; Rice, G.P.; Confavreux, C.; Paty, D.W.; Stewart, J.A.; Scheyer, R.; Teriflunomide Multiple Sclerosis Trial Group; et al. A Phase II study of the safety and efficacy of teriflunomide in multiple sclerosis with relapses. Neurology 2006, 66, 894–900. [Google Scholar] [CrossRef]
- Vermersch, P.; Czlonkowska, A.; Grimaldi, L.M.; Confavreux, C.; Comi, G.; Kappos, L.; Olsson, T.P.; Benamor, M.; Bauer, D.; Truffinet, P.; et al. Teriflunomide versus subcutaneous interferon beta-1a in patients with relapsing multiple sclerosis: A randomised, controlled phase 3 trial. Mult. Scler. 2014, 20, 705–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, A.E. Oral teriflunpmide in the treatment of relapsing forms of multiple sclerosis: Clinical evidence and long-term experience. Ther. Adv. Neurol. Disord. 2017, 10, 381–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wostradowski, T.; Prajeeth, C.K.; Gudi, V.; Kronenberg, J.; Witte, S.; Brieskorn, M. In vitro evaluation of physiologically relevant cocentrations of teriflunomide on activation and proliferation of primary rodent microglia. J. Neuroinflamm. 2016, 13, 250. [Google Scholar] [CrossRef] [Green Version]
- Manna, S.K.; Aggarwal, B.B. Immunosuppressive leflunomide metabolite (A77 1726) blocks TNF-dependent nuclear factor-kappa B activation and gene expression. J. Immunol. Baltim. 1999, 162, 2095–2102. [Google Scholar]
- Gonzalez-Alvaro, I.; Ortiz, A.M.; Dominguez-Jimenez, C.; Aragon-Bodi, A.; Diaz Sanchez, B.; Sanchez-Madrid, F. Inhibition of tumour necrosis factor and IL-17 production by leflunomide involves the JAK/STAT pathway. Ann. Rheum. Dis. 2009, 68, 1644–1650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bilger, A.; Plowshay, J.; Ma, S.; Nawandar, D.; Barlow, E.A.; Romero-Masters, J.C. Leflunomide/teriflunomide inhibit Epstein-Barr virus (EBV)-induced lymphoproliferative disease and lytic viral replication. Oncotarget 2017, 8, 44266–44280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Modica, C.M.; Schweser, F.; Sudyn, M.L.; Bertolino, N.; Preda, M.; Polak, P. Effect of teriflunomide on cortex-basal ganglia-thalamus (CxBGTh) circuit glutamatergic dysregulation in the Theiler’s Murine Encephalomyelitis Virus mouse model of multiple sclerosis. PLoS ONE 2017, 12, e0182729. [Google Scholar] [CrossRef] [Green Version]
- Göttle, P.; Manousi, A.; Kremer, D.; Reiche, L.; Hartung, H.-P.; Küry, P. Teriflunomide promotes oligodendroglial differentiation and myelination. J. Neuroinflamm. 2018, 15, 76. [Google Scholar] [CrossRef] [Green Version]
- Groh, J.; Hörner, M.; Martini, R. Teriflunomide attenuates neuroinflammation-related neural damage in mice carrying human PLP1 mutations. J. Neuroinflamm. 2018, 15, 194. [Google Scholar] [CrossRef]
- AUBAGIO (Teriflunomide). Prescribing Information; Genzyme Canada Inc.: Mississauga, ON, Canada, 2016. [Google Scholar]
- Bar-Or, A.; Pachner, A.; Menguy-Vacheron, F.; Kaplan, J.; Wiendl, H. Teriflunomide and its mechanism of action in multiple sclerosis. Drugs 2014, 74, 659–674. [Google Scholar] [CrossRef] [Green Version]
- Comi, G.; Miller, A.E.; Benamor, M.; Truffinet, P.; Poole, E.M.; Freedman, M.S. Characterizing lymphocyte counts and infection rates with long-term teriflunomide treatment: Pooled analysis of clinical trials. Mult. Scler. 2020, 26, 1083–1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bar-Or, A.; Wiendl, H.; Miller, B. Randomized study of teriflunomide effects on immune response to neoantigen and recall antigens. Neurol.-Neuroimmunol. Neuroinflamm. 2015, 2, e70. [Google Scholar] [CrossRef] [Green Version]
- Kappos, L.; Bar-Or, A.; Cree, B.; Fox, R.; Giovannoni, G.; Gold, R.; Vermersch, P.; Arnould, S.; Sidorenko, T.; Wolf, C.; et al. Efficacy of Siponimod in Secondary Progressive Multiple Sclerosis: Results of the Phase 3 Study (CT.002). Neurology 2017, 88, CT.002. [Google Scholar]
- Nofer, J.R.; Bot, M.; Brodde, M. FTY720, a synthetic sphingosine 1 phosphate analogue, inhibits development of atherosclerosis in low-density lipoprotein receptor-deficient mice. Circulation 2007, 115, 501–508. [Google Scholar] [CrossRef] [Green Version]
- Baumruker, T.; Billich, A.; Brinkmann, V. FTY720, an immunomodulatory sphingolipid mimetic: Translation of a novel mechanism into clinical benefit in multiple sclerosis. Expert Opin. Investig. Drugs 2007, 16, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Mizugishi, K.; Yamashita, T.; Olivera, A.; Miller, G.F.; Spiegel, S.; Proia, R.L. Essential role for sphingosine kinases in neural and vascular development. Mol. Cell. Biol. 2005, 25, 11113–11121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandala, S.; Hajdu, R.; Bergstrom, J. Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science 2002, 296, 346–349. [Google Scholar] [CrossRef] [PubMed]
- Matloubian, M.; Lo, C.G.; Cinamon, G. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 2004, 427, 355–360. [Google Scholar] [CrossRef]
- Mehling, M.; Lindberg, R.; Kuhle, J. Oral fingolimod (FTY720) treatment reduces peripheral IL-17-producing TH17 cells in patients with multiple sclerosis. Mult. Scler. 2008, 14, 234. [Google Scholar]
- Brinkmann, V.; Cyster, J.G.; Hla, T. FTY720: Sphingosine 1-phosphate receptor-1 in the control of lymphocyte egress and endothelial barrier function. Am. J. Transplant. 2004, 4, 1019–1025. [Google Scholar] [CrossRef]
- Graler, M.H.; Goetzl, E.J. The immunosuppressant FTY720 down-regulates sphingosine 1-phosphate G-protein-coupled receptors. FASEB J. 2004, 18, 551–553. [Google Scholar] [CrossRef]
- Dev, K.K.; Mullershausen, F.; Mattes, H. Brain sphingosine-1-phosphate receptors: Implication for FTY720 in the treatment of multiple sclerosis. Pharmacol. Ther. 2008, 117, 77–93. [Google Scholar] [CrossRef]
- Spohr, T.C.; Choi, J.W.; Gardell, S.E.; Herr, D.R.; Rehen, S.K.; Gomes, F.C.; Chun, J. Lysophosphatidic acid receptor-dependent secondary effects via astrocytes promote neuronal differentiation. J. Biol. Chem. 2008, 283, 7470–7479. [Google Scholar] [CrossRef] [Green Version]
- Brinkmann, V. Sphingosine 1-phosphate receptors in health and disease: Mechanistic insights from gene deletion studies and reverse pharmacology. Pharmacol. Ther. 2007, 115, 84–105. [Google Scholar] [CrossRef]
- Brinkmann, V.; Baumruker, T. Pulmonary and vascular pharmacology of sphingosine 1-phosphate. Curr. Opin. Pharmacol. 2006, 6, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V. FTY720 (fingolimod) in Multiple Sclerosis: Therapeutic effects in the immune and the central nervous system. Br. J. Pharmacol. 2009, 158, 1173–1182. [Google Scholar] [CrossRef] [Green Version]
- Comi, G.; Hartung, H.P.; Bakshi, R.; Williams, I.M.; Wiendl, H. Benefit-Risk Profile of Sphingosine-1-Phosphate Receptor Modulators in Relapsing and Secondary Progressive Multiple Sclerosis. Drugs 2017, 77, 1755–1768. [Google Scholar] [CrossRef]
- Hjorth, M.; Dandu, N.; Mellergård, J. Treatment effects of fingolimod in multiple sclerosis: Selective changes in peripheral blood lymphocyte subsets. PLoS ONE 2020, 15, e0228380. [Google Scholar] [CrossRef] [PubMed]
- Warnke, C.; Dehmel, T.; Ramanujam, R. Initial lymphocyte count and low BMI may affect fingolimod-induced lymphopenia. Neurology 2014, 83, 2153–2157. [Google Scholar] [CrossRef]
- Francis, G.; Kappos, L.; O’Connor, P. Temporal profile of lymphocyte counts and relationship with infections with fingolimod therapy. Mult. Scler. 2014, 20, 471–480. [Google Scholar] [CrossRef]
- Ohtani, R.; Mori, M.; Uchida, T.; Uzawa, A.; Masuda, H.; Liu, J.; Kuwabara, S. Risk factors for fingolimod-induced lymphopenia in multiple sclerosis. Mult. Scler. J. Exp. Transl. Clin. 2018, 4, 2055217318759692. [Google Scholar] [CrossRef] [Green Version]
- Naldini, A.; Fleischmann, W.R.J. In vivo myelosuppression by combination interferon treatment: Antagonism of MuIFN-gamma and MuIFN-beta myelosuppressive effects. J. Biol. Response Modif. 1987, 6, 546–555. [Google Scholar]
- FDA. Gilenya (Fingolimod) Label. 9 December 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/022527s008lbl.pdf (accessed on 9 December 2019).
- Cavone, L.; Felici, R.; Lapucci, A.; Buonvicino, D.; Pratesi, S.; Muzzi, M.; Hakiki, B.; Maggi, L.; Peruzzi, B.; Caporale, R.; et al. Dysregulation of sphingosine 1 phosphate receptor-1 (S1P1) signaling and regulatory lymphocyte-dependent immunosuppression in a model of post-fingolimod MS rebound. Brain Behav. Immun. 2015, 50, 78–86. [Google Scholar] [CrossRef]
- Giordana, M.; Cavalla, P.; Uccelli, A.; Laroni, A.; Bandini, F.; Vercellino, M.; Mancardi, G. Overexpression of sphingosine-1-phosphate receptors on reactive astrocytes drives neuropathology of multiple sclerosis rebound after fingolimod discontinuation. Mult. Scler. J. 2018, 24, 1133–1137. [Google Scholar] [CrossRef]
- Barry, B.; Erwin, A.A.; Stevens, J.; Tornatore, C. Fingolimod Rebound: A Review of the Clinical Experience and Management Considerations. Neurol. Ther. 2019, 8, 241–250. [Google Scholar] [CrossRef] [Green Version]
- Hatcher, S.E.; Waubant, E.; Nourbakhsh, B.; Crabtree-Hartman, E.; Graves, J.S. Rebound syndrome in patients with multiple sclerosis after cessation of fingolimod treatment. JAMA Neurol. 2016, 73, 790–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamout, B.I.; Zeineddine, M.M.; Sawaya, R.A.; Khoury, S.J. Safety and efficacy of reduced fingolimod dosage treatment. J. Neuroimmunol. 2015, 285, 13–15. [Google Scholar] [CrossRef] [PubMed]
- Kappos, L.; Bar-Or, A.; Cree, B.A.C.; Fox, R.J.; Giovannoni, G.; Gold, R.; Vermersch, P.; Arnold, D.L.; Arnould, S.; Scherz, T.; et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): A double-blind, randomised, phase 3 study. Lancet 2018, 391, 1263–1273. [Google Scholar] [CrossRef]
- Swallow, E.; Patterson-Lomba, O.; Yin, L.; Mehta, R.; Pelletier, C.; Kao, D.; Sheffield, J.K.; Stonehouse, T.; Signorovitch, J. Comparative safety and efficacy of ozanimod versus fingolimod for relapsing multiple sclerosis. J. Comp. Eff. Res. 2020, 9, 275–285. [Google Scholar] [CrossRef] [Green Version]
- Cohen, J.A.; Comi, G.; Arnold, D.L.; Bar-Or, A.; Selmaj, K.W.; Steinman, L.; Havrdová, E.K.; Cree, B.A.; Montalbán, X.; Hartung, H.P.; et al. Efficacy and safety of ozanimod in multiple sclerosis: Dose-blinded extension of a randomized phase II study. Mult. Scler. 2019, 25, 1255–1262. [Google Scholar] [CrossRef]
- Jurcevic, S.; Juif, P.E.; Hamid, C.; Greenlaw, R.; D’Ambrosio, D.; Dingemanse, J. Effects of multiple-dose ponesimod, a selective S1P1 receptor modulator, on lymphocyte subsets in healthy humans. Drug Des. Dev. Ther. 2016, 11, 123–131. [Google Scholar] [CrossRef] [Green Version]
- Kompetenznetz Multiple Sklerose, Qualitätshandbuch. 2019. Available online: https://www.kompetenznetz-multiplesklerose.de/wp-content/uploads/2019/09/KKNMS_Qualit%C3%A4tshandbuch-MSNMOSD_2019_webfrei.pdf (accessed on 19 December 2020).
- Polman, C.H.; O’Connor, P.W.; Hawrdova, E.; Hutchinson, M.; Kappos, L.; Miller, D.H. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N. Engl. J. Med. 2006, 354, 899–910. [Google Scholar] [CrossRef] [Green Version]
- Rudick, R.A.; Stuart, W.H.; Calabresi, P.A.; Confavreux, C.; Galetta, S.L.; Radue, E.-W. Natalizumab plus interferon beta-1a for relapsing multiple sclerosis. N. Engl. J. Med. 2006, 354, 911–923. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, R.; Ho, P.-R.; Campbell, N.; Chang, I.; Deykin, A.; Forrestal, F. Effect of natalizumab on disease progression in secondary progressive multiple sclerosis (ASCEND): A phase 3, randomised, double-blind, placebo-controlled trial with an open-label extension. Lancet Neurol. 2018, 17, 405–415. [Google Scholar] [CrossRef]
- Stüve, O.; Bennett, J.L. Pharmacological properties, toxicology and specific rationale for the use of natalizumab (Tysabri) in inflammatory diseases. CNS Drug Rev. 2007, 13, 79–95. [Google Scholar] [CrossRef]
- Ali, R.; Nicholas, R.S.J.; Muraro, P.A. Drugs in development for relapsing multiple sclerosis. Drugs 2013, 73, 625–650. [Google Scholar] [CrossRef]
- Mountain, A.; Adair, J.R. Engineering antibodies for therapy. Biotechnol. Genet. Eng. Rev. 1992, 10, 1–142. [Google Scholar] [CrossRef]
- Theien, B.E.; Vanderlugt, C.L.; Eagar, T.N.; Nickerson-Nutter, C.; Nazareno, R.; Kuchroo, V.K.; Miller, S.D. Discordant effects of anti-VLA-4 treatment before and after onset of relapsing experimental autoimmune encephalomyelitis. J. Clin. Investig. 2001, 107, 995–1006. [Google Scholar] [CrossRef] [Green Version]
- Zohen, F.; Toutzaris, D.; Klarner, V.; Hartung, H.P.; Kieseier, B.; Haas, R. The monoclonal anti-VLA-4 antibody natalizumab mobilizes CD34+ hematopoietic progenitor cells in humans. Blood 2008, 111, 3893–3895. [Google Scholar] [CrossRef] [Green Version]
- Stüve, O.; Gold, R.; Chan, A.; Mix, E.; Zettl, U.; Kieseier, B.C. Alpha4-Integrin antagonism with natalizumab: Effects and adverse effects. J. Neurol. 2008, 255, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Theien, B.E.; Vanderlugt, C.L.; Nickerson-Nutter, C.; Cornebise, M.; Scott, D.M.; Perper, S.J.; Whalley, E.T.; Miller, S.D. Differential effects of treatment with a small-molecule VLA-4 antagonist before and after onset of relapsing EAE. Blood 2003, 102, 4464–4471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vajkoczy, P.; Laschinger, M.; Engelhardt, B. Alpha4-integrin-VCAM-1 binding mediates G protein-independent capture of encephalitogenic Tcell blasts to CNS white matter micro-vessels. J. Clin. Investig. 2001, 108, 557–565. [Google Scholar] [CrossRef] [PubMed]
- Stuve, O.; Marra, C.M.; Jerome, K.R.; Cook, L.; Cravens, P.D.; Cepok, S.; Frohman, E.M.; Phillips, J.T.; Arendt, G.; Hemmer, B.; et al. Immune surveillance in multiple sclerosis patients treated with natalizumab. Ann. Neurol. 2006, 59, 743–747. [Google Scholar] [CrossRef]
- Link, J.; Ramanujam, R.; Auer, M.; Ryner, M.; Hässler, S.; Bachelet, D. Clinical practice of analysis of anti-drug antibodies against interferon beta and natalizumab in multiple sclerosis patients in Europe: A descriptive study of test results. PLoS ONE 2017, 12, e0170395. [Google Scholar] [CrossRef]
- Kaufmann, M.; Haase, R.; Proschmann, U.; Ziemssen, T.; Akgün, K. Real-World Lab Data in Natalizumab Treated Multiple Sclerosis Patients Up to 6 Years Long-Term Follow Up. Front. Neurol. 2018, 9, 1071. [Google Scholar] [CrossRef]
- Metze, C.; Winkelmann, A.; Loebermann, M.; Hecker, M.; Schweiger, B.; Reisinger, E.C.; Zettl, U.K. Immunogenicity and Predictors of Response to a Single Dose Trivalent Seasonal Influenza Vaccine in Multiple Sclerosis Patients Receiving Disease-Modifying Therapies. CNS Neurosci. Ther. 2019, 25, 245–254. [Google Scholar] [CrossRef]
- Kaufman, M.; Pardo, G.; Rossman, H.; Sweetser, M.T.; Forrestal, F.; Duda, P. Natalizumab Treatment Shows No Clinically Meaningful Effects on Immunization Responses in Patients with Relapsing-Remitting Multiple Sclerosis. J. Neurol. Sci. 2014, 341, 22–27. [Google Scholar] [CrossRef]
- Lehmann-Horn, K.; Kinzel, S.; Weber, M.S. Deciphering the role of B cells in multiple sclerosis towards specific targeting of pathogenic function. Int. J. Mol. Sci. 2017, 18, 2048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauser, S.L.; Bar-Or, A.; Comi, G.; Giovannoni, G.; Hartung, H.P.; Hemmer, B.; Lublin , F.; Montalban, X.; Rammohan, K.W.; Selmaj , K.; et al. Ocrelizumab versus Interferon Beta-1a in Relapsing Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Mayer, L.; Kappos, L.; Racke, M.K.; Rammohan, K.; Traboulsee, A.; Hauser, S.L.; Julian, L.; Köndgen, H.; Li, C.; Napieralski, J.; et al. Ocrelizumab infusion experience in patients with relapsing and primary progressive multiple sclerosis: Results from the phase 3 randomized OPERA I, OPERA II, and ORATORIO studies. Mult. Scler. Relat. Disord. 2019, 30, 236–243. [Google Scholar] [CrossRef]
- European Medicines Agency (EMA). Ocrevus 300 mg Concentrate for Solution for Infusion: EU Summary of Product Characteristics; European Medicines Agency (EMA): Amsterdam, The Netherlands, 2018. [Google Scholar]
- Sorensen, P.S.; Blinkenberg, M. The potential role for ocrelizumab in the treatment of multiple sclerosis: Current evidence and future prospects. Ther. Adv. Neurol. Disord. 2016, 9, 44–52. [Google Scholar] [CrossRef] [Green Version]
- Laurent, S.; Michel, B.; Wu, H. Effect of ocrelizumab on B and T cell immune repertoires in patients with relapsing multiple sclerosis (abstract P693). Mult. Scler. J. 2017, 23, 337. [Google Scholar]
- Gelfand, J.M.; Cree, B.A.C.; Hauser, S.L. Ocrelizumab and Other CD20+ B-Cell-Depleting Therapies in Multiple Sclerosis. Neurotherapeutics. 2017, 14, 835–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- European Medicines Agency (EMA). Ocrevus: Assessment Report; European Medicines Agency (EMA): Amsterdam, The Netherlands, 2018. [Google Scholar]
- Kappos, L.; Li, D.; Calabresi, P.A. Ocrelizumab in relapsing-remitting multiple sclerosis: A phase 2, randomised, placebo-controlled, multicentre trial. Lancet 2011, 378, 1779–1787. [Google Scholar] [CrossRef]
- Baker, D.; Pryce, G.; James, L.K.; Marta, M.; Schmierer, K. The ocrelizumab phase II extension trial suggests the potential to improve the risk: Benefit balance in multiple sclerosis. Mult. Scler. Relat. Disord. 2020, 44, 102279. [Google Scholar] [CrossRef]
- Häusler, D.; Häusser-Kinzel, S.; Feldmann, L.; Torke, S.; Lepennetier, G.; Bernard, C.C.A. Functional characterization of reappearing B cells after anti-CD20 treatment of CNS autoimmune disease. Proc. Natl. Acad. Sci. USA 2018, 115, 9773–9778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bar-Or, A.; Grove, R.A.; Austin, D.J.; Tolson, J.M.; VanMeter, S.A.; Lewis, E.W. Subcutaneous ofatunumab in patients with relapsing-remitting multiple sclerosis: The MIRROR study. Neurology 2018, 90, e1805–e1814. [Google Scholar] [CrossRef]
- EMA Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/ocrevus-epar-product-information_en.pdf (accessed on 9 December 2020).
- Montalban, X.; Hauser, S.L.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Comi, G.; de Seze, J.; Giovannoni, G.; Hartung, H.P.; Hemmer, B.; et al. Ocrelizumab versus Placebo in Primary Progressive Multiple Sclerosis. N. Engl. J. Med. 2017, 19, 376. [Google Scholar] [CrossRef] [PubMed]
- Soleimani, B.; Murray, K.; Hunt, D. Established and Emerging Immunological Complications of Biological Therapeutics in Multiple Sclerosis. Drug Saf. 2019, 42(8), 941–956. [Google Scholar] [CrossRef] [PubMed]
- Bar-Or, A.; Calkwood, J.C.; Chognot, C.; Evershed, J.; Fox, E.J.; Herman, A.; Manfrini, M.; McNamara, J.; Robertson, D.S.; Stokmaier, D.; et al. Effect of ocrelizumab on vaccine responses in patients with multiple sclerosis: The VELOCE study. Neurology 2020, 95, e1999–e2008. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.A.; Coles, A.J.; Arnold, D.J.; Confavreux, C.; Fox, E.J.; Hartung, H.P.; Havrdova, E.; Selmaj, K.W.; Weiner, H.L.; Fisher, E.; et al. Alemtuzumab versus interferon beta 1 a as first-line treatment for patients with relapsing-remitting multiple sclerosis: A randomised controlled phase 3 trial. Lancet 2012, 380, 1819–1828. [Google Scholar] [CrossRef]
- Coles, A.J.; Twyman, C.L.; Arnold, D.L.; Cohen, J.A.; Confavreux, C.; Fox, E.J.; Hartung, H.P.; Havrdova, E.; Selmaj, K.W.; Weiner, H.L.; et al. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: A randomised controlled phase 3 trial. Lancet 2012, 380, 1829–1839. [Google Scholar] [CrossRef]
- Zhang, X.; Tao, Y.; Chopra, M.; Ahn, M.; Marcus, K.L.; Choudhary, N.; Zhu, H.; Markovic-Plese, S. Differential reconstitution of T cell subsets following immunodepleting treatment with alemtuzumab (anti-CD52 monoclonal antibody) in patients with relapsing-remitting multiple sclerosis. J. Immunol. 2013, 191, 5867–5874. [Google Scholar] [CrossRef] [Green Version]
- Baker, D.; Herrod, S.S.; Alvarez-Gonzalez, C.; Giovannoni, G.; Schmierer, K. Interpreting Lymphocyte Reconstitution Data From the Pivotal Phase 3 Trials of Alemtuzumab. JAMA Neurol. 2017, 74, 961–969. [Google Scholar] [CrossRef]
- Akgün, K.; Blankenburg, J.; Marggraf, M.; Haase, R.; Ziemssen, T. Event-Driven Immunoprofiling Predicts Return of Disease Activity in Alemtuzumab-Treated Multiple Sclerosis. Front Immunol. 2020, 11, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Turner, M.J.; Shields, J.; Gale, M.S.; Hutto, E.; Roberts, B.J.; Siders, W.M.; Kaplan, J.M. Investigation of the mechanism of action of alemtuzumab in a human CD52 transgenic mouse model. Immunology 2009, 128, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.A.; Jones, J.J.; Cox, A.L.; Compston, D.A.; Coles, A.J. B-cell reconstitution and BAFF after alemtuzumab (Campath-1H) treatment of multiple sclerosis. J. Clin. Immunol. 2010, 30, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Coles, A.J.; Compston, D.A.; Selmaj, K.W.; Lake, S.L.; Moran, S.; Margolin, D.H.; Norris, K.; Tandon, P.K. Alemtuzumab vs. Interferon beta-1a in early mutiple sclerosis. N. Engl. J. Med. 2008, 359, 1786–1801. [Google Scholar]
- Wray, S.; Havrdova, E.; Snydman, D.R.; Arnold, D.L.; Cohen, J.A.; Coles, A.; Hartung, H.P.; Selmaj, K.W.; Weiner, H.L.; Daizadeh, N.; et al. Infection risk with alemtuzumab decreases over time: Pooled analysis of 6-year data from the CAMMS223, CARE-MS I, and CARE-MS II studies and the CAMMS03409 extension study. Mult. Scler. 2019, 25, 1605–1617. [Google Scholar] [CrossRef]
- Dubuisson, N.; Baker, D.; Kang, A.S.; Pryce, G.; Marta, M.; Visser, L.H.; Hofmann, W.E.; Gnanapavan, S.; Giovannoni, G.; Schmierer, K. Alemtuzumab depletion failure can occur in multiple sclerosis. Immunology 2018, 154, 253–260. [Google Scholar] [CrossRef] [Green Version]
- Gleeson, P.A.; Toh, B.H.; van Driel, I.R. Organ-specific autoimmunity induced by lymphopenia. Immunol. Rev. 1996, 149, 97–125. [Google Scholar] [CrossRef]
- Zandman-Goddard, G.; Shoenfeld, Y. HIV and autoimmunity. Autoimmun. Rev. 2002, 1, 329–337. [Google Scholar] [CrossRef]
- Khoruts, A.; Fraser, J. A causal link between lymphopenia and autoimmunity. Immunol. Lett. 2005, 98, 23–31. [Google Scholar] [CrossRef]
- Baccala, R.; Theofilopoulos, A.N. The new paradigm of T cell homeostatic proliferation-induced autoimmunity. Trends Immunol. 2005, 26, 5–8. [Google Scholar] [CrossRef]
- Krupica, T., Jr.; Fry, T.J.; Mackall, C.L. Autoimmunity during lymphopenia: A two-hit model. Clin. Immunol. 2006, 120, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Romine, J.S.; Sipe, J.C.; Koziol, J.A.; Zyroff, J.; Beutler, E. A double-blind, placebo-controlled, randomized trial of cladribine in relapsing-remitting multiple sclerosis. Proc. Assoc. Am. Phys. 1999, 111, 35–44. [Google Scholar] [CrossRef]
- Beutler, E.; Sipe, J.C.; Romine, J.S.; Koziol, J.A.; McMillan, R.; Zyroff, J. The treatment of chronic progressive multiple sclerosis with cladribine. Proc. Natl. Acad. Sci. USA 1996, 93, 1716–1720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giovannoni, G.; Comi, G.; Cook, S.; Rammohan, K.; Rieckmann, P.; Soelberg Sørensen, P.; Vermersch, P.; Chang, P.; Hamlett, A.; Musch, B.; et al. A placebo-controlled trial of oral cladribine for relapsing multiple sclerosis. N. Engl. J. Med. 2010, 362, 416–426. [Google Scholar] [CrossRef] [Green Version]
- Leist, T.P.; Weissert, R. Cladribine: Mode of action and implications for treatment of multiple sclerosis. Clin. Neuropharmacol. 2011, 34, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Brousil, J.A.; Roberts, R.J.; Schlein, A.L. Cladribine: An investigational immunomodulatory agent for multiple sclerosis. Ann. Pharmacother. 2006, 40, 1814–1821. [Google Scholar] [CrossRef]
- Genini, D.; Budihardjo, I.; Plunkett, W. Nucleotide requirements for the in vitro activation of the apoptosis protein-activating factor-1-mediated caspase pathway. J. Biol. Chem. 2000, 275, 29–34. [Google Scholar] [CrossRef] [Green Version]
- Sorensen, P.S.; Sellebjerg, F. Pulsed immune reconstitution therapy in multiple sclerosis. Ther. Adv. Neurol. Disord. 2019, 12, 1756286419836913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, D.; Herrod, S.S.; Alvarez-Gonzalez, C. Both cladribine and alemtuzumab may effect MS via B-cell depletion. Neurol.-Neuroimmunol. Neuroinflamm. 2017, 4, e360. [Google Scholar] [CrossRef] [Green Version]
- Beutler, E. Cladribine (2-chlorodeoxyadenosine). Lancet 1992, 340, 952–956. [Google Scholar] [CrossRef]
- Comi, G.; Cook, S.; Giovannoni, G.; Rieckmann, P.; Sørensen, P.S.; Vermersch, P.; Galazka, A.; Nolting, A.; Hicking, C.; Dangond, F. Effect of cladribine tablets on lymphocyte reduction and repopulation dynamics in patients with relapsing multiple sclerosis. Mult. Scler. Relat. Disord. 2019, 29, 168–174. [Google Scholar] [CrossRef] [Green Version]
- Wiendl, H.; Carraro, M.; Comi, G.; Izquierdo, G.; Kim, H.J.; Sharrack, B.; Tornatore, C.; Daizadeh, N.; Chung, L.; Jacobs, A.K.; et al. Lymphocyte pharmacodynamics are not associated with autoimmunity or efficacy after alemtuzumab. Neurol.-Neuroimmunol. Neuroinflamm. 2019, 7, e635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacco, K.A.; Abraham, R.S. Consequences of B-cell-depleting therapy: Hypogammaglobulinemia and impaired B-cell reconstitution. Immunotherapy 2018, 10, 713–728. [Google Scholar] [CrossRef]
- García-Merino, A. Bruton’s Tyrosine Kinase Inhibitors: A New Generation of Promising Agents for Multiple Sclerosis Therapy. Cells 2021, 10, 2560. [Google Scholar] [CrossRef]
- Neys, S.F.H.; Rip, J.; Hendriks, R.W.; Corneth, O.B.J. Bruton’s Tyrosine Kinase Inhibition as an Emerging Therapy in Systemic Autoimmune Disease. Drugs 2021, 81, 1605–1626. [Google Scholar] [CrossRef] [PubMed]
- Montalban, X.; Arnold, D.L.; Weber, M.S.; Staikov, I.; Piasecka-Stryczynska, K.; Willmer, J.; Martin, E.C.; Dangond, F.; Syed, S.; Wolinsky, J.S.; et al. Placebo-Controlled Trial of an Oral BTK Inhibitor in Multiple Sclerosis. N. Engl. J. Med. 2019, 380, 2406–2417. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Orelabrutinib: First Approval. Drugs 2021, 81, 503–507. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Grade | NCI-CTAE Definitions of Severity for Adverse Reactions | Leukocyte Count | Lymphocyte Count | CD4 Lymphocyte Count |
|---|---|---|---|---|
| 1 | Mild, with no or mild symptoms; no interventions required | LLN–3.0 GPt/L | LLN–0.8 GPt/L | LLN–0.5 GPt/L |
| 2 | Moderate; minimal intervention indicated; some limitation of activities | <3.0–2.0 GPt/L | <0.8–0.5 GPt/L | <0.5–0.2 GPt/L |
| 3 | Severe but not life-threatening; hospitalization required; limitation of patient’s ability to care for him/herself | <2.0–1.0 GPt/L | <0.5–0.2 GPt/L | <0.2–0.05 GPt/L |
| 4 | Life-threatening; urgent intervention required | <1.0 GPt/L | <0.2 GPt/L | <0.05 GPt/L |
| 5 | Death related to adverse event |
| Drug Name | Recommendations for Lymphocyte Cut-Off Values | |
|---|---|---|
| Oral therapies | Dimethyl fumarate | Complete blood count every 6–8 weeks in first year of treatment, subsequently every 3–6 months, discontinuation of therapy in case of leukopenia of <3.0 GPt/L or lymphopenia of <0.5 GPt/L, in case of grade 2 lymphopenia (0.5–0.8 GPt/L) continuous control of blood counts and high vigilance for opportunistic infections |
| Teriflunomide | Regular check of blood counts every second month in the first six months, subsequently every three months in the case of normal lymphocyte and leukocyte counts; therapy discontinuation in case of lymphocyte decrease < 0.5 GPt/L | |
| Fingolimod Siponimod Ozanimod Ponesimod | Regular check of blood counts 4 weeks after starting therapy, subsequently in case of normal lymphocyte and leukocyte counts, every 3–6 months; in case of repeated peripheral lymphopenia < 0.2 GPt/L, therapy discontinuation until lymphocyte counts reach levels > 0.6 GPt/L | |
| Cladribine | Regular complete blood count prior to cladribine intake and 2 and 6 months after start of treatment in each treatment year, in case of lymphocytopenia < 0.8 GPt/L, the next cladribine pulse must not be started and active monitoring is required until values increase again; in case of not reaching a lymphocyte count of at least 0.8 GPt/L within 18 months after cladribine start, continuation is not recommended | |
| Injectables | Glatiramer acetate | Regular check of blood counts at least 3 monthly in first year of therapy, subsequently once or twice a year; in case of lymphopenia < 0.5 GPt/L discontinuation of therapy |
| Interferons | Regular check of blood counts at least 3 monthly in first year of therapy, subsequently once or twice a year; in case of lymphopenia < 0.5 GPt/L discontinuation of therapy | |
| Infusion therapies | Ocrelizumab Ofatumumab | Regular check of blood counts 3 monthly, including status of peripheral T and B cell subtypes as well as immunoglobulin levels, relevant humoral immunoglobulin deficiency (Ig < 3 g/L), and significant decrease of CD4+ T cells (<0.250 GPt/L) should be ruled out |
| Natalizumab | Regular check of blood counts every 3–6 months, peripheral increase of absolute leukocyte and lymphocyte count can serve as a biomarker, indicating sufficient VLA-4 antagonism | |
| Alemtuzumab | Regular complete blood count monthly in the course of at least 48 months after last alemtuzumab application |
| Months of Treatment | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Drug Name | Predose | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | Post Month 12 | |
| Oral therapies | Dimethyl fumarate | X | X | X | X | X | X | X | every 3–6 months | ||||||
| Teriflunomide | X | X | X | X | X | X | every 3 months | ||||||||
| Fingolimod Siponimod Ozanimod Ponesimod | X a,b | X | X | X | X | X | every 3–6 months | ||||||||
| Cladribine | X c | X | X | X | before initiating treatment in year 2, 2 and 6 months after start of treatment cycle in each year d | ||||||||||
| Injectables | Glatiramer acetate | X | X | X | X | X | once or twice a year | ||||||||
| Interferons | X | X | X | X | X | once or twice a year | |||||||||
| Infusion Therapies | Ocrelizumab Ofatumumab | X | X | X | X | X | every 3 months | ||||||||
| Natalizumab | X a | X | X | X | X | every 3–6 months | |||||||||
| Alemtuzumab | X | X | X | X | X | X | X | X | X | X | X | X | X | monthly for at least 48 months after last application | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fischer, S.; Proschmann, U.; Akgün, K.; Ziemssen, T. Lymphocyte Counts and Multiple Sclerosis Therapeutics: Between Mechanisms of Action and Treatment-Limiting Side Effects. Cells 2021, 10, 3177. https://doi.org/10.3390/cells10113177
Fischer S, Proschmann U, Akgün K, Ziemssen T. Lymphocyte Counts and Multiple Sclerosis Therapeutics: Between Mechanisms of Action and Treatment-Limiting Side Effects. Cells. 2021; 10(11):3177. https://doi.org/10.3390/cells10113177
Chicago/Turabian StyleFischer, Stefanie, Undine Proschmann, Katja Akgün, and Tjalf Ziemssen. 2021. "Lymphocyte Counts and Multiple Sclerosis Therapeutics: Between Mechanisms of Action and Treatment-Limiting Side Effects" Cells 10, no. 11: 3177. https://doi.org/10.3390/cells10113177
APA StyleFischer, S., Proschmann, U., Akgün, K., & Ziemssen, T. (2021). Lymphocyte Counts and Multiple Sclerosis Therapeutics: Between Mechanisms of Action and Treatment-Limiting Side Effects. Cells, 10(11), 3177. https://doi.org/10.3390/cells10113177