
Revisiting the Role of GSK3, A Modulator of Innate Immunity, in Idiopathic Inclusion Body Myositis

 , , ,
, , ,  and
and
Abstract
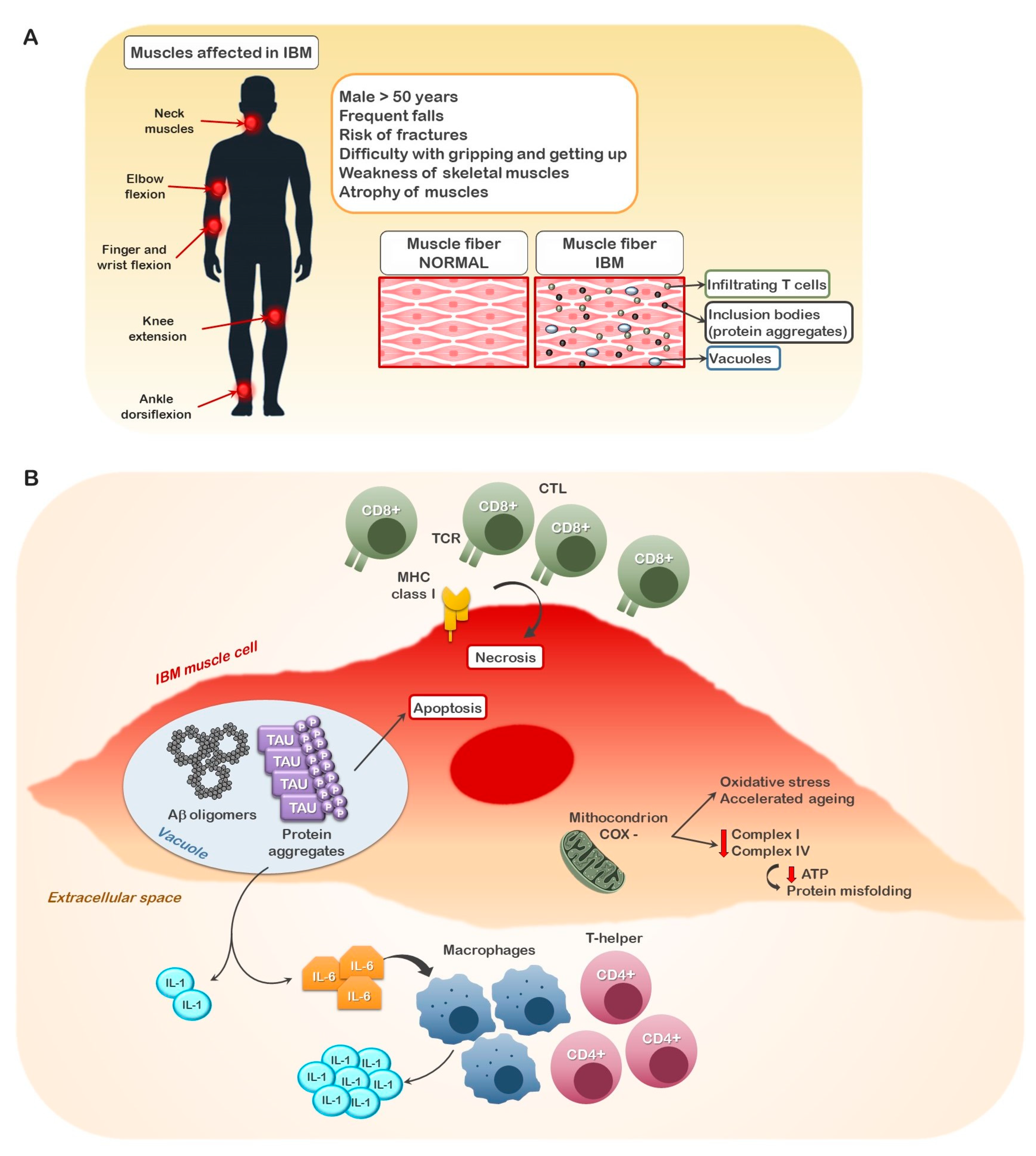
:1. Introduction
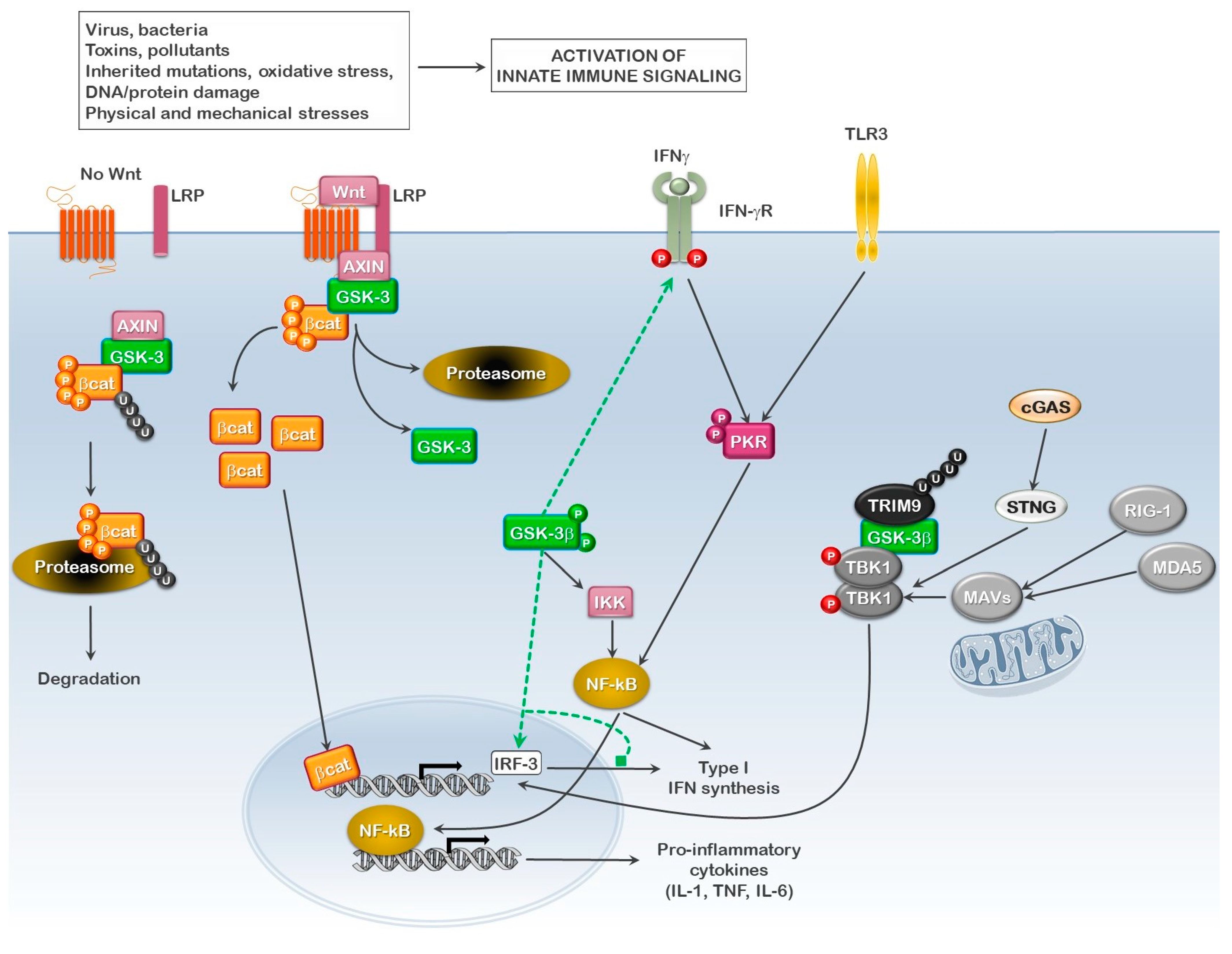
2. GSK3 Signaling
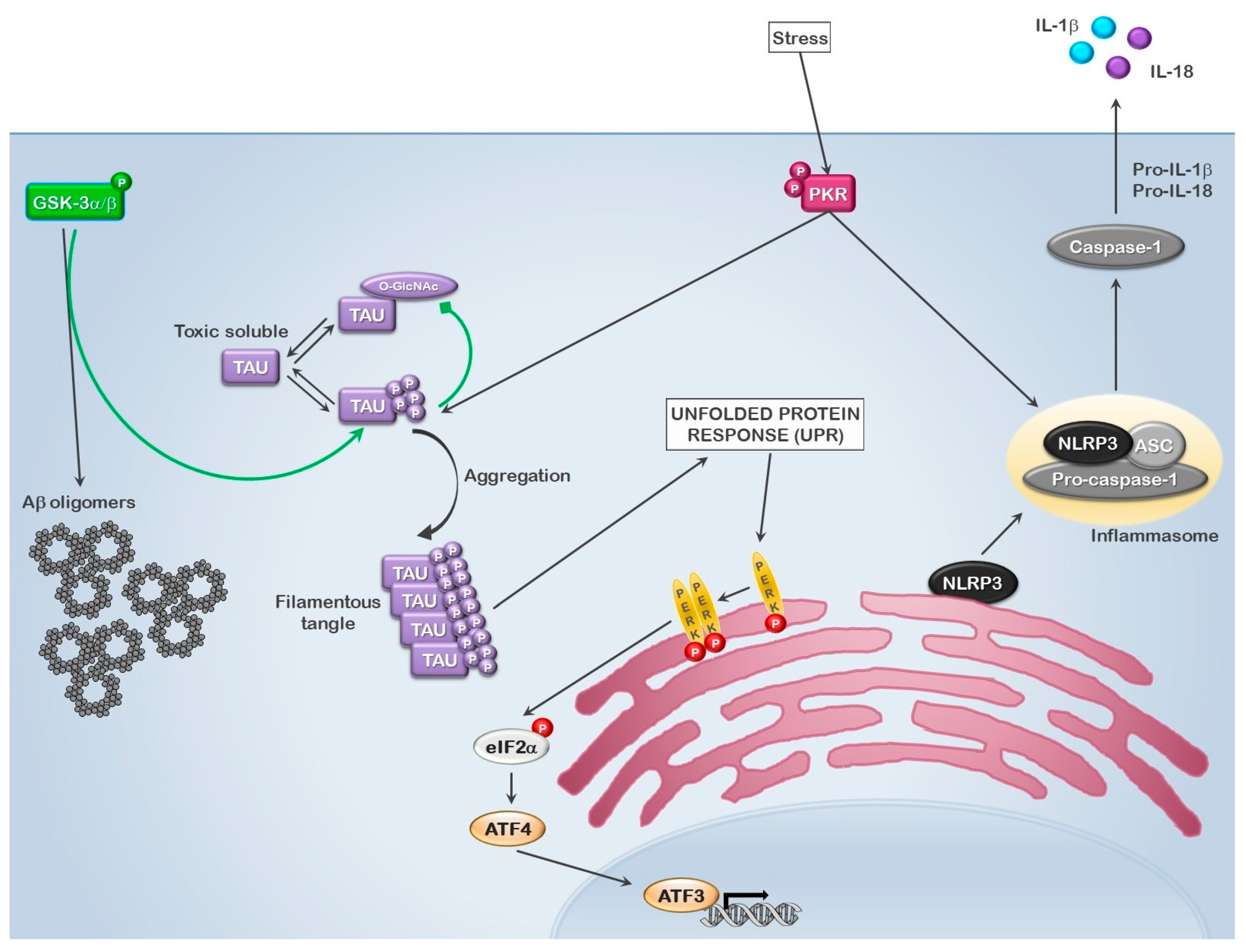
3. GSK3 and Inclusions
4. GSK3 as a Mediator of Innate Immunity in Inclusion Body Myositis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Panginikkod, S.; Musa, R. Inclusion Body Myositis; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Greenberg, S.A. Inclusion body myositis: Clinical features and pathogenesis. Nat. Rev. Rheumatol. 2019, 15, 257–272. [Google Scholar] [CrossRef]
- Fratta, P.; Engel, W.K.; McFerrin, J.; Davies, K.J.; Lin, S.W.; Askanas, V. Proteasome inhibition and aggresome formation in sporadic inclusion-body myositis and in amyloid-beta precursor protein-overexpressing cultured human muscle fibers. Am. J. Pathol. 2005, 167, 517–526. [Google Scholar] [CrossRef]
- Greenberg, S.A. Theories of the pathogenesis of inclusion body myositis. Curr. Rheumatol. Rep. 2010, 12, 221–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedberg-Oldfors, C.; Lindgren, U.; Basu, S.; Visuttijai, K.; Lindberg, C.; Falkenberg, M.; Larsson Lekholm, E.; Oldfors, A. Mitochondrial DNA variants in inclusion body myositis characterized by deep sequencing. Brain Pathol. 2021, 31, e12931. [Google Scholar] [CrossRef]
- Dalakas, M.C.; Rakocevic, G.; Schmidt, J.; Salajegheh, M.; McElroy, B.; Harris-Love, M.O.; Shrader, J.A.; Levy, E.W.; Dambrosia, J.; Kampen, R.L.; et al. Effect of Alemtuzumab (CAMPATH 1-H) in patients with inclusion-body myositis. Brain A J. Neurol. 2009, 132, 1536–1544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alemtuzumab to Treat Sporadic Inclusion Body Myositis. ClincalTrials.gov; US Library of Medicine. Available online: https://clinicaltrials.gov/ct2/show/NCT00079768 (accessed on 15 September 2021).
- Leff, R.L.; Miller, F.W.; Hicks, J.; Fraser, D.D.; Plotz, P.H. The treatment of inclusion body myositis: A retrospective review and a randomized, prospective trial of immunosuppressive therapy. Medicine 1993, 72, 225–235. [Google Scholar] [CrossRef]
- Soueidan, S.A.; Dalakas, M.C. Treatment of inclusion-body myositis with high-dose intravenous immunoglobulin. Neurology 1993, 43, 876–879. [Google Scholar] [CrossRef]
- Amato, A.A.; Barohn, R.J.; Jackson, C.E.; Pappert, E.J.; Sahenk, Z.; Kissel, J.T. Inclusion body myositis: Treatment with intravenous immunoglobulin. Neurology 1994, 44, 1516–1518. [Google Scholar] [CrossRef]
- Barohn, R.J.; Amato, A.A.; Sahenk, Z.; Kissel, J.T.; Mendell, J.R. Inclusion body myositis: Explanation for poor response to immunosuppressive therapy. Neurology 1995, 45, 1302–1304. [Google Scholar] [CrossRef]
- Lindberg, C.; Trysberg, E.; Tarkowski, A.; Oldfors, A. Anti-T-lymphocyte globulin treatment in inclusion body myositis: A randomized pilot study. Neurology 2003, 61, 260–262. [Google Scholar] [CrossRef]
- Dalakas, M.C.; Sonies, B.; Dambrosia, J.; Sekul, E.; Cupler, E.; Sivakumar, K. Treatment of inclusion-body myositis with IVIg: A double-blind, placebo-controlled study. Neurology 1997, 48, 712–716. [Google Scholar] [CrossRef]
- Walter, M.C.; Lochmuller, H.; Toepfer, M.; Schlotter, B.; Reilich, P.; Schroder, M.; Muller-Felber, W.; Pongratz, D. High-dose immunoglobulin therapy in sporadic inclusion body myositis: A double-blind, placebo-controlled study. J. Neurol. 2000, 247, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Dalakas, M.C.; Koffman, B.; Fujii, M.; Spector, S.; Sivakumar, K.; Cupler, E. A controlled study of intravenous immunoglobulin combined with prednisone in the treatment of IBM. Neurology 2001, 56, 323–327. [Google Scholar] [CrossRef]
- Badrising, U.A.; Maat-Schieman, M.L.; Ferrari, M.D.; Zwinderman, A.H.; Wessels, J.A.; Breedveld, F.C.; van Doorn, P.A.; van Engelen, B.G.; Hoogendijk, J.E.; Howeler, C.J.; et al. Comparison of weakness progression in inclusion body myositis during treatment with methotrexate or placebo. Ann. Neurol. 2002, 51, 369–372. [Google Scholar] [CrossRef]
- Kyriazopoulou, E.; Panagopoulos, P.; Metallidis, S.; Dalekos, G.N.; Poulakou, G.; Gatselis, N.; Karakike, E.; Saridaki, M.; Loli, G.; Stefos, A.; et al. An open label trial of anakinra to prevent respiratory failure in COVID-19. eLife 2021, 10, e66125. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; Machado, P.M.; Miller, A.; Spicer, C.; Herbelin, L.; He, J.; Noel, J.; Wang, Y.; McVey, A.L.; Pasnoor, M.; et al. Targeting protein homeostasis in sporadic inclusion body myositis. Sci. Transl. Med. 2016, 8, 331ra341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arimoclomol in Sporadic Inclusion Body Myositis—Open Label Extension Trial. ClincalTrials.gov; US Library of Medicine. Available online: https://clinicaltrials.gov/ct2/show/NCT04049097 (accessed on 15 September 2021).
- Arimoclomol in Sporadic Inclusion Body Myositis. ClincalTrials.gov; US Library of Medicine. Available online: https://clinicaltrials.gov/ct2/show/study/NCT00769860 (accessed on 15 September 2021).
- Study of Arimoclomol in Inclusion Body Myositis (IBM). ClincalTrials.gov; US Library of Congress. Available online: https://clinicaltrials.gov/ct2/show/NCT02753530 (accessed on 15 September 2021).
- A Randomised, Double-Blind Placebo-Controlled Pilot Study Assessing the Safety and Tolerability of Arimoclomol in Adult with Sporadic Inclusion Body Myositis. EU Clinical Trials Register. European Medicines Agency. Available online: https://www.clinicaltrialsregister.eu/ctr-search/trial/2008-008208-42/results (accessed on 15 September 2021).
- Kosmidis, M.L.; Alexopoulos, H.; Tzioufas, A.G.; Dalakas, M.C. The effect of anakinra, an IL1 receptor antagonist, in patients with sporadic inclusion body myositis (sIBM): A small pilot study. J. Neurol. Sci. 2013, 334, 123–125. [Google Scholar] [CrossRef]
- Anakinra in Myositis. ClincalTrials.gov; US Library of Medicine. Available online: https://clinicaltrials.gov/ct2/show/NCT01165008 (accessed on 16 September 2021).
- Oikawa, Y.; Izumi, R.; Koide, M.; Hagiwara, Y.; Kanzaki, M.; Suzuki, N.; Kikuchi, K.; Matsuhashi, T.; Akiyama, Y.; Ichijo, M.; et al. Mitochondrial dysfunction underlying sporadic inclusion body myositis is ameliorated by the mitochondrial homing drug MA-5. PLoS ONE 2020, 15, e0231064. [Google Scholar] [CrossRef] [PubMed]
- A Phase 1 Study of ABC008 in Adult Patients With Inclusion Body Myositis (IBM). ClinicalTrials.gov; US National Library of Medicine. Available online: https://clinicaltrials.gov/ct2/show/NCT04659031 (accessed on 16 September 2021).
- An Open-Label, Non-Randomized Trial to Investigate the Efficacy and Safety of Early Versus Delayed Start of Arimoclomol in Patients with Sporadic Inclusion Body Myositis Who have Completed the IBM4809 Trial. EU Clinical Trials Register. European Medicines Agency. Available online: https://www.clinicaltrialsregister.eu/ctr-search/trial/2019-000749-11/GB (accessed on 15 September 2021).
- Phase 2/3 Study of Arimoclomol in Inclusion Body Myositis (IBM). EU Clincal Trials Register. European Medicines Agency. Available online: https://www.clinicaltrialsregister.eu/ctr-search/trial/2017-004903-33/GB (accessed on 15 September 2021).
- A Randomised, Phase IIa Treatment Delayed-Start Trial of the Oral JAK 1/2 Inhibitor, Baricitinib, in the Treatment of Adult Idiopathic Inflammatory Myopathy. EU Clinical Trials Register. European Medicines Agency. Available online: https://www.clinicaltrialsregister.eu/ctr-search/trial/2019-003868-42/GB (accessed on 17 September 2021).
- Can Local Intramuscular Botulinum Toxin Improve Dysphagia in Patients with Myopathic Dysphagia and Constriction of the Cricoid Muscle? EU Clinical Trials Register. European Medicines Agency. Available online: https://www.clinicaltrialsregister.eu/ctr-search/trial/2014-002210-23/results (accessed on 17 September 2021).
- Extension of the CBYM338B2203 Phase IIb/III Study to Evaluate the Long-Term Efficacy, Safety and Tolerability of Intravenous BYM338 in Patients with Sporadic Inclusion Body Myositis. EU Clincal Trials Register. European Medicines Agency. Available online: https://www.clinicaltrialsregister.eu/ctr-search/trial/2015-001411-12/results (accessed on 17 September 2021).
- An Extension Study of the Efficacy, Safety and Tolerability of BYM338 (Bimagrumab) in Patients With Sporadic Inclusion Body Myositis Who Previously Participated in the Core Study CBYM338B2203. ClinicalTrials.gov; US National Library of Medicine. Available online: https://clinicaltrials.gov/ct2/show/NCT02573467 (accessed on 17 September 2021).
- Amato, A.A.; Hanna, M.G.; Machado, P.M.; Badrising, U.A.; Chinoy, H.; Benveniste, O.; Karanam, A.K.; Wu, M.; Tanko, L.B.; Schubert-Tennigkeit, A.A.; et al. Efficacy and Safety of Bimagrumab in Sporadic Inclusion Body Myositis: Long-term Extension of RESILIENT. Neurology 2021, 96, e1595–e1607. [Google Scholar] [CrossRef]
- Study of Long-term Safety, Efficacy Tolerability of BYM338 in Patients With Sporadic Inclusion Body Myositis (BYM338). ClinicalTrials.gov; US National Library of Medicine. Available online: https://clinicaltrials.gov/ct2/show/NCT02250443 (accessed on 17 September 2021).
- Sivakumar, K.; Cochrane, T.I.; Sloth, B.; Ashar, H.; Laurent, D.; Tanko, L.B.; Amato, A.A. Long-term safety and tolerability of bimagrumab (BYM338) in sporadic inclusion body myositis. Neurology 2020, 95, e1971–e1978. [Google Scholar] [CrossRef]
- A Randomized, Double-Blind, Placebo-Controlled, Multicenter, Parallel Group, Dose-Finding, Pivotal, Phase 2b/3 Study to Evaluate the Efficacy, Safety, and Tolerability of Intravenous BYM338 at 52 Weeks on Physical Function, Muscle Strength, and Mobility and Additional Long-Term Safety Up to 2 Years in Patients with Sporadic Inclusion Body Myositis. EU Clinical Trials Register. European Medicines Agency. Available online: https://www.clinicaltrialsregister.eu/ctr-search/trial/2013-000705-23/results (accessed on 17 September 2021).
- Efficacy and Safety of Bimagrumab/BYM338 at 52 Weeks on Physical Function, Muscle Strength, Mobility in sIBM Patients (RESILIENT). ClinicalTrials.gov; US National Library of Medicine. Available online: https://clinicaltrials.gov/ct2/show/NCT01925209 (accessed on 17 September 2021).
- Hanna, M.G.; Badrising, U.A.; Benveniste, O.; Lloyd, T.E.; Needham, M.; Chinoy, H.; Aoki, M.; Machado, P.M.; Liang, C.; Reardon, K.A.; et al. Safety and efficacy of intravenous bimagrumab in inclusion body myositis (RESILIENT): A randomised, double-blind, placebo-controlled phase 2b trial. Lancet. Neurol. 2019, 18, 834–844. [Google Scholar] [CrossRef]
- Efficacy, Safety and Tolerability of BYM338 in Patients With Sporadic Inclusion Body Myositis. ClinicalTrials.gov; US Nationa Library of Medicine. Available online: https://clinicaltrials.gov/ct2/show/NCT01423110 (accessed on 17 September 2021).
- Amato, A.A.; Sivakumar, K.; Goyal, N.; David, W.S.; Salajegheh, M.; Praestgaard, J.; Lach-Trifilieff, E.; Trendelenburg, A.U.; Laurent, D.; Glass, D.J.; et al. Treatment of sporadic inclusion body myositis with bimagrumab. Neurology 2014, 83, 2239–2246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Double-blind, Randomized, Placebo-controlled Trial of Etanercept for 12 Months in Subjects With Inclusion Body Myositis. ClinicalTrials.gov; US National Library of Medicine. Available online: https://clinicaltrials.gov/ct2/show/NCT00802815 (accessed on 20 September 2021).
- Barohn, R.J.; Herbelin, L.; Kissel, J.T.; King, W.; McVey, A.L.; Saperstein, D.S.; Mendell, J.R. Pilot trial of etanercept in the treatment of inclusion-body myositis. Neurology 2006, 66, S123–S124. [Google Scholar] [CrossRef]
- Muscle Study, G. Randomized pilot trial of betaINF1a (Avonex) in patients with inclusion body myositis. Neurology 2001, 57, 1566–1570. [Google Scholar] [CrossRef]
- Muscle Study, G. Randomized pilot trial of high-dose betaINF-1a in patients with inclusion body myositis. Neurology 2004, 63, 718–720. [Google Scholar] [CrossRef]
- Intravenousimmunoglobulin (IVIg) for the Treatment of Inflammatory Myopathies. ClinicalTrials.gov; US National Library of Medicine. Available online: https://clinicaltrials.gov/ct2/show/record/NCT00001261 (accessed on 20 September 2021).
- Lithium in Inclusion Body Myositis (IBM) (Li-IBM). ClinicalTrials.gov; US National Library of Medicine. Available online: https://clinicaltrials.gov/ct2/show/NCT00917956 (accessed on 20 September 2021).
- Natalizumab in Inclusion Body Myositis (IBM) (IBM-NAT). ClinicalTrials.gov; US National Library of Medicine. Available online: https://clinicaltrials.gov/ct2/show/NCT02483845 (accessed on 20 September 2021).
- Rutkove, S.B.; Parker, R.A.; Nardin, R.A.; Connolly, C.E.; Felice, K.J.; Raynor, E.M. A pilot randomized trial of oxandrolone in inclusion body myositis. Neurology 2002, 58, 1081–1087. [Google Scholar] [CrossRef]
- Safety and Tolerability of Phenylbutyrate in Inclusion Body Myositis. ClinicalTrials.gov; US National Library of Medicine. Available online: https://clinicaltrials.gov/ct2/show/NCT04421677 (accessed on 21 September 2021).
- Study of Pioglitazone in Sporadic Inclusion Body Myositis. ClinicalTrials.gov; US National Library of Medicine. Available online: https://clinicaltrials.gov/ct2/show/NCT03440034 (accessed on 21 September 2021).
- Rapamycine vs Placebo for the Treatment of Inclusion Body Myositis (RAPAMI). ClinicalTrials.gov; US National Library of Medicine. Available online: https://clinicaltrials.gov/ct2/show/NCT02481453 (accessed on 21 September 2021).
- Simvastatin treatment in inclusion body myositis (IBM). EU Clinical Trials Register. European Medicines Agency. Available online: https://www.clinicaltrialsregister.eu/ctr-search/trial/2007-004359-12/IT (accessed on 21 September 2021).
- Simvastatin therapy in IBM. EU Clinical Trials Register. European Medicines Agency. Available online: https://www.clinicaltrialsregister.eu/ctr-search/trial/2006-005942-35/IT (accessed on 21 September 2021).
- Phase III Trial of Sirolimus in IBM. ClinicalTrials.gov; US National Library of Medicine. Available online: https://clinicaltrials.gov/ct2/show/NCT04789070 (accessed on 21 September 2021).
- Inclusion Body Myositis Treatment With Celution Processed Adipose Derived Regenerative Cells. ClinicalTrials.gov; US National Library of Medicine. Available online: https://clinicaltrials.gov/ct2/show/NCT04975841 (accessed on 14 September 2021).
- Cell Therapy for IBM by Muscle Injection of ADSVF: A Phase I Trial (ADSVF-in-IBM). ClinicalTrials.gov; US National Library of Medicine. Available online: https://clinicaltrials.gov/ct2/show/NCT05032131 (accessed on 14 September 2021).
- Follistatin Gene Transfer to Patients With Becker Muscular Dystrophy and Sporadic Inclusion Body Myositis. ClinicalTrials.gov; US National Library of Medicine. Available online: https://clinicaltrials.gov/ct2/show/NCT01519349 (accessed on 14 September 2021).
- Mendell, J.R.; Sahenk, Z.; Al-Zaidy, S.; Rodino-Klapac, L.R.; Lowes, L.P.; Alfano, L.N.; Berry, K.; Miller, N.; Yalvac, M.; Dvorchik, I.; et al. Follistatin Gene Therapy for Sporadic Inclusion Body Myositis Improves Functional Outcomes. Mol. Ther. J. Am. Soc. Gene Ther. 2017, 25, 870–879. [Google Scholar] [CrossRef] [Green Version]
- Nagini, S.; Sophia, J.; Mishra, R. Glycogen synthase kinases: Moonlighting proteins with theranostic potential in cancer. Semin. Cancer Biol. 2019, 56, 25–36. [Google Scholar] [CrossRef]
- Mancinelli, R.; Carpino, G.; Petrungaro, S.; Mammola, C.L.; Tomaipitinca, L.; Filippini, A.; Facchiano, A.; Ziparo, E.; Giampietri, C. Multifaceted Roles of GSK-3 in Cancer and Autophagy-Related Diseases. Oxidative Med. Cell. Longev. 2017, 2017, 4629495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef] [Green Version]
- Cole, A.; Frame, S.; Cohen, P. Further evidence that the tyrosine phosphorylation of glycogen synthase kinase-3 (GSK3) in mammalian cells is an autophosphorylation event. Biochem. J. 2004, 377, 249–255. [Google Scholar] [CrossRef] [Green Version]
- Takahashi-Yanaga, F.; Shiraishi, F.; Hirata, M.; Miwa, Y.; Morimoto, S.; Sasaguri, T. Glycogen synthase kinase-3beta is tyrosine-phosphorylated by MEK1 in human skin fibroblasts. Biochem. Biophys. Res. Commun. 2004, 316, 411–415. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Bertrand, F.E.; Davis, N.M.; Sokolosky, M.; Abrams, S.L.; Montalto, G.; D’Assoro, A.B.; Libra, M.; Nicoletti, F.; et al. GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget 2014, 5, 2881–2911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayas, C.L.; Ariaens, A.; Ponsioen, B.; Moolenaar, W.H. GSK-3 is activated by the tyrosine kinase Pyk2 during LPA1-mediated neurite retraction. Mol. Biol. Cell 2006, 17, 1834–1844. [Google Scholar] [CrossRef] [Green Version]
- Forde, J.E.; Dale, T.C. Glycogen synthase kinase 3: A key regulator of cellular fate. Cell. Mol. Life Sci. CMLS 2007, 64, 1930–1944. [Google Scholar] [CrossRef]
- Park, S.A.; Ahn, S.I.; Gallo, J.M. Tau mis-splicing in the pathogenesis of neurodegenerative disorders. BMB Rep. 2016, 49, 405–413. [Google Scholar] [CrossRef] [Green Version]
- Balaji, V.; Kaniyappan, S.; Mandelkow, E.; Wang, Y.; Mandelkow, E.M. Pathological missorting of endogenous MAPT/Tau in neurons caused by failure of protein degradation systems. Autophagy 2018, 14, 2139–2154. [Google Scholar] [CrossRef]
- Bose, A.; Mouton-Liger, F.; Paquet, C.; Mazot, P.; Vigny, M.; Gray, F.; Hugon, J. Modulation of tau phosphorylation by the kinase PKR: Implications in Alzheimer’s disease. Brain Pathol. 2011, 21, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Hugon, J.; Mouton-Liger, F.; Dumurgier, J.; Paquet, C. PKR involvement in Alzheimer’s disease. Alzheimer Res. Ther. 2017, 9, 83. [Google Scholar] [CrossRef]
- Llorens-Martin, M.; Jurado, J.; Hernandez, F.; Avila, J. GSK-3beta, a pivotal kinase in Alzheimer disease. Front. Mol. Neurosci. 2014, 7, 46. [Google Scholar] [CrossRef] [Green Version]
- Singh, T.J.; Grundke-Iqbal, I.; Wu, W.Q.; Chauhan, V.; Novak, M.; Kontzekova, E.; Iqbal, K. Protein kinase C and calcium/calmodulin-dependent protein kinase II phosphorylate three-repeat and four-repeat tau isoforms at different rates. Mol. Cell. Biochem. 1997, 168, 141–148. [Google Scholar] [CrossRef]
- Wang, J.Z.; Wu, Q.; Smith, A.; Grundke-Iqbal, I.; Iqbal, K. Tau is phosphorylated by GSK-3 at several sites found in Alzheimer disease and its biological activity markedly inhibited only after it is prephosphorylated by A-kinase. FEBS Lett. 1998, 436, 28–34. [Google Scholar] [CrossRef] [Green Version]
- Mailliot, C.; Podevin-Dimster, V.; Rosenthal, R.E.; Sergeant, N.; Delacourte, A.; Fiskum, G.; Buee, L. Rapid tau protein dephosphorylation and differential rephosphorylation during cardiac arrest-induced cerebral ischemia and reperfusion. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2000, 20, 543–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Illenberger, S.; Zheng-Fischhofer, Q.; Preuss, U.; Stamer, K.; Baumann, K.; Trinczek, B.; Biernat, J.; Godemann, R.; Mandelkow, E.M.; Mandelkow, E. The endogenous and cell cycle-dependent phosphorylation of tau protein in living cells: Implications for Alzheimer’s disease. Mol. Biol. Cell 1998, 9, 1495–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, A.; Biernat, J.; von Bergen, M.; Mandelkow, E.; Mandelkow, E.M. Phosphorylation that detaches tau protein from microtubules (Ser262, Ser214) also protects it against aggregation into Alzheimer paired helical filaments. Biochemistry 1999, 38, 3549–3558. [Google Scholar] [CrossRef]
- Liu, F.; Li, B.; Tung, E.J.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Site-specific effects of tau phosphorylation on its microtubule assembly activity and self-aggregation. Eur. J. Neurosci. 2007, 26, 3429–3436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Liang, Z.; Shi, J.; Yin, D.; El-Akkad, E.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. PKA modulates GSK-3beta- and cdk5-catalyzed phosphorylation of tau in site- and kinase-specific manners. FEBS Lett. 2006, 580, 6269–6274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Gong, C.X. Involvement of aberrant glycosylation in phosphorylation of tau by cdk5 and GSK-3beta. FEBS Lett. 2002, 530, 209–214. [Google Scholar] [CrossRef] [Green Version]
- Leroy, A.; Landrieu, I.; Huvent, I.; Legrand, D.; Codeville, B.; Wieruszeski, J.M.; Lippens, G. Spectroscopic studies of GSK3{beta} phosphorylation of the neuronal tau protein and its interaction with the N-terminal domain of apolipoprotein E. J. Biol. Chem. 2010, 285, 33435–33444. [Google Scholar] [CrossRef] [Green Version]
- Qian, W.; Shi, J.; Yin, X.; Iqbal, K.; Grundke-Iqbal, I.; Gong, C.X.; Liu, F. PP2A regulates tau phosphorylation directly and also indirectly via activating GSK-3beta. J. Alzheimer Dis. JAD 2010, 19, 1221–1229. [Google Scholar] [CrossRef]
- Sengupta, A.; Novak, M.; Grundke-Iqbal, I.; Iqbal, K. Regulation of phosphorylation of tau by cyclin-dependent kinase 5 and glycogen synthase kinase-3 at substrate level. FEBS Lett. 2006, 580, 5925–5933. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, A.; Kabat, J.; Novak, M.; Wu, Q.; Grundke-Iqbal, I.; Iqbal, K. Phosphorylation of tau at both Thr 231 and Ser 262 is required for maximal inhibition of its binding to microtubules. Arch. Biochem. Biophys. 1998, 357, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Moszczynski, A.J.; Gohar, M.; Volkening, K.; Leystra-Lantz, C.; Strong, W.; Strong, M.J. Thr175-phosphorylated tau induces pathologic fibril formation via GSK3beta-mediated phosphorylation of Thr231 in vitro. Neurobiol. Aging 2015, 36, 1590–1599. [Google Scholar] [CrossRef] [PubMed]
- Tatebayashi, Y.; Planel, E.; Chui, D.H.; Sato, S.; Miyasaka, T.; Sahara, N.; Murayama, M.; Kikuchi, N.; Yoshioka, K.; Rivka, R.; et al. c-jun N-terminal kinase hyperphosphorylates R406W tau at the PHF-1 site during mitosis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2006, 20, 762–764. [Google Scholar] [CrossRef] [PubMed]
- Azorsa, D.O.; Robeson, R.H.; Frost, D.; Meec Hoovet, B.; Brautigam, G.R.; Dickey, C.; Beaudry, C.; Basu, G.D.; Holz, D.R.; Hernandez, J.A.; et al. High-content siRNA screening of the kinome identifies kinases involved in Alzheimer’s disease-related tau hyperphosphorylation. BMC Genom. 2010, 11, 25. [Google Scholar] [CrossRef] [Green Version]
- Ho, Y.S.; Yang, X.; Lau, J.C.; Hung, C.H.; Wuwongse, S.; Zhang, Q.; Wang, J.; Baum, L.; So, K.F.; Chang, R.C. Endoplasmic reticulum stress induces tau pathology and forms a vicious cycle: Implication in Alzheimer’s disease pathogenesis. J. Alzheimer Dis. JAD 2012, 28, 839–854. [Google Scholar] [CrossRef] [Green Version]
- Nijholt, D.A.; van Haastert, E.S.; Rozemuller, A.J.; Scheper, W.; Hoozemans, J.J. The unfolded protein response is associated with early tau pathology in the hippocampus of tauopathies. J. Pathol. 2012, 226, 693–702. [Google Scholar] [CrossRef]
- Abisambra, J.F.; Jinwal, U.K.; Blair, L.J.; O’Leary, J.C., III; Li, Q.; Brady, S.; Wang, L.; Guidi, C.E.; Zhang, B.; Nordhues, B.A.; et al. Tau accumulation activates the unfolded protein response by impairing endoplasmic reticulum-associated degradation. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 9498–9507. [Google Scholar] [CrossRef] [Green Version]
- Lu, B.; Nakamura, T.; Inouye, K.; Li, J.; Tang, Y.; Lundback, P.; Valdes-Ferrer, S.I.; Olofsson, P.S.; Kalb, T.; Roth, J.; et al. Novel role of PKR in inflammasome activation and HMGB1 release. Nature 2012, 488, 670–674. [Google Scholar] [CrossRef] [Green Version]
- Van Zeller, M.; Dias, D.M.; Sebastiao, A.M.; Valente, C.A. NLRP3 Inflammasome: A Starring Role in Amyloid-beta- and Tau-Driven Pathological Events in Alzheimer’s Disease. J. Alzheimer Dis. JAD 2021, 83, 939–961. [Google Scholar] [CrossRef] [PubMed]
- Reimer, L.; Betzer, C.; Kofoed, R.H.; Volbracht, C.; Fog, K.; Kurhade, C.; Nilsson, E.; Overby, A.K.; Jensen, P.H. PKR kinase directly regulates tau expression and Alzheimer’s disease-related tau phosphorylation. Brain Pathol. 2021, 31, 103–119. [Google Scholar] [CrossRef]
- Piazzi, M.; Bavelloni, A.; Faenza, I.; Blalock, W. Glycogen synthase kinase (GSK)-3 and the double-strand RNA-dependent kinase, PKR: When two kinases for the common good turn bad. Biochim. Et Biophys. Acta Mol. Cell Res. 2020, 1867, 118769. [Google Scholar] [CrossRef]
- Takashima, A. GSK-3 is essential in the pathogenesis of Alzheimer’s disease. J. Alzheimer’s Dis. JAD 2006, 9, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Sayas, C.L.; Avila, J. GSK-3 and Tau: A Key Duet in Alzheimer’s Disease. Cells 2021, 10, 721. [Google Scholar] [CrossRef] [PubMed]
- Chahin, N.; Engel, A.G. Correlation of muscle biopsy, clinical course, and outcome in PM and sporadic IBM. Neurology 2008, 70, 418–424. [Google Scholar] [CrossRef]
- Temiz, P.; Weihl, C.C.; Pestronk, A. Inflammatory myopathies with mitochondrial pathology and protein aggregates. J. Neurol. Sci. 2009, 278, 25–29. [Google Scholar] [CrossRef]
- Kitazawa, M.; Green, K.N.; Caccamo, A.; LaFerla, F.M. Genetically augmenting Abeta42 levels in skeletal muscle exacerbates inclusion body myositis-like pathology and motor deficits in transgenic mice. Am. J. Pathol. 2006, 168, 1986–1997. [Google Scholar] [CrossRef] [Green Version]
- Kitazawa, M.; Trinh, D.N.; LaFerla, F.M. Inflammation induces tau pathology in inclusion body myositis model via glycogen synthase kinase-3beta. Ann. Neurol. 2008, 64, 15–24. [Google Scholar] [CrossRef]
- Nicot, A.S.; Lo Verso, F.; Ratti, F.; Pilot-Storck, F.; Streichenberger, N.; Sandri, M.; Schaeffer, L.; Goillot, E. Phosphorylation of NBR1 by GSK3 modulates protein aggregation. Autophagy 2014, 10, 1036–1053. [Google Scholar] [CrossRef] [Green Version]
- Chou, S.M. Myxovirus-like structures in a case of human chronic polymyositis. Science 1967, 158, 1453–1455. [Google Scholar] [CrossRef]
- Ciaccio, M.; Lo Sasso, B.; Scazzone, C.; Gambino, C.M.; Ciaccio, A.M.; Bivona, G.; Piccoli, T.; Giglio, R.V.; Agnello, L. COVID-19 and Alzheimer’s Disease. Brain Sci. 2021, 11, 305. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Xu, J.; Hou, Y.; Leverenz, J.B.; Kallianpur, A.; Mehra, R.; Liu, Y.; Yu, H.; Pieper, A.A.; Jehi, L.; et al. Network medicine links SARS-CoV-2/COVID-19 infection to brain microvascular injury and neuroinflammation in dementia-like cognitive impairment. Alzheimer Res. Ther. 2021, 13, 110. [Google Scholar] [CrossRef]
- Ioannidou, A.; Goulielmaki, E.; Garinis, G.A. DNA Damage: From Chronic Inflammation to Age-Related Deterioration. Front. Genet. 2016, 7, 187. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Byrne, M.; Brown, K.D.; Konopleva, M.Y.; Kornblau, S.M.; Bennett, R.L.; May, W.S. PKR inhibits the DNA damage response, and is associated with poor survival in AML and accelerated leukemia in NHD13 mice. Blood 2015, 126, 1585–1594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, H.H.; Yang, Y.X.; Meng, X.; Luo, X.Y.; Li, X.M.; Shuai, Z.W.; Ye, D.Q.; Pan, H.F. NLRP3: A promising therapeutic target for autoimmune diseases. Autoimmun. Rev. 2018, 17, 694–702. [Google Scholar] [CrossRef]
- Wang, H.; Brown, J.; Martin, M. Glycogen synthase kinase 3: A point of convergence for the host inflammatory response. Cytokine 2011, 53, 130–140. [Google Scholar] [CrossRef] [Green Version]
- Beurel, E.; Michalek, S.M.; Jope, R.S. Innate and adaptive immune responses regulated by glycogen synthase kinase-3 (GSK3). Trends Immunol. 2010, 31, 24–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, R.; Park, J.H.; Ha, H.; Choi, Y.; Lee, S.Y. Glycogen synthase kinase 3beta ubiquitination by TRAF6 regulates TLR3-mediated pro-inflammatory cytokine production. Nat. Commun. 2015, 6, 6765. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Wang, H.; Ni, M.; Yue, S.; Xia, Y.; Busuttil, R.W.; Kupiec-Weglinski, J.W.; Lu, L.; Wang, X.; Zhai, Y. Glycogen synthase kinase 3beta promotes liver innate immune activation by restraining AMP-activated protein kinase activation. J. Hepatol. 2018, 69, 99–109. [Google Scholar] [CrossRef]
- Prantner, D.; Perkins, D.J.; Vogel, S.N. AMP-activated Kinase (AMPK) Promotes Innate Immunity and Antiviral Defense through Modulation of Stimulator of Interferon Genes (STING) Signaling. J. Biol. Chem. 2017, 292, 292–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marineau, A.; Khan, K.A.; Servant, M.J. Roles of GSK-3 and beta-Catenin in Antiviral Innate Immune Sensing of Nucleic Acids. Cells 2020, 9, 897. [Google Scholar] [CrossRef] [Green Version]
- Du, X.; He, W.; He, H.; Wang, H. Beta-catenin inhibits bovine parainfluenza virus type 3 replication via innate immunity pathway. BMC Vet. Res. 2020, 16, 72. [Google Scholar] [CrossRef] [Green Version]
- Qin, Y.; Liu, Q.; Tian, S.; Xie, W.; Cui, J.; Wang, R.F. TRIM9 short isoform preferentially promotes DNA and RNA virus-induced production of type I interferon by recruiting GSK3beta to TBK1. Cell Res. 2016, 26, 613–628. [Google Scholar] [CrossRef]
- Khan, K.A.; Do, F.; Marineau, A.; Doyon, P.; Clement, J.F.; Woodgett, J.R.; Doble, B.W.; Servant, M.J. Fine-Tuning of the RIG-I-Like Receptor/Interferon Regulatory Factor 3-Dependent Antiviral Innate Immune Response by the Glycogen Synthase Kinase 3/beta-Catenin Pathway. Mol. Cell. Biol. 2015, 35, 3029–3043. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.T.; Chang, L.S.; Chen, C.J.; Doong, S.L.; Chang, C.W.; Chen, M.R. Glycogen synthase kinase 3 negatively regulates IFN regulatory factor 3 transactivation through phosphorylation at its linker region. Innate Immun. 2014, 20, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Stamos, J.L.; Weis, W.I. The beta-catenin destruction complex. Cold Spring Harb. Perspect. Biol. 2013, 5, a007898. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chen, H.; Chen, L.; Chen, Y.; Liu, X.; Mo, D. miR-709 modulates LPS-induced inflammatory response through targeting GSK-3beta. Int. Immunopharmacol. 2016, 36, 333–338. [Google Scholar] [CrossRef]
- Londino, J.D.; Gulick, D.L.; Lear, T.B.; Suber, T.L.; Weathington, N.M.; Masa, L.S.; Chen, B.B.; Mallampalli, R.K. Post-translational modification of the interferon-gamma receptor alters its stability and signaling. Biochem. J. 2017, 474, 3543–3557. [Google Scholar] [CrossRef] [Green Version]
- Beurel, E.; Jope, R.S. Glycogen synthase kinase-3 promotes the synergistic action of interferon-gamma on lipopolysaccharide-induced IL-6 production in RAW264.7 cells. Cell. Signal. 2009, 21, 978–985. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Paik, P.K.; Chen, J.; Yarilina, A.; Kockeritz, L.; Lu, T.T.; Woodgett, J.R.; Ivashkiv, L.B. IFN-gamma suppresses IL-10 production and synergizes with TLR2 by regulating GSK3 and CREB/AP-1 proteins. Immunity 2006, 24, 563–574. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.F.; Tsai, C.C.; Huang, W.C.; Wang, C.Y.; Tseng, H.C.; Wang, Y.; Kai, J.I.; Wang, S.W.; Cheng, Y.L. IFN-gamma synergizes with LPS to induce nitric oxide biosynthesis through glycogen synthase kinase-3-inhibited IL-10. J. Cell. Biochem. 2008, 105, 746–755. [Google Scholar] [CrossRef]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.C.; Uhl, S.; Hoagland, D.; Moller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045. [Google Scholar] [CrossRef]
- Rudd, C.E. GSK-3 Inhibition as a Therapeutic Approach Against SARs CoV2: Dual Benefit of Inhibiting Viral Replication While Potentiating the Immune Response. Front. Immunol. 2020, 11, 1638. [Google Scholar] [CrossRef]
- del Ser, T.; Steinwachs, K.C.; Gertz, H.J.; Andres, M.V.; Gomez-Carrillo, B.; Medina, M.; Vericat, J.A.; Redondo, P.; Fleet, D.; Leon, T. Treatment of Alzheimer’s disease with the GSK-3 inhibitor tideglusib: A pilot study. J. Alzheimer Dis. JAD 2013, 33, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Lovestone, S.; Boada, M.; Dubois, B.; Hull, M.; Rinne, J.O.; Huppertz, H.J.; Calero, M.; Andres, M.V.; Gomez-Carrillo, B.; Leon, T.; et al. A phase II trial of tideglusib in Alzheimer’s disease. J. Alzheimer Dis. JAD 2015, 45, 75–88. [Google Scholar] [CrossRef]
- Hostiuc, S.; Perlea, P.; Marinescu, M.; Dogaroiu, C.; Drima, E. GSK-3 Inhibitors and Tooth Repair: An Ethical Analysis. Front. Pharmacol. 2018, 9, 1495. [Google Scholar] [CrossRef] [PubMed]
- Mettey, Y.; Gompel, M.; Thomas, V.; Garnier, M.; Leost, M.; Ceballos-Picot, I.; Noble, M.; Endicott, J.; Vierfond, J.M.; Meijer, L. Aloisines, a new family of CDK/GSK-3 inhibitors. SAR study, crystal structure in complex with CDK2, enzyme selectivity, and cellular effects. J. Med. Chem. 2003, 46, 222–236. [Google Scholar] [CrossRef]
- Selenica, M.L.; Jensen, H.S.; Larsen, A.K.; Pedersen, M.L.; Helboe, L.; Leist, M.; Lotharius, J. Efficacy of small-molecule glycogen synthase kinase-3 inhibitors in the postnatal rat model of tau hyperphosphorylation. Br. J. Pharmacol. 2007, 152, 959–979. [Google Scholar] [CrossRef] [Green Version]
- Ring, D.B.; Johnson, K.W.; Henriksen, E.J.; Nuss, J.M.; Goff, D.; Kinnick, T.R.; Ma, S.T.; Reeder, J.W.; Samuels, I.; Slabiak, T.; et al. Selective glycogen synthase kinase 3 inhibitors potentiate insulin activation of glucose transport and utilization in vitro and in vivo. Diabetes 2003, 52, 588–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Sun, W.; Zhang, Y.; Wei, W.; Ambasudhan, R.; Xia, P.; Talantova, M.; Lin, T.; Kim, J.; Wang, X.; et al. Rapid induction and long-term self-renewal of primitive neural precursors from human embryonic stem cells by small molecule inhibitors. Proc. Natl. Acad. Sci. USA 2011, 108, 8299–8304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mussmann, R.; Geese, M.; Harder, F.; Kegel, S.; Andag, U.; Lomow, A.; Burk, U.; Onichtchouk, D.; Dohrmann, C.; Austen, M. Inhibition of GSK3 promotes replication and survival of pancreatic beta cells. J. Biol. Chem. 2007, 282, 12030–12037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, S.; Wu, T.Y.; Brinker, A.; Peters, E.C.; Hur, W.; Gray, N.S.; Schultz, P.G. Synthetic small molecules that control stem cell fate. Proc. Natl. Acad. Sci. USA 2003, 100, 7632–7637. [Google Scholar] [CrossRef] [Green Version]
- Muralidharan, S.; Hanley, P.J.; Liu, E.; Chakraborty, R.; Bollard, C.; Shpall, E.; Rooney, C.; Savoldo, B.; Rodgers, J.; Dotti, G. Activation of Wnt signaling arrests effector differentiation in human peripheral and cord blood-derived T lymphocytes. J. Immunol. 2011, 187, 5221–5232. [Google Scholar] [CrossRef]
- Ugolkov, A.V.; Bondarenko, G.I.; Dubrovskyi, O.; Berbegall, A.P.; Navarro, S.; Noguera, R.; O’Halloran, T.V.; Hendrix, M.J.; Giles, F.J.; Mazar, A.P. 9-ING-41, a small-molecule glycogen synthase kinase-3 inhibitor, is active in neuroblastoma. Anti-Cancer Drugs 2018, 29, 717–724. [Google Scholar] [CrossRef]
- Wu, X.; Stenson, M.; Abeykoon, J.; Nowakowski, K.; Zhang, L.; Lawson, J.; Wellik, L.; Li, Y.; Krull, J.; Wenzl, K.; et al. Targeting glycogen synthase kinase 3 for therapeutic benefit in lymphoma. Blood 2019, 134, 363–373. [Google Scholar] [CrossRef]
- Cross, D.A.; Culbert, A.A.; Chalmers, K.A.; Facci, L.; Skaper, S.D.; Reith, A.D. Selective small-molecule inhibitors of glycogen synthase kinase-3 activity protect primary neurones from death. J. Neurochem. 2001, 77, 94–102. [Google Scholar] [CrossRef]
- Gurrieri, C.; Piazza, F.; Gnoato, M.; Montini, B.; Biasutto, L.; Gattazzo, C.; Brunetta, E.; Cabrelle, A.; Cinetto, F.; Niero, R.; et al. 3-(2,4-dichlorophenyl)-4-(1-methyl-1H-indol-3-yl)-1H-pyrrole-2,5-dione (SB216763), a glycogen synthase kinase-3 inhibitor, displays therapeutic properties in a mouse model of pulmonary inflammation and fibrosis. J. Pharmacol. Exp. Ther. 2010, 332, 785–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickey, A.; Schleicher, S.; Leahy, K.; Hu, R.; Hallahan, D.; Thotala, D.K. GSK-3beta inhibition promotes cell death, apoptosis, and in vivo tumor growth delay in neuroblastoma Neuro-2A cell line. J. Neuro-Oncol. 2011, 104, 145–153. [Google Scholar] [CrossRef] [Green Version]
- Piazza, F.; Manni, S.; Tubi, L.Q.; Montini, B.; Pavan, L.; Colpo, A.; Gnoato, M.; Cabrelle, A.; Adami, F.; Zambello, R.; et al. Glycogen Synthase Kinase-3 regulates multiple myeloma cell growth and bortezomib-induced cell death. BMC Cancer 2010, 10, 526. [Google Scholar] [CrossRef]
- Pizarro, J.G.; Yeste-Velasco, M.; Rimbau, V.; Casadesus, G.; Smith, M.A.; Pallas, M.; Folch, J.; Camins, A. Neuroprotective effects of SB-415286 on hydrogen peroxide-induced cell death in B65 rat neuroblastoma cells and neurons. Int. J. Dev. Neurosci. Off. J. Int. Soc. Dev. Neurosci. 2008, 26, 269–276. [Google Scholar] [CrossRef]
- Whittle, B.J.; Varga, C.; Posa, A.; Molnar, A.; Collin, M.; Thiemermann, C. Reduction of experimental colitis in the rat by inhibitors of glycogen synthase kinase-3beta. Br. J. Pharmacol. 2006, 147, 575–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coghlan, M.P.; Culbert, A.A.; Cross, D.A.; Corcoran, S.L.; Yates, J.W.; Pearce, N.J.; Rausch, O.L.; Murphy, G.J.; Carter, P.S.; Roxbee Cox, L.; et al. Selective small molecule inhibitors of glycogen synthase kinase-3 modulate glycogen metabolism and gene transcription. Chem. Biol. 2000, 7, 793–803. [Google Scholar] [CrossRef] [Green Version]
- Sato, N.; Meijer, L.; Skaltsounis, L.; Greengard, P.; Brivanlou, A.H. Maintenance of pluripotency in human and mouse embryonic stem cells through activation of Wnt signaling by a pharmacological GSK-3-specific inhibitor. Nat. Med. 2004, 10, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Sklirou, A.D.; Gaboriaud-Kolar, N.; Papassideri, I.; Skaltsounis, A.L.; Trougakos, I.P. 6-bromo-indirubin-3’-oxime (6BIO), a Glycogen synthase kinase-3beta inhibitor, activates cytoprotective cellular modules and suppresses cellular senescence-mediated biomolecular damage in human fibroblasts. Sci. Rep. 2017, 7, 11713. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Li, J.; Wu, X.; Chen, M.; Luo, F.; Li, J. GSK-3beta inhibitor 6-bromo-indirubin-3’-oxime promotes both adhesive activity and drug resistance in colorectal cancer cells. Int. J. Oncol. 2017, 51, 1821–1830. [Google Scholar] [CrossRef] [Green Version]
- Leclerc, S.; Garnier, M.; Hoessel, R.; Marko, D.; Bibb, J.A.; Snyder, G.L.; Greengard, P.; Biernat, J.; Wu, Y.Z.; Mandelkow, E.M.; et al. Indirubins inhibit glycogen synthase kinase-3 beta and CDK5/p25, two protein kinases involved in abnormal tau phosphorylation in Alzheimer’s disease. A property common to most cyclin-dependent kinase inhibitors? J. Biol. Chem. 2001, 276, 251–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.J.; Kim, M.Y.; Mo, J.S.; Ann, E.J.; Seo, M.S.; Hong, J.A.; Kim, Y.C.; Park, H.S. Indirubin-3’-monoxime, a derivative of a Chinese anti-leukemia medicine, inhibits Notch1 signaling. Cancer Lett. 2008, 265, 215–225. [Google Scholar] [CrossRef]
- Gonzalez Arbelaez, L.F.; Perez Nunez, I.A.; Mosca, S.M. Gsk-3beta inhibitors mimic the cardioprotection mediated by ischemic pre- and postconditioning in hypertensive rats. BioMed Res. Int. 2013, 2013, 317456. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Hoi, P.M.; Chan, J.Y.; Lee, S.M. New perspective on the dual functions of indirubins in cancer therapy and neuroprotection. Anti-Cancer Agents Med. Chem. 2014, 14, 1213–1219. [Google Scholar] [CrossRef]
- Zhang, N.; Zhong, R.; Yan, H.; Jiang, Y. Structural features underlying selective inhibition of GSK3beta by dibromocantharelline: Implications for rational drug design. Chem. Biol. Drug Des. 2011, 77, 199–205. [Google Scholar] [CrossRef]
- Kramer, T.; Schmidt, B.; Lo Monte, F. Small-Molecule Inhibitors of GSK-3: Structural Insights and Their Application to Alzheimer’s Disease Models. Int. J. Alzheimer Dis. 2012, 2012, 381029. [Google Scholar] [CrossRef]
- Meijer, L.; Thunnissen, A.M.; White, A.W.; Garnier, M.; Nikolic, M.; Tsai, L.H.; Walter, J.; Cleverley, K.E.; Salinas, P.C.; Wu, Y.Z.; et al. Inhibition of cyclin-dependent kinases, GSK-3beta and CK1 by hymenialdisine, a marine sponge constituent. Chem. Biol. 2000, 7, 51–63. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, D.; Jin, H.; Ye, Z.; Wang, C.; Chen, K.; Kuek, V.; Xu, K.; Qiu, H.; Chen, P.; et al. Hymenialdisine: A Marine Natural Product That Acts on Both Osteoblasts and Osteoclasts and Prevents Estrogen-Dependent Bone Loss in Mice. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2020, 35, 1582–1596. [Google Scholar] [CrossRef] [PubMed]
- Kalinichev, M.; Dawson, L.A. Evidence for antimanic efficacy of glycogen synthase kinase-3 (GSK3) inhibitors in a strain-specific model of acute mania. Int. J. Neuropsychopharmacol. 2011, 14, 1051–1067. [Google Scholar] [CrossRef] [Green Version]
- Tatebayashi, Y.; Haque, N.; Tung, Y.C.; Iqbal, K.; Grundke-Iqbal, I. Role of tau phosphorylation by glycogen synthase kinase-3beta in the regulation of organelle transport. J. Cell Sci. 2004, 117, 1653–1663. [Google Scholar] [CrossRef] [Green Version]
- Cui, C.; Wang, Y.; Wang, Y.; Zhao, M.; Peng, S. Alsterpaullone, a Cyclin-Dependent Kinase Inhibitor, Mediated Toxicity in HeLa Cells through Apoptosis-Inducing Effect. J. Anal. Methods Chem. 2013, 2013, 602091. [Google Scholar] [CrossRef] [Green Version]
- Faria, C.C.; Agnihotri, S.; Mack, S.C.; Golbourn, B.J.; Diaz, R.J.; Olsen, S.; Bryant, M.; Bebenek, M.; Wang, X.; Bertrand, K.C.; et al. Identification of alsterpaullone as a novel small molecule inhibitor to target group 3 medulloblastoma. Oncotarget 2015, 6, 21718–21729. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, T.; Sato, Y.; Masud, H.; Takayama, M.; Matsuda, H.; Hara, Y.; Yanagi, Y.; Yoshida, M.; Goshima, F.; Murata, T.; et al. Antitumor activity of cyclin-dependent kinase inhibitor alsterpaullone in Epstein-Barr virus-associated lymphoproliferative disorders. Cancer Sci. 2020, 111, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Stukenbrock, H.; Mussmann, R.; Geese, M.; Ferandin, Y.; Lozach, O.; Lemcke, T.; Kegel, S.; Lomow, A.; Burk, U.; Dohrmann, C.; et al. 9-cyano-1-azapaullone (cazpaullone), a glycogen synthase kinase-3 (GSK-3) inhibitor activating pancreatic beta cell protection and replication. J. Med. Chem. 2008, 51, 2196–2207. [Google Scholar] [CrossRef]
- Zaharevitz, D.W.; Gussio, R.; Leost, M.; Senderowicz, A.M.; Lahusen, T.; Kunick, C.; Meijer, L.; Sausville, E.A. Discovery and initial characterization of the paullones, a novel class of small-molecule inhibitors of cyclin-dependent kinases. Cancer Res. 1999, 59, 2566–2569. [Google Scholar]
- Yoshida, H.; Kotani, H.; Kondo, T.; Tani, I.; Wei, X.; Tsuruta, S.; Kimura, A.; Asakawa, M.; Ito, M.; Nagai, S.; et al. CDK inhibitors suppress Th17 and promote iTreg differentiation, and ameliorate experimental autoimmune encephalomyelitis in mice. Biochem. Biophys. Res. Commun. 2013, 435, 378–384. [Google Scholar] [CrossRef]
- Joo, H.C.; Choi, J.W.; Moon, H.; Lee, C.Y.; Yoo, K.J.; Kim, S.W.; Hwang, K.C. Protective effects of kenpaullone on cardiomyocytes following H2O2-induced oxidative stress are attributed to inhibition of connexin 43 degradation by SGSM3. Biochem. Biophys. Res. Commun. 2018, 499, 368–373. [Google Scholar] [CrossRef]
- Kitabayashi, T.; Dong, Y.; Furuta, T.; Sabit, H.; Jiapaer, S.; Zhang, J.; Zhang, G.; Hayashi, Y.; Kobayashi, M.; Domoto, T.; et al. Identification of GSK3beta inhibitor kenpaullone as a temozolomide enhancer against glioblastoma. Sci. Rep. 2019, 9, 10049. [Google Scholar] [CrossRef] [PubMed]
- Martins, D.F.; Rosa, A.O.; Gadotti, V.M.; Mazzardo-Martins, L.; Nascimento, F.P.; Egea, J.; Lopez, M.G.; Santos, A.R. The antinociceptive effects of AR-A014418, a selective inhibitor of glycogen synthase kinase-3 beta, in mice. J. Pain 2011, 12, 315–322. [Google Scholar] [CrossRef]
- Koh, S.H.; Kim, Y.; Kim, H.Y.; Hwang, S.; Lee, C.H.; Kim, S.H. Inhibition of glycogen synthase kinase-3 suppresses the onset of symptoms and disease progression of G93A-SOD1 mouse model of ALS. Exp. Neurol. 2007, 205, 336–346. [Google Scholar] [CrossRef]
- Bhat, R.; Xue, Y.; Berg, S.; Hellberg, S.; Ormo, M.; Nilsson, Y.; Radesater, A.C.; Jerning, E.; Markgren, P.O.; Borgegard, T.; et al. Structural insights and biological effects of glycogen synthase kinase 3-specific inhibitor AR-A014418. J. Biol. Chem. 2003, 278, 45937–45945. [Google Scholar] [CrossRef] [Green Version]
- Georgievska, B.; Sandin, J.; Doherty, J.; Mortberg, A.; Neelissen, J.; Andersson, A.; Gruber, S.; Nilsson, Y.; Schott, P.; Arvidsson, P.I.; et al. AZD1080, a novel GSK3 inhibitor, rescues synaptic plasticity deficits in rodent brain and exhibits peripheral target engagement in humans. J. Neurochem. 2013, 125, 446–456. [Google Scholar] [CrossRef]
- Eldar-Finkelman, H.; Martinez, A. GSK-3 Inhibitors: Preclinical and Clinical Focus on CNS. Front. Mol. Neurosci. 2011, 4, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bidon-Chanal, A.; Fuertes, A.; Alonso, D.; Perez, D.I.; Martinez, A.; Luque, F.J.; Medina, M. Evidence for a new binding mode to GSK-3: Allosteric regulation by the marine compound palinurin. Eur. J. Med. Chem. 2013, 60, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Perez, D.I.; Conde, S.; Perez, C.; Gil, C.; Simon, D.; Wandosell, F.; Moreno, F.J.; Gelpi, J.L.; Luque, F.J.; Martinez, A. Thienylhalomethylketones: Irreversible glycogen synthase kinase 3 inhibitors as useful pharmacological tools. Bioorganic Med. Chem. 2009, 17, 6914–6925. [Google Scholar] [CrossRef]
- Rosa, A.O.; Egea, J.; Martinez, A.; Garcia, A.G.; Lopez, M.G. Neuroprotective effect of the new thiadiazolidinone NP00111 against oxygen-glucose deprivation in rat hippocampal slices: Implication of ERK1/2 and PPARgamma receptors. Exp. Neurol. 2008, 212, 93–99. [Google Scholar] [CrossRef]
- Luna-Medina, R.; Cortes-Canteli, M.; Alonso, M.; Santos, A.; Martinez, A.; Perez-Castillo, A. Regulation of inflammatory response in neural cells in vitro by thiadiazolidinones derivatives through peroxisome proliferator-activated receptor gamma activation. J. Biol. Chem. 2005, 280, 21453–21462. [Google Scholar] [CrossRef] [Green Version]
- Rosa, A.O.; Kaster, M.P.; Binfare, R.W.; Morales, S.; Martin-Aparicio, E.; Navarro-Rico, M.L.; Martinez, A.; Medina, M.; Garcia, A.G.; Lopez, M.G.; et al. Antidepressant-like effect of the novel thiadiazolidinone NP031115 in mice. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2008, 32, 1549–1556. [Google Scholar] [CrossRef]
- Dominguez, J.M.; Fuertes, A.; Orozco, L.; del Monte-Millan, M.; Delgado, E.; Medina, M. Evidence for irreversible inhibition of glycogen synthase kinase-3beta by tideglusib. J. Biol. Chem. 2012, 287, 893–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sereno, L.; Coma, M.; Rodriguez, M.; Sanchez-Ferrer, P.; Sanchez, M.B.; Gich, I.; Agullo, J.M.; Perez, M.; Avila, J.; Guardia-Laguarta, C.; et al. A novel GSK-3beta inhibitor reduces Alzheimer’s pathology and rescues neuronal loss in vivo. Neurobiol. Dis. 2009, 35, 359–367. [Google Scholar] [CrossRef]
- Luna-Medina, R.; Cortes-Canteli, M.; Sanchez-Galiano, S.; Morales-Garcia, J.A.; Martinez, A.; Santos, A.; Perez-Castillo, A. NP031112, a thiadiazolidinone compound, prevents inflammation and neurodegeneration under excitotoxic conditions: Potential therapeutic role in brain disorders. J. Neurosci. Off. J. Soc. Neurosci. 2007, 27, 5766–5776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzman, M.L.; Li, X.; Corbett, C.A.; Rossi, R.M.; Bushnell, T.; Liesveld, J.L.; Hebert, J.; Young, F.; Jordan, C.T. Rapid and selective death of leukemia stem and progenitor cells induced by the compound 4-benzyl, 2-methyl, 1,2,4-thiadiazolidine, 3,5 dione (TDZD-8). Blood 2007, 110, 4436–4444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, A.; Alonso, M.; Castro, A.; Perez, C.; Moreno, F.J. First non-ATP competitive glycogen synthase kinase 3 beta (GSK-3beta) inhibitors: Thiadiazolidinones (TDZD) as potential drugs for the treatment of Alzheimer’s disease. J. Med. Chem. 2002, 45, 1292–1299. [Google Scholar] [CrossRef] [PubMed]
- Hamann, M.; Alonso, D.; Martin-Aparicio, E.; Fuertes, A.; Perez-Puerto, M.J.; Castro, A.; Morales, S.; Navarro, M.L.; Del Monte-Millan, M.; Medina, M.; et al. Glycogen synthase kinase-3 (GSK-3) inhibitory activity and structure-activity relationship (SAR) studies of the manzamine alkaloids. Potential for Alzheimer’s disease. J. Nat. Prod. 2007, 70, 1397–1405. [Google Scholar] [CrossRef]
- Radwan, M.; Hanora, A.; Khalifa, S.; Abou-El-Ela, S.H. Manzamines: A potential for novel cures. Cell Cycle 2012, 11, 1765–1772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaidanovich-Beilin, O.; Milman, A.; Weizman, A.; Pick, C.G.; Eldar-Finkelman, H. Rapid antidepressive-like activity of specific glycogen synthase kinase-3 inhibitor and its effect on beta-catenin in mouse hippocampus. Biol. Psychiatry 2004, 55, 781–784. [Google Scholar] [CrossRef]
- Chen, G.; Bower, K.A.; Ma, C.; Fang, S.; Thiele, C.J.; Luo, J. Glycogen synthase kinase 3beta (GSK3beta) mediates 6-hydroxydopamine-induced neuronal death. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2004, 18, 1162–1164. [Google Scholar] [CrossRef]
- Kaidanovich-Beilin, O.; Eldar-Finkelman, H. Long-term treatment with novel glycogen synthase kinase-3 inhibitor improves glucose homeostasis in ob/ob mice: Molecular characterization in liver and muscle. J. Pharmacol. Exp. Ther. 2006, 316, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Avrahami, L.; Farfara, D.; Shaham-Kol, M.; Vassar, R.; Frenkel, D.; Eldar-Finkelman, H. Inhibition of glycogen synthase kinase-3 ameliorates beta-amyloid pathology and restores lysosomal acidification and mammalian target of rapamycin activity in the Alzheimer disease mouse model: In vivo and in vitro studies. J. Biol. Chem. 2013, 288, 1295–1306. [Google Scholar] [CrossRef] [Green Version]
- Beurel, E.; Kaidanovich-Beilin, O.; Yeh, W.I.; Song, L.; Palomo, V.; Michalek, S.M.; Woodgett, J.R.; Harrington, L.E.; Eldar-Finkelman, H.; Martinez, A.; et al. Regulation of Th1 cells and experimental autoimmune encephalomyelitis by glycogen synthase kinase-3. J. Immunol. 2013, 190, 5000–5011. [Google Scholar] [CrossRef] [Green Version]
- Rippin, I.; Bonder, K.; Joseph, S.; Sarsor, A.; Vaks, L.; Eldar-Finkelman, H. Inhibition of GSK-3 ameliorates the pathogenesis of Huntington’s disease. Neurobiol. Dis. 2021, 154, 105336. [Google Scholar] [CrossRef] [PubMed]
- Licht-Murava, A.; Paz, R.; Vaks, L.; Avrahami, L.; Plotkin, B.; Eisenstein, M.; Eldar-Finkelman, H. A unique type of GSK-3 inhibitor brings new opportunities to the clinic. Sci. Signal. 2016, 9, ra110. [Google Scholar] [CrossRef]
- Motawi, T.M.; Bustanji, Y.; El-Maraghy, S.A.; Taha, M.O.; Al Ghussein, M.A. Naproxen and cromolyn as new glycogen synthase kinase 3beta inhibitors for amelioration of diabetes and obesity: An investigation by docking simulation and subsequent in vitro/in vivo biochemical evaluation. J. Biochem. Mol. Toxicol. 2013, 27, 425–436. [Google Scholar] [CrossRef]
- Alhusaini, A.; Fadda, L.; Hasan, I.H.; Zakaria, E.; Alenazi, A.M.; Mahmoud, A.M. Curcumin Ameliorates Lead-Induced Hepatotoxicity by Suppressing Oxidative Stress and Inflammation, and Modulating Akt/GSK-3beta Signaling Pathway. Biomolecules 2019, 9, 703. [Google Scholar] [CrossRef] [Green Version]
- McCubrey, J.A.; Lertpiriyapong, K.; Steelman, L.S.; Abrams, S.L.; Cocco, L.; Ratti, S.; Martelli, A.M.; Candido, S.; Libra, M.; Montalto, G.; et al. Regulation of GSK-3 activity by curcumin, berberine and resveratrol: Potential effects on multiple diseases. Adv. Biol. Regul. 2017, 65, 77–88. [Google Scholar] [CrossRef]
- Jeong, C.W.; Yoo, K.Y.; Lee, S.H.; Jeong, H.J.; Lee, C.S.; Kim, S.J. Curcumin protects against regional myocardial ischemia/reperfusion injury through activation of RISK/GSK-3beta and inhibition of p38 MAPK and JNK. J. Cardiovasc. Pharmacol. Ther. 2012, 17, 387–394. [Google Scholar] [CrossRef]
- Yu, W.; Wu, J.; Cai, F.; Xiang, J.; Zha, W.; Fan, D.; Guo, S.; Ming, Z.; Liu, C. Curcumin alleviates diabetic cardiomyopathy in experimental diabetic rats. PLoS ONE 2012, 7, e52013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.C.; Xu, K.; Jiang, Z.F. Curcumin-mediated neuroprotection against amyloid-beta-induced mitochondrial dysfunction involves the inhibition of GSK-3beta. J. Alzheimer Dis. JAD 2012, 32, 981–996. [Google Scholar] [CrossRef]
- Xiong, Z.; Hongmei, Z.; Lu, S.; Yu, L. Curcumin mediates presenilin-1 activity to reduce beta-amyloid production in a model of Alzheimer’s Disease. Pharmacol. Rep. PR 2011, 63, 1101–1108. [Google Scholar] [CrossRef]
- Bustanji, Y.; Taha, M.O.; Almasri, I.M.; Al-Ghussein, M.A.; Mohammad, M.K.; Alkhatib, H.S. Inhibition of glycogen synthase kinase by curcumin: Investigation by simulated molecular docking and subsequent in vitro/in vivo evaluation. J. Enzym. Inhib. Med. Chem. 2009, 24, 771–778. [Google Scholar] [CrossRef]
- Mohammad, M.; Al-Masri, I.M.; Issa, A.; Al-Ghussein, M.A.; Fararjeh, M.; Alkhatib, H.; Taha, M.O.; Bustanji, Y. Famotidine inhibits glycogen synthase kinase-3beta: An investigation by docking simulation and experimental validation. J. Enzym. Inhib. Med. Chem. 2013, 28, 690–694. [Google Scholar] [CrossRef]
- Unal, G.; Dokumaci, A.H.; Ozkartal, C.S.; Yerer, M.B.; Aricioglu, F. Famotidine has a neuroprotective effect on MK-801 induced toxicity via the Akt/GSK-3beta/beta-catenin signaling pathway in the SH-SY5Y cell line. Chem.-Biol. Interact. 2019, 314, 108823. [Google Scholar] [CrossRef] [PubMed]
- Beurel, E.; Jope, R.S. Inflammation and lithium: Clues to mechanisms contributing to suicide-linked traits. Transl. Psychiatry 2014, 4, e488. [Google Scholar] [CrossRef]
- Won, E.; Kim, Y.K. An Oldie but Goodie: Lithium in the Treatment of Bipolar Disorder through Neuroprotective and Neurotrophic Mechanisms. Int. J. Mol. Sci. 2017, 18, 2679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morlet, E.; Hozer, F.; Costemale-Lacoste, J.F. Neuroprotective effects of lithium: What are the implications in humans with neurodegenerative disorders? Geriatr. Et Psychol. Neuropsychiatr. Du Vieil. 2018, 16, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Lista, S.; Mango, D.; Nistico, R.; Perry, G.; Avila, J.; Hernandez, F.; Geerts, H.; Vergallo, A.; Alzheimer Precision Medicine, I. Lithium as a Treatment for Alzheimer’s Disease: The Systems Pharmacology Perspective. J. Alzheimer Dis. JAD 2019, 69, 615–629. [Google Scholar] [CrossRef] [PubMed]
- Snitow, M.E.; Bhansali, R.S.; Klein, P.S. Lithium and Therapeutic Targeting of GSK-3. Cells 2021, 10, 255. [Google Scholar] [CrossRef]
- Mohammad, M.K.; Al-Masri, I.M.; Taha, M.O.; Al-Ghussein, M.A.; Alkhatib, H.S.; Najjar, S.; Bustanji, Y. Olanzapine inhibits glycogen synthase kinase-3beta: An investigation by docking simulation and experimental validation. Eur. J. Pharmacol. 2008, 584, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Rosborough, K.M.; Friedman, A.B.; Zhu, W.; Roth, K.A. Regulation of mouse brain glycogen synthase kinase-3 by atypical antipsychotics. Int. J. Neuropsychopharmacol. 2007, 10, 7–19. [Google Scholar] [CrossRef]
- Li, X.; Liu, M.; Cai, Z.; Wang, G.; Li, X. Regulation of glycogen synthase kinase-3 during bipolar mania treatment. Bipolar Disord. 2010, 12, 741–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilouz, R.; Kaidanovich, O.; Gurwitz, D.; Eldar-Finkelman, H. Inhibition of glycogen synthase kinase-3beta by bivalent zinc ions: Insight into the insulin-mimetic action of zinc. Biochem. Biophys. Res. Commun. 2002, 295, 102–106. [Google Scholar] [CrossRef]
- Szewczyk, B.; Poleszak, E.; Sowa-Kucma, M.; Siwek, M.; Dudek, D.; Ryszewska-Pokrasniewicz, B.; Radziwon-Zaleska, M.; Opoka, W.; Czekaj, J.; Pilc, A.; et al. Antidepressant activity of zinc and magnesium in view of the current hypotheses of antidepressant action. Pharmacol. Rep. PR 2008, 60, 588–589. [Google Scholar]
- Chanoit, G.; Lee, S.; Xi, J.; Zhu, M.; McIntosh, R.A.; Mueller, R.A.; Norfleet, E.A.; Xu, Z. Exogenous zinc protects cardiac cells from reperfusion injury by targeting mitochondrial permeability transition pore through inactivation of glycogen synthase kinase-3beta. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H1227–H1233. [Google Scholar] [CrossRef]
- Stelmashook, E.V.; Isaev, N.K.; Genrikhs, E.E.; Amelkina, G.A.; Khaspekov, L.G.; Skrebitsky, V.G.; Illarioshkin, S.N. Role of zinc and copper ions in the pathogenetic mechanisms of Alzheimer’s and Parkinson’s diseases. Biochemistry. Biokhimiia 2014, 79, 391–396. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Therapeutic | Start Date | Completion Date | Publication Date | Type of Trial | End Point | Ref. |
|---|---|---|---|---|---|---|
| Pharmacological | ||||||
| ABC008 | 2021 | N/A * | N/A * | Phase I; open-label | Safety and tolerability | [26] |
| Alemtuzumab | 2004 | 2007 | 2009 | Phase II | Efficacy and safety | [6,7] |
| Anakinra | 2003 | 2008 | N/A | Phase II/III; non-randomized, open-label, non-placebo-controlled | Efficacy | [24] |
| Anakinra | N/A | N/A | 2013 | Pilot study; open-label, uncontrolled | Efficacy | [23] |
| Antithymocyte Ig+MTX versus MTX alone | N/A | N/A | 2003 | Pilot study; randomized, open-label, non-placebo-controlled | Efficacy | [12] |
| Arimoclomol | 2019 | N/A * | N/A * | Phase III (extension); open-label non-randomized | Efficacy | [19] |
| Arimoclomol | 2019 | N/A * | N/A * | Phase III; open-label, non-randomized | Efficacy and safety of early vs late start of therapy | [27] |
| Arimoclomol | 2018 | N/A * | N/A * | Phase II/III; randomized, double-blind, placebo-controlled | Efficacy | [28] |
| Arimoclomol | 2017 | 2021 | N/A | Phase II; randomized, double-blind, placebo-controlled | Efficacy | [21] |
| Arimoclomol | 2009 | 2012 | 2017 | Phase II; randomized, double-blind, placebo-controlled | Safety and tolerability | [22] |
| Arimoclomol | 2008 | 2012 | 2017 | Phase II/III; randomized, double-blind, placebo-controlled | Efficacy and safety | [18,20] |
| Baricitinib | 2020 | N/A * | N/A * | Phase IIa; randomized, controlled | Assessment of clinical response across 12- and 24-week treatment arms | [29] |
| Botulism | 2014 | 2018 | 2021 | Phase II; open-label | Alleviating dysphagia | [30] |
| BYM338 (Bimagrumab) | 2015 | 2017 | 2018 | Phase IIb/III; randomized, double-blind, placebo-controlled (extension) | Efficacy, safety and tolerability | [31] |
| BYM338 | 2015 | 2016 | 2018 | Phase IIb/III (extension); non-random, double-blind, placebo-controlled | Efficacy, safety and tolerability | [32,33] |
| BYM338 | 2014 | 2016 | 2018 | Phase II/III; open-label | Efficacy, safety and tolerability | [34,35] |
| BYM338 | 2014 | 2016 | 2017 | Phase IIb/III; randomized, double-blind, placebo-controlled | Efficacy, safety and tolerability | [36] |
| BYM338 | 2013 | 2016 | 2017 | Phase II/III; randomized, double-blind, placebo-controlled | Efficacy | [37,38] |
| BYM338 | 2011 | 2012 | 2014 | Phase II; randomized, double-blind, placebo-controlled | Efficacy, safety and tolerability | [39,40] |
| Etanercept | 2005 | 2014 | N/A | Phase I; randomized, double-blind, placebo-controlled | Efficacy | [41] |
| Etanercept | N/A | N/A | 2006 | Pilot study; non-randomized open-label, non-placebo-controlled | Efficacy | [42] |
| IFNβ1a (low-dose) | N/A | N/A | 2001 | Pilot study; randomized, double-blind, placebo-controlled | Efficacy, safety and tolerability | [43] |
| IFNβ1a (high-dose) | N/A | N/A | 2004 | Pilot study; randomized, double-blind, placebo-controlled | Efficacy, safety and tolerability | [44] |
| IVIg + prednisone | N/A | N/A | 2001 | Phase II; randomized, double-blind, placebo-controlled | Efficacy and safety | [15] |
| IVIg | 1990 | 2002 | 1997, 2001 | Phase II; double-blind, placebo-controlled | Efficacy and safety | [13,14,45] |
| Lithium | 2008 | 2009 | N/A | Pilot study; cohort | Efficacy | [46] |
| MTX | 1996 | 2000 | 2002 | Pilot study; randomized, double-blind, placebo-controlled | Efficacy | [16] |
| Natalizumab | 2013 | N/A | N/A | Phase I; open-label, non-placebo-controlled | Efficacy and safety | [47] |
| Oxandrolone | N/A | N/A | 2002 | Pilot study; randomized, double-blind, placebo-controlled | Efficacy | [48] |
| Phenylbutyrate | 2020 | N/A * | N/A * | Phase I; open-label | Efficacy, safety and tolerability | [49] |
| Pioglitazone | 2018 | 2020 | N/A | Pilot study; open-label, non-randomized, non-placebo-controlled | Efficacy | [50] |
| Rapamycin | 2015 | 2018 | N/A | Phase II/III; randomized, double-blind, placebo-controlled | Efficacy | [51] |
| Simvastatin | 2007 | N/A | N/A | Phase III; randomized, controlled | Efficacy, safety and tolerability | [52] |
| Simvastatin | 2007 | N/A | N/A | Phase III; randomized, controlled, | Efficacy, safety and tolerability | [53] |
| Sirolimus | N/A * | N/A * | N/A * | Phase III; randomized, double-blind, placebo-controlled | Efficacy | [54] |
| Cell-based | ||||||
| Adipose-derived stem cells | 2021 | N/A * | N/A * | Open-label; non-random | Efficacy and safety | [55] |
| Adipose-derived stromal vascular fraction | N/A * | N/A * | N/A * | Phase I; open-label, non-placebo-controlled | Efficacy, safety and tolerability | [56] |
| Gene-based | ||||||
| Follistatin | 2012 | 2017 | N/A | Phase I; non-randomized, open-label, non-placebo-controlled | Efficacy and safety | [57,58] |
| GSK3α | GSK3β | Modification | Enzyme or Treatment/Significance |
|---|---|---|---|
| T19 | - | phosphorylation | Unknown; induces inhibition of kinase activity |
| S21 | S9 | phosphorylation | AKT/PKC/RSK/Aurora (GSK3β only); induces inhibition of kinase activity |
| S41 | - | phosphorylation | MG132 withdrawal; unknown |
| S52 | - | phosphorylation | MEK inhibition; unknown |
| S63 | - | phosphorylation | MEK inhibition; unknown |
| - | S21 | phosphorylation | 5, 7-dihydroxyflavone (chrysin); apoptosis induced |
| - | S25 | phosphorylation | MG132 withdrawal; unknown |
| S97 | S35 | phosphorylation | MG132 withdrawal; unknown |
| - | T43 | phosphorylation | p38α/ERK; activation of enzymatic activity |
| - | Y56 | phosphorylation | MET; activation of enzymatic activity |
| Y134 | Y71 | phosphorylation | Unknown; unknown |
| - | K86 | ubiquitination | Unknown; activation of enzymatic activity |
| - | S147 | phosphorylation | PKCζ; activation of enzymatic activity |
| K246 | K183 | ubiquitination | Unknown; degradation of protein |
| K260 | K197 | ubiquitination | Unknown; inhibition of enzymatic activity |
| K268 | K205 | ubiquitination; acetylation | siRNA; subcellular localization and phosphorylation altered |
| S278 | S215 | phosphorylation | IL3, serum; unknown |
| Y279 | Y216 | phosphorylation | GSK3/MEK; activation of enzymatic activity |
| S282 | S219 | phosphorylation | MEK inhibition; unknown |
| Y284 | Y221 | phosphorylation | Unknown; unknown |
| Y285 | Y222 | phosphorylation | Unknown; unknown |
| K355 | K292 | ubiquitination; sumoylation | Unknown; unknown |
| - | S389 | phosphorylation | p38α; intracellular localization |
| - | T390 | phosphorylation | p38α; activation of enzymatic activity |
| - | T392 | phosphorylation | Nocodazole; unknown |
| - | T402 | phosphorylation | Nocodazole; unknown |
| - | T420 | phosphorylation | MG132 withdrawal; unknown |
| Isoform | Phosphorylation Site | Ref. | ||
|---|---|---|---|---|
| GSK3α | GSK3β | |||
| TAU isoform 2 | S307 (S713) | X | [73] | |
| TAU isoform 2 | S315 (S721) | X | [73] | |
| TAU isoform 5 | T181 (T498) | X | [74] | |
| TAU isoform 5 | S184 (S501) | X | [74] | |
| TAU isoform 5 | S195 (S512) | X | [74] | |
| TAU isoform 5 | S198 (S515) | X | [74] | |
| TAU isoform 5 | S199 (S516) | X | [74] | |
| TAU isoform 5 | S202 (S519) | X | [74] | |
| TAU isoform 5 | T205 (T522) | X | [74] | |
| TAU isoform 5 | T231 (T548) | X | [73,74] | |
| TAU isoform 5 | S235 (S552) | X | [73,74] | |
| TAU isoform 5 | S262 (S575) | X | [74] | |
| TAU isoform 5 | S325 (S673) | X | [74] | |
| TAU isoform 5 | S365 (S713) | X | [73] | |
| TAU isoform 5 | S369 (S717) | X | [74] | |
| TAU isoform 5 | S373 (S721) | X | [73,74] | |
| TAU isoform 6 | T173 (T548) | X | [73,75] | |
| TAU isoform 6 | S177 (S552) | X | [73] | |
| TAU isoform 6 | S338 (S713) | X | [73,75] | |
| TAU isoform 6 | S346 (S721) | X | [73,75] | |
| TAU isoform 8 | S46 (S46) | X | [76] | |
| TAU isoform 8 | T50 (T50) | X | [76] | |
| TAU isoform 8 | T153 (T470) | X | [77] | |
| TAU isoform 8 | T175 (T492) | X | [77] | |
| TAU isoform 8 | T181 (T498) | X | [78,79,80] | |
| TAU isoform 8 | S195 (S512) | X | [81] | |
| TAU isoform 8 | S199 (S516) | X | [81,82] | |
| TAU isoform 8 | S202 (S519) | X | [78,82] | |
| TAU isoform 8 | T205 (T522) | X | [81,82] | |
| TAU isoform 8 | S210 (S527) | X | [81] | |
| TAU isoform 8 | T212 (T529) | X | [78,82] | |
| TAU isoform 8 | S214 (S531) | X | [79,81] | |
| TAU isoform 8 | T217 (T534) | X | [78,82] | |
| TAU isoform 8 | T231 (T548) | X | X | [73,78,83,84,85] |
| TAU isoform 8 | S235 (S552) | X | X | [73,77,84] |
| TAU isoform 8 | S262 (S579) | X | X | [79,82,83,84,86] |
| TAU isoform 8 | S396 (S713) | X | X | [73,81,82] |
| TAU isoform 8 | S400 (S717) | X | [81,87] | |
| TAU isoform 8 | S404 (S721) | X | X | [73,81,82] |
| TAU isoform 8 | S409 (S726) | X | [79] | |
| TAU isoform 8 | S422 (S739) | X | [79] | |
| Inhibitor | Mode of Inhibition | Clinically Approved | IC50 | Ref |
|---|---|---|---|---|
| Specific Inhibition | ||||
| Aloisines | ATP competitive | Pre-clinical; inhibits cell proliferation. | 0.5–1.5 µM | [129] |
| -Aminopyrimidines: | ||||
| CT98014 | Pre-clinical; potentiates insulin activation and glucose metabolism; reduced TAU hyperphosphorylation. | 0.6–7 nM | [130,131] | |
| CT98023 | Pre-clinical; supported self-renewal of ESCs; reduced TAU phosphorylation. | 0.6–7 nM | [130,132] | |
| CT99021 | Pre-clinical; potentiates insulin activation and glucose metabolism; promotes replication and survival of pancreatic β-cells. | 0.6–7 nM | [131,133] | |
| TWS119 | Pre-clinical; supported self-renewal of ESCs; induced neuronal differentiation; arrest effector T-cell differentiation. | 0.6–7 nM | [134,135] | |
| -Arylindolemaleimide: | ||||
| 9-ING-41 | Phase I/II; anti-tumor activity in diverse advanced cancers | 710 nM | [136,137] | |
| SB-216763 | Pre-clinical; neuroprotective; beneficial in AD models; anti-inflammatory | 34 nM | [138,139] | |
| SB-415286 | Pre-clinical; neuroprotective; beneficial in AD models, antitumorigenic, anti-inflammatory | 77 nM | [138,140,141,142,143,144] | |
| -Indirubins: | ||||
| 6-BIO | Pre-clinical; neuro-/cytoprotection; maintenance of ESC pluripotency; may promote tumorigenic characteristics | 1.5 nM | [145,146,147] | |
| Indirubin | Pre-clinical; reduced TAU phosphorylation, cardioprotection, neuroprotection, antitumorigenic | 0.6–5 µM | [148,149,150,151] | |
| -Marine spong- derived | ||||
| Dibromocantharelline | Pre-clinical | 3 µM | [152,153] | |
| Hymenialdisine | Pre-clinical; reduces estrogen-dependent bone loss; reduces TAU phosphorylation; neuroprotection. | 10 nM | [154,155] | |
| -Paullones: | ||||
| Alsterpaullone | Pre-clinical; inhibits TAU hyperphosphorylation, antitumorigenic, antimanic. | 4–80 nM | [156,157,158,159,160] | |
| Cazpaullone | Pre-clinical; protects pancreatic β-cells. | 4–80 nM | [161] | |
| Kenpaullone | Pre-clinical; neuroprotective, reduces Abeta production and TAU phoshphorylation; cardioprotective; reduces inflammation and autoinflammation; chemotherapeutic enhancer in glioblastoma. | 4–80 nM | [162,163,164,165] | |
| -Thiazoles: | ||||
| AR-A014418 | Pre-clinical; neuroprotective, beneficial in AD and ALS models; blocks TAU phosphorylation; inhibits neurodegeneration; inhibits pain and inflammation. | 104 nM | [166,167,168] | |
| AZD-1080 | Withdrawn from phase I trials; neuroprotective, beneficial in AD (pre-clinical) | 6.9–31 nM | [169] | |
| -Furanosesquiterpenes: | Non-ATP competitive | |||
| Palinurin | Pre-clinical; decreased TAU phosphorylation | 4.5 µM | [170,171] | |
| Tricantin | Pre-clinical; decreased TAU phosphorylation | 7.5 µM | [170] | |
| -Halomethylketones: | ||||
| HMK-32 | Pre-clinical; neuroprotective, decreased TAU phosphorylation | 1.5 µM | [172] | |
| -Thiadiazolidindiones: | ||||
| NP00111 | Pre-clinical; neuroprotection, anti-inflammatory effects | 2 µM | [173,174] | |
| NP031115 | Pre-clinical; antidepressant-like effects | 4 µM | [175] | |
| Tideglusib (NP031112) | Phase II (orphan drug status); neuroprotection, decreased TAU phosphorylation; decreased Abeta plaque formation and gliosis; reduces inflammation | 60 nM | [176,177,178] | |
| TDZD-8 | Pre-clinical; neuroprotection, decreased TAU phosphorylation; promotes leukemic stem and progenitor cell death. | 2 µM | [179,180] | |
| -Manzamines | ||||
| Manzamine A | Pre-clinical; decreased TAU phosphorylation | 1.5 µM | [181,182] | |
| -Petides: | ||||
| L803-mts | Pre-clinical; neuroprotective; acts as an antidepressant; inhibits Abeta phosphorylation and neurotoxicity; improves glucose homeostasis, reduces autoinflammation. | 20 µM | [183,184,185,186,187] | |
| L807-mts | Pre-clinical; neuroprotective; inhibits protein aggregates, reduces inflammation, enhances autophagy. | 1 µM | [188,189] | |
| Non-specific Inhibition | ||||
| Cromolyn | Steric hindrance of the binding pocket | Non-steroidal, anti-inflammatory; Diabetes mellitus | 2.0 µM | [190] |
| Curcumin | Steric hindrance of the binding pocket | Dietary spice with a wide range of pharmacological activities reported; cardioprotective, neuroprotective, anti-inflammatory. | 66.3 nM | [191,192,193,194,195,196,197] |
| Famotidine | Steric hindrance of the binding pocket | H2-receptor antagonist used to treat gastric reflux disease and peptic ulcer; has neuroprotective effects | 1.44 µM | [198,199] |
| Lithium (Li++) | Unknown | Diabetes mellitus; bipolar disorder; Alzheimer’s and other neurodegenerative diseases; neuroprotective effects, anti-inflammatory effects. | 2 mM | [200,201,202,203,204] |
| Naproxen | Steric hindrance of the binding pocket | Non-steroidal, anti-inflammatory; Diabetes mellitus | 1.5 µM | [190] |
| Olanzapine | Steric hindrance of the binding pocket | Antipsychotic used for schizophrenia, bipolar disorder and anxiety; alters glucose metabolism | 91.0 nM | [205,206,207] |
| Zinc (Zn++) | Unknown | Antidepressant; alters glucose metabolism, cardioprotective effects, neurotoxic effects | 15 µM | [208,209,210,211] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piazzi, M.; Bavelloni, A.; Cenni, V.; Faenza, I.; Blalock, W.L. Revisiting the Role of GSK3, A Modulator of Innate Immunity, in Idiopathic Inclusion Body Myositis. Cells 2021, 10, 3255. https://doi.org/10.3390/cells10113255
Piazzi M, Bavelloni A, Cenni V, Faenza I, Blalock WL. Revisiting the Role of GSK3, A Modulator of Innate Immunity, in Idiopathic Inclusion Body Myositis. Cells. 2021; 10(11):3255. https://doi.org/10.3390/cells10113255
Chicago/Turabian StylePiazzi, Manuela, Alberto Bavelloni, Vittoria Cenni, Irene Faenza, and William L. Blalock. 2021. "Revisiting the Role of GSK3, A Modulator of Innate Immunity, in Idiopathic Inclusion Body Myositis" Cells 10, no. 11: 3255. https://doi.org/10.3390/cells10113255
APA StylePiazzi, M., Bavelloni, A., Cenni, V., Faenza, I., & Blalock, W. L. (2021). Revisiting the Role of GSK3, A Modulator of Innate Immunity, in Idiopathic Inclusion Body Myositis. Cells, 10(11), 3255. https://doi.org/10.3390/cells10113255