
Post-Translational Modification of HMGB1 Disulfide Bonds in Stimulating and Inhibiting Inflammation


Abstract
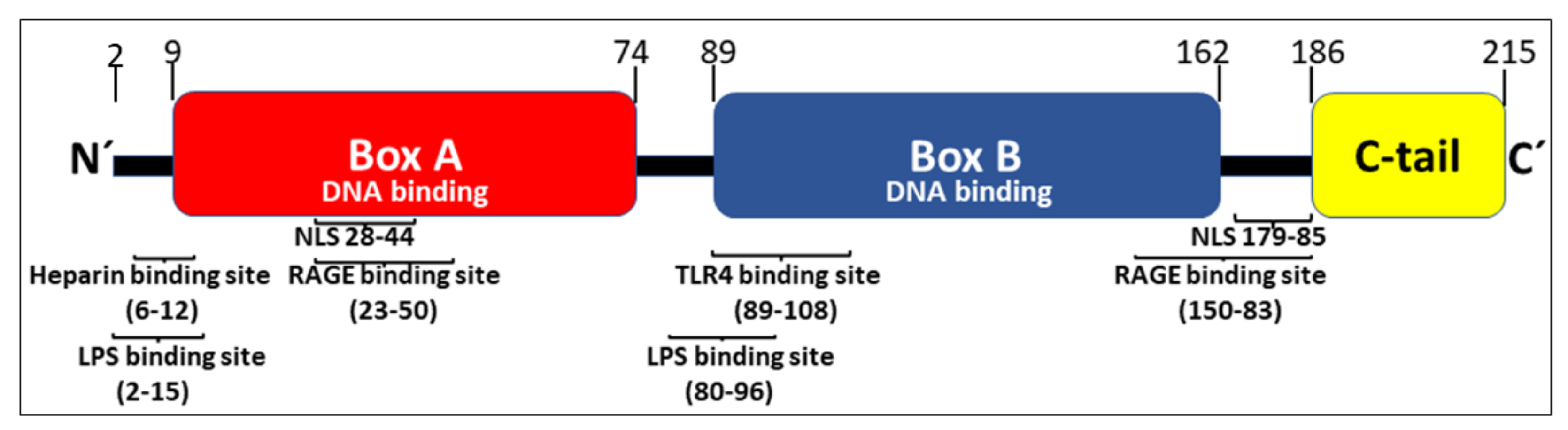
:1. Introduction
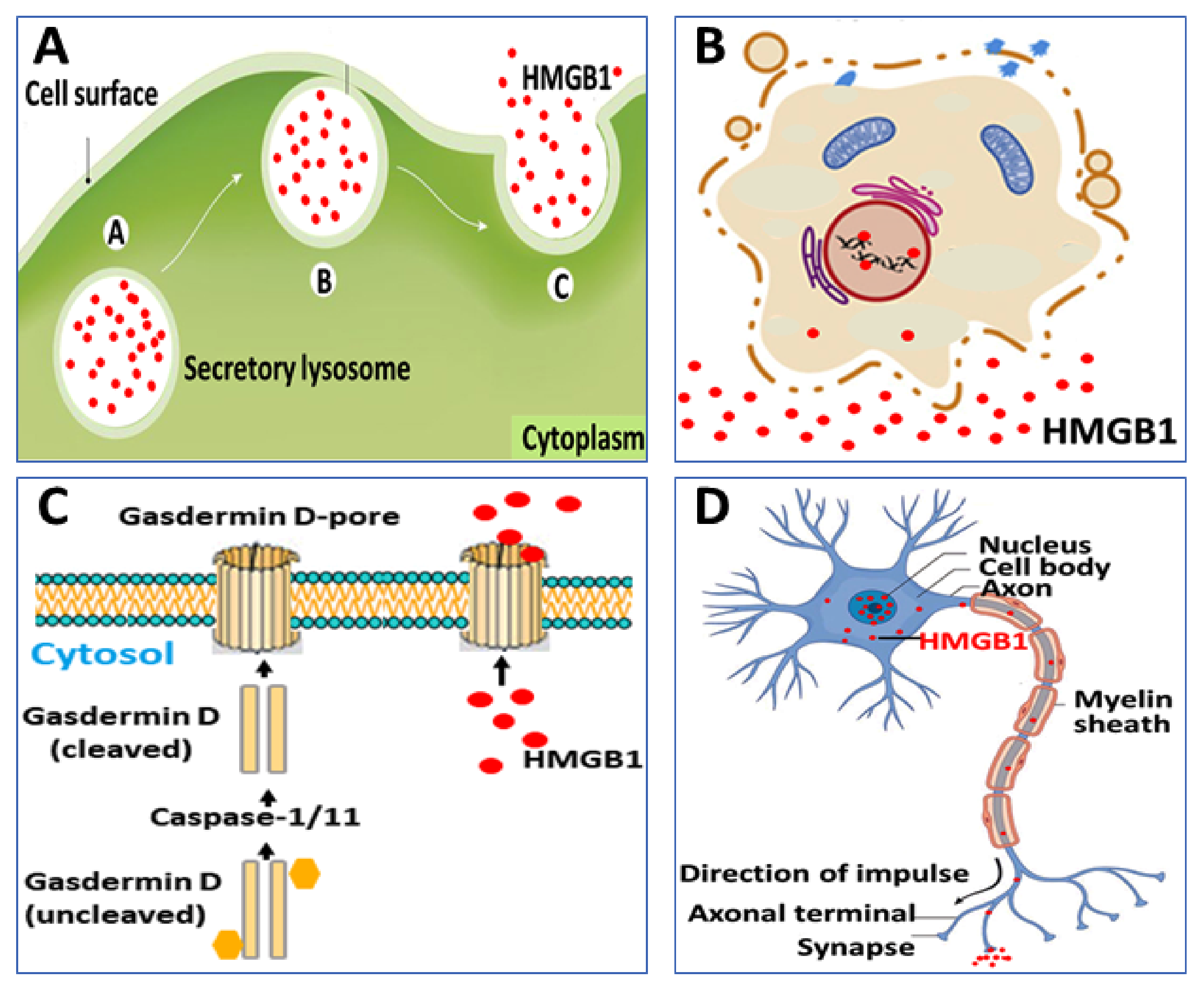
2. Extracellular HMGB1 Release
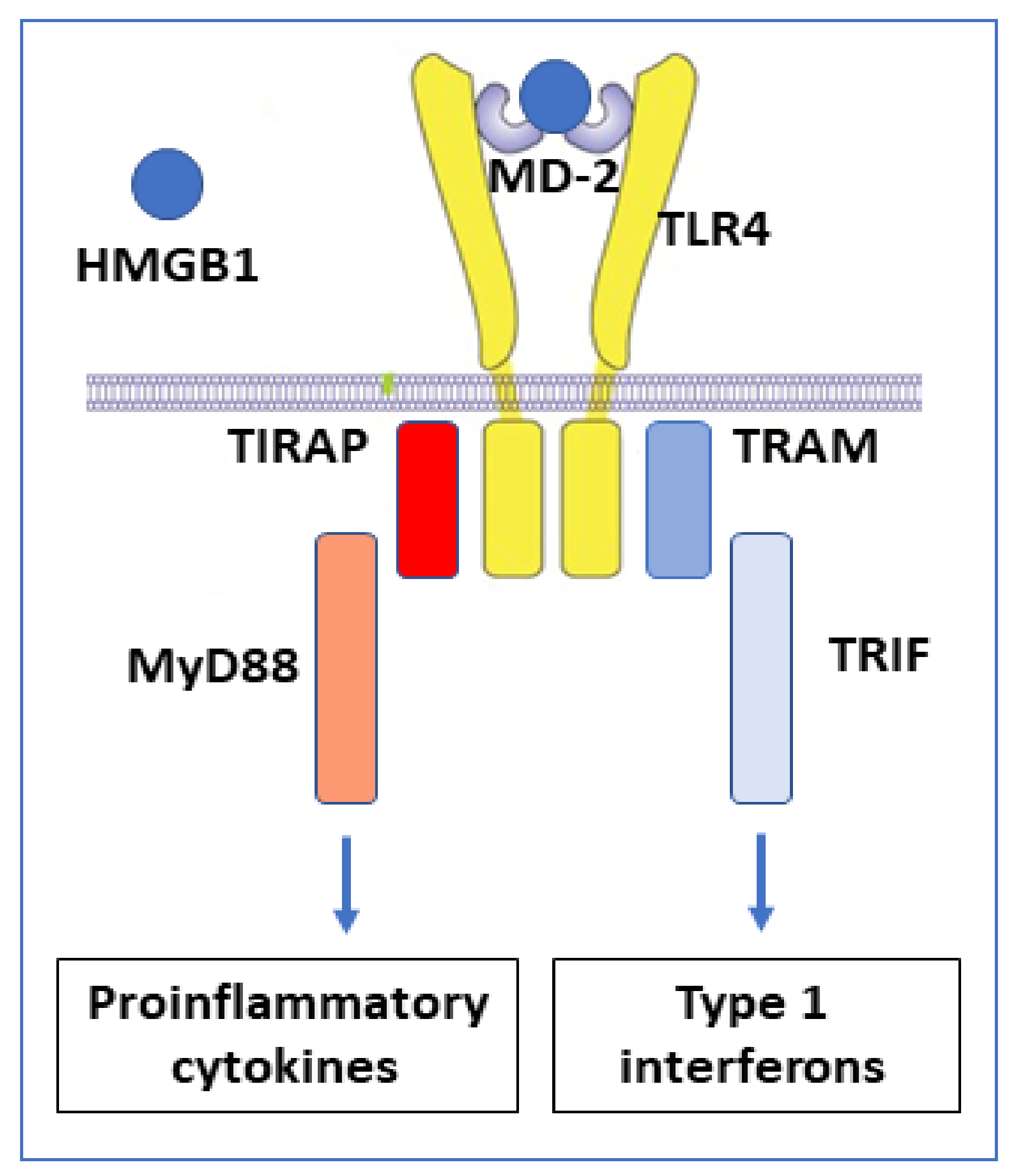
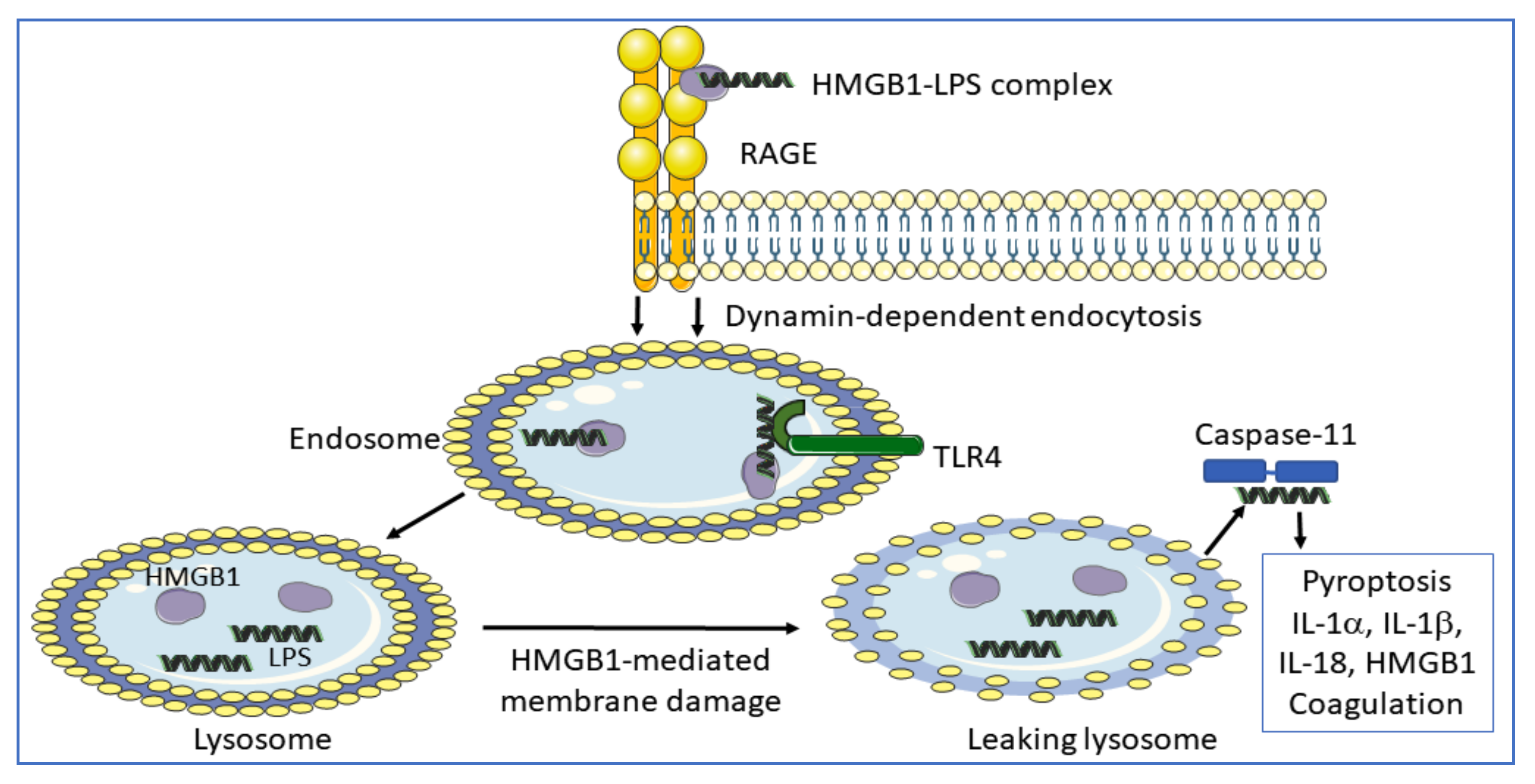
3. HMGB1 Receptor Usage
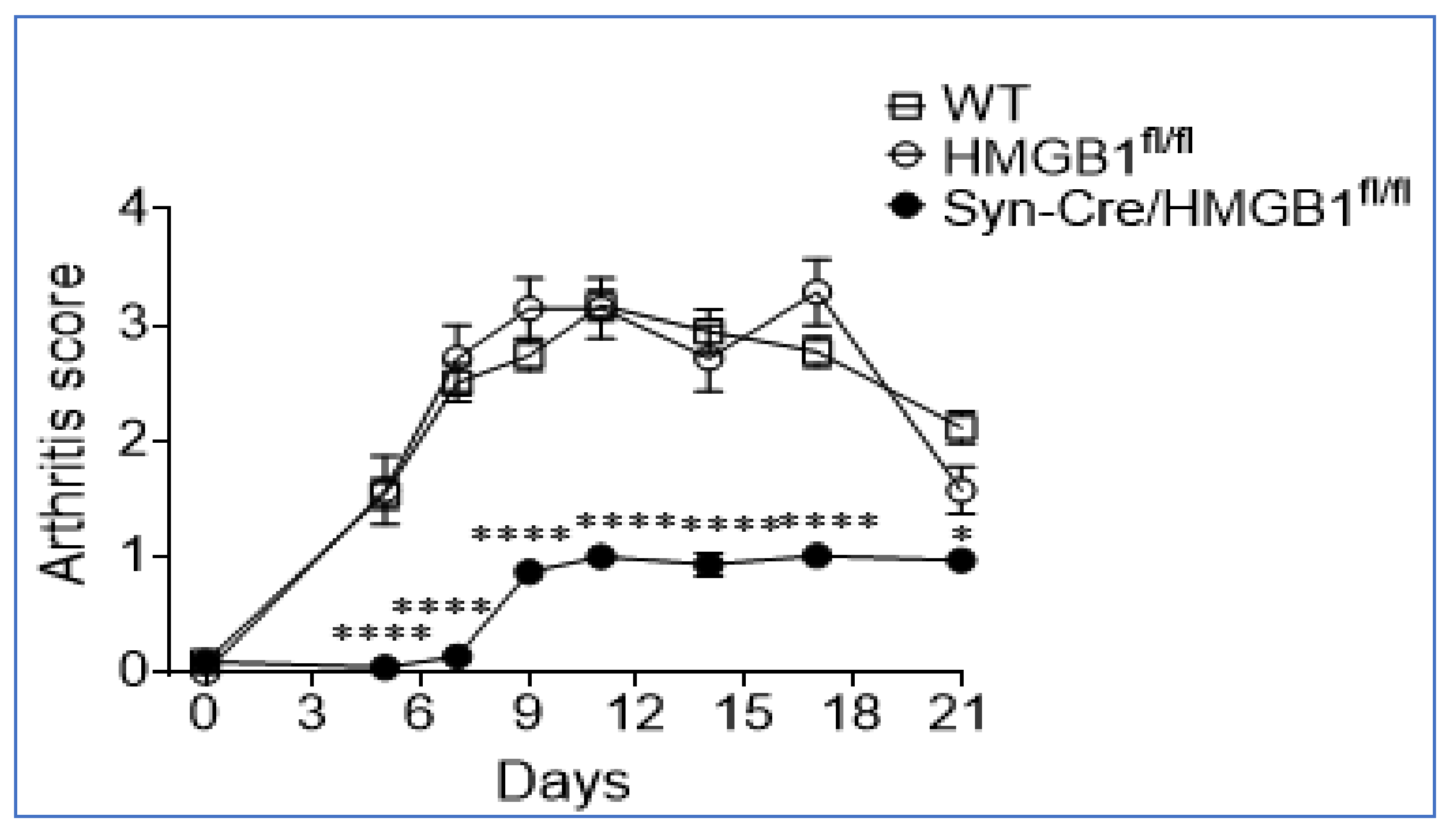
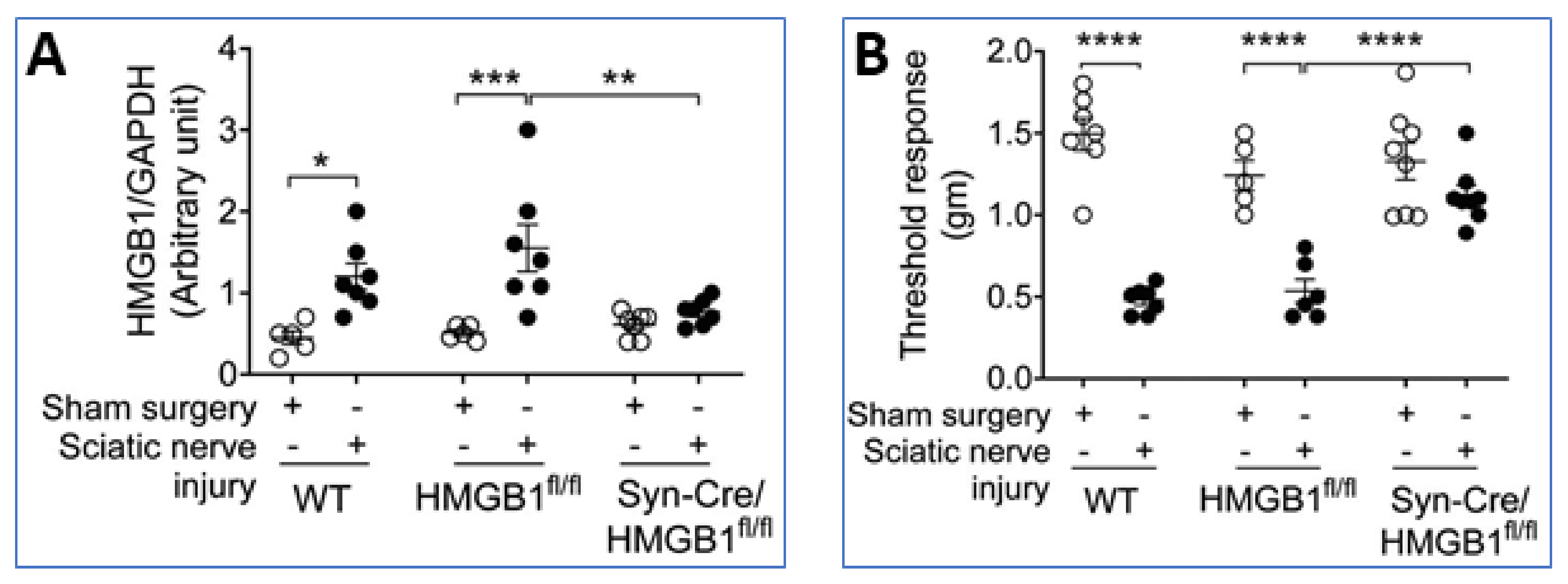
4. Sensory Neurons Direct Inflammation via HMGB1 Release
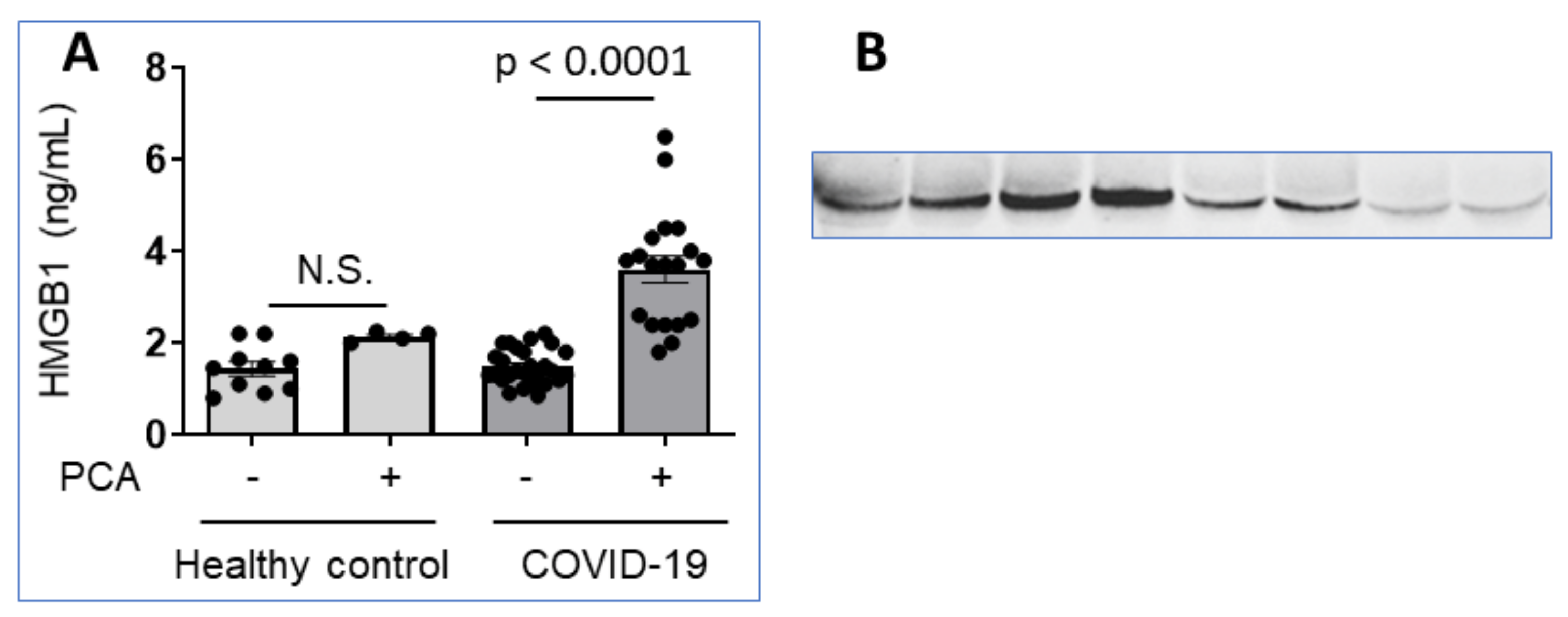
5. HMGB1 in COVID-19
6. HMGB1 and Acetylcholine-Potent Antagonists Balancing Inflammation
7. Key Challenges in the HMGB1 Field
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| HMGB1 | high mobility group box 1 protein |
| RAGE | receptor for advanced glycation end-product |
| TLR4 | toll-like receptor 4 |
| MD-2 | myeloid differentiation factor 2 |
| TIRAP | TIR domain containing adaptor protein |
| TRAM | toll-receptor-associated molecule |
| MyD88 | myeloid differentiation primary response 88 |
| TRIF | TIR-domain-containing adapter-inducing interferon-β |
| CXCL12 | C-X-C Motif Chemokine Ligand 12 |
| CXCR4 | C-X-C chemokine receptor type 4 |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| LPS | lipopolysaccharide |
| DAMP | damage-associated molecular pattern molecule |
| PAMP | pathogen-associated molecular pattern molecule |
| HDAC | histone-deacetylase |
| HAT | histone-acetylase |
| SIRT1 | sirtuin 1 |
| CRMI | nuclear exportin chromosome-region maintenance 1 |
| NLS | nuclear localization site |
| SHP-1 | Src homology 2 domain-containing protein tyrosine phosphatase 1 |
| a7nAChR | a7-nicotinic acetylcholine receptor |
| MDSC | myeloid-derived suppressor cells |
| Treg cell | regulatory T lymphocyte |
| M1 macrophage | proinflammatory macrophage |
| M2 macrophage | anti-inflammatory macrophage |
| Syn-Cre/HMGB1fl/fl mice | neuronally HMGB1 gene-deficient mice |
| CD24 | cluster of differentiation 24 |
| Siglec-10 | Sialic acid-binding Ig-like lectin 10 |
References
- Kang, R.; Chen, R.; Zhang, Q.; Hou, W.; Wu, S.; Cao, L.; Huang, J.; Yu, Y.; Fan, X.-G.; Yan, Z.; et al. HMGB1 in health and disease. Mol. Asp. Med. 2014, 40, 1–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oppenheim, J.J.; Yang, D. Alarmins: Chemotactic activators of immune responses. Curr. Opin. Immunol. 2005, 17, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Andersson, U.; Tracey, K.J. HMGB1 Is a Therapeutic Target for Sterile Inflammation and Infection. Annu. Rev. Immunol. 2011, 29, 139–162. [Google Scholar] [CrossRef] [Green Version]
- Andersson, U.; Yang, H.; Harris, H. High-mobility group box 1 protein (HMGB1) operates as an alarmin outside as well as inside cells. Semin. Immunol. 2018, 38, 40–48. [Google Scholar] [CrossRef]
- Kwak, M.S.; Rhee, W.J.; Lee, Y.J.; Kim, H.S.; Kim, Y.H.; Kwon, M.K.; Shin, J.-S. Reactive oxygen species induce Cys106-mediated anti-parallel HMGB1 dimerization that protects against DNA damage. Redox Biol. 2021, 40, 101858. [Google Scholar] [CrossRef]
- Lu, B.; Nakamura, T.; Inouye, K.; Li, J.; Tang, Y.; Lundbäck, P.; Valdés-Ferrer, S.I.; Olofsson, P.S.; Kalb, T.; Roth, J.; et al. Novel role of PKR in inflammasome activation and HMGB1 release. Nature 2012, 488, 670–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonaldi, T.; Talamo, F.; Scaffidi, P.; Ferrera, D.; Porto, A.; Bachi, A.; Rubartelli, A.; Agresti, A.; Bianchi, M.E. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003, 22, 5551–5560. [Google Scholar] [CrossRef] [Green Version]
- Evankovich, J.; Cho, S.W.; Zhang, R.; Cardinal, J.; Dhupar, R.; Zhang, L.; Klune, J.R.; Zlotnicki, J.; Billiar, T.; Tsung, A. High Mobility Group Box 1 Release from Hepatocytes during Ischemia and Reperfusion Injury Is Mediated by Decreased Histone Deacetylase Activity. J. Biol. Chem. 2010, 285, 39888–39897. [Google Scholar] [CrossRef] [Green Version]
- Zou, J.Y.; Crews, F.T. Release of Neuronal HMGB1 by Ethanol through Decreased HDAC Activity Activates Brain Neuroimmune Signaling. PLoS ONE 2014, 9, e87915. [Google Scholar] [CrossRef] [Green Version]
- Hwang, J.S.; Choi, H.S.; Ham, S.A.; Yoo, T.; Lee, W.J.; Paek, K.S.; Seo, H.G. Deacetylation-mediated interaction of SIRT1-HMGB1 improves survival in a mouse model of endotoxemia. Sci. Rep. 2015, 5, 15791. [Google Scholar] [CrossRef] [Green Version]
- Zhao, P.; Xu, S.; Huang, Z.; Jiang, G.; Deng, P.; Zhang, Y. Hyperbaric Oxygen via Mediating SIRT1-Induced Deacetylation of HMGB1 Improved Cerebral Ischemia/Reperfusion injury. Eur. J. Neurosci. 2021, 54, 7318–7331. [Google Scholar] [CrossRef]
- Karkischenko, V.N.; Skvortsova, V.I.; Gasanov, M.T.; Fokin, Y.V.; Nesterov, M.S.; Petrova, N.V.; Alimkina, O.V.; Pomytkin, I.A. Inhaled [D-Ala2]-Dynorphin 1-6 Prevents Hyperacetylation and Release of High Mobility Group Box 1 in a Mouse Model of Acute Lung Injury. J. Immunol. Res. 2021, 2021, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Le, K.; Daliv, E.C.; Wu, S.; Qian, F.; Ali, A.I.; Yu, D.; Guo, Y. SIRT1-regulated HMGB1 release is partially involved in TLR4 signal transduction: A possible anti-neuroinflammatory mechanism of resveratrol in neonatal hypoxic-ischemic brain injury. Int. Immunopharmacol. 2019, 75, 105779. [Google Scholar] [CrossRef]
- Rabadi, M.M.; Xavier, S.; Vasko, R.; Kaur, K.; Goligorksy, M.S.; Ratliff, B.B. High-mobility group box 1 is a novel deacetylation target of Sirtuin1. Kidney Int. 2015, 87, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Zeng, Z.; Zhao, M.; Huang, Q.; Gao, Y.; Dai, X.; Lu, J.; Huang, W.; Zhao, K. Evidence for SIRT1 Mediated HMGB1 Release from Kidney Cells in the Early Stages of Hemorrhagic Shock. Front. Physiol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Chen, W.; Ni, X.; Little, P.J.; Xu, S.; Tang, L.; Weng, J. Metformin, Macrophage Dysfunction and Atherosclerosis. Front. Immunol. 2021, 12, 682853. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Zhou, X.; Xiang, H.; Wang, S.; Cui, Z.; Zhou, J. Resveratrol Reduced Liver Damage After Liver Resection in a Rat Model by Upregulating Sirtuin 1 (SIRT1) and Inhibiting the Acetylation of High Mobility Group Box 1 (HMGB1). Med Sci. Monit. 2019, 25, 3212–3220. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Wu, X.; Peng, B.; Zou, H.; Li, S.; Wang, J.; Cao, J. Curcumin improves necrotising microscopic colitis and cell pyroptosis by activating SIRT1/NRF2 and inhibiting the TLR4 signalling pathway in newborn rats. Innate Immun. 2020, 26, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Guarente, L. Sirtuins, Aging, and Medicine. N. Engl. J. Med. 2011, 364, 2235–2244. [Google Scholar] [CrossRef] [PubMed]
- Dobbin, M.M.; Madabhushi, R.; Pan, L.; Chen, Y.; Kim, D.; Gao, J.; Ahanonu, B.; Pao, P.-C.; Qiu, Y.; Zhao, Y.; et al. SIRT1 collaborates with ATM and HDAC1 to maintain genomic stability in neurons. Nat. Neurosci. 2013, 16, 1008–1015. [Google Scholar] [CrossRef]
- Hubbard, B.P.; Sinclair, D.A. Small molecule SIRT1 activators for the treatment of aging and age-related diseases. Trends Pharmacol. Sci. 2014, 35, 146–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sixto-López, Y.; Rosales-Hernández, M.C.; De Oca, A.C.-M.; Fragoso-Morales, L.G.; Mendieta-Wejebe, J.E.; Correa-Basurto, A.M.; Abarca-Rojano, E.; Correa-Basurto, J. N-(2′-Hydroxyphenyl)-2-Propylpentanamide (HO-AAVPA) Inhibits HDAC1 and Increases the Translocation of HMGB1 Levels in Human Cervical Cancer Cells. Int. J. Mol. Sci. 2020, 21, 5873. [Google Scholar] [CrossRef]
- Kwak, M.S.; Kim, H.S.; Lee, B.; Kim, Y.H.; Son, M.; Shin, J.-S. Immunological Significance of HMGB1 Post-Translational Modification and Redox Biology. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Gardella, S.; Andrei, C.; Ferrera, D.; Lotti, L.V.; Torrisi, M.R.; Bianchi, M.E.; Rubartelli, A. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep. 2002, 3, 995–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mobarrez, F.; Vikerfors, A.; Gustafsson, J.T.; Gunnarsson, I.; Zickert, A.; Larsson, A.; Pisetsky, D.S.; Wallén, H.; Svenungsson, E. Microparticles in the blood of patients with systemic lupus erythematosus (SLE): Phenotypic characterization and clinical associations. Sci. Rep. 2016, 6, 36025. [Google Scholar] [CrossRef] [PubMed]
- Pisetsky, D.S.; Gauley, J.; Ullal, A. HMGB1 and Microparticles as Mediators of the Immune Response to Cell Death. Antioxidants Redox Signal. 2011, 15, 2209–2219. [Google Scholar] [CrossRef] [Green Version]
- Vogel, S.; Bodenstein, R.; Chen, Q.; Feil, S.; Feil, R.; Rheinlaender, J.; Schäffer, T.; Bohn, E.; Frick, J.-S.; Borst, O.; et al. Platelet-derived HMGB1 is a critical mediator of thrombosis. J. Clin. Investig. 2015, 125, 4638–4654. [Google Scholar] [CrossRef] [Green Version]
- Stark, K.; Philippi, V.; Stockhausen, S.; Busse, J.; Antonelli, A.; Miller, M.; Schubert, I.; Hoseinpour, P.; Chandraratne, S.; Von Brühl, M.-L.; et al. Disulfide HMGB1 derived from platelets coordinates venous thrombosis in mice. Blood 2016, 128, 2435–2449. [Google Scholar] [CrossRef]
- Maugeri, N.; Capobianco, A.; Rovere-Querini, P.; Ramirez, G.A.; Tombetti, E.; Della Valle, P.; Monno, A.; D’Alberti, V.; Gasparri, A.M.; Franchini, S.; et al. Platelet microparticles sustain autophagy-associated activation of neutrophils in systemic sclerosis. Sci. Transl. Med. 2018, 10, eaao3089. [Google Scholar] [CrossRef] [Green Version]
- Lamkanfi, M.; Sarkar, A.; Walle, L.V.; Vitari, A.C.; Amer, A.O.; Wewers, M.D.; Tracey, K.J.; Kanneganti, T.-D.; Dixit, V.M. Inflammasome-Dependent Release of the Alarmin HMGB1 in Endotoxemia. J. Immunol. 2010, 185, 4385–4392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, B.; Wang, H.; Andersson, U.; Tracey, K.J. Regulation of HMGB1 release by inflammasomes. Protein Cell 2013, 4, 163–167. [Google Scholar] [CrossRef] [Green Version]
- Deng, M.; Scott, M.J.; Fan, J.; Billiar, T.R. Location is the key to function: HMGB1 in sepsis and trauma-induced inflammation. J. Leukoc. Biol. 2019, 106, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Deng, M.; Loughran, P.A.; Yang, M.; Lin, M.; Yang, C.; Gao, W.; Jin, S.; Li, S.; Cai, J.; et al. LPS Induces Active HMGB1 Release from Hepatocytes Into Exosomes Through the Coordinated Activities of TLR4 and Caspase-11/GSDMD Signaling. Front. Immunol. 2020, 11, 229. [Google Scholar] [CrossRef] [PubMed]
- Deng, M.; Tang, Y.; Li, W.; Wang, X.; Zhang, R.; Zhang, X.; Zhao, X.; Liu, J.; Tang, C.; Liu, Z.; et al. The Endotoxin Delivery Protein HMGB1 Mediates Caspase-11-Dependent Lethality in Sepsis. Immunity 2018, 49, 740–753.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Zeng, Q.; Silverman, H.A.; Gunasekaran, M.; George, S.J.; Devarajan, A.; Addorisio, M.E.; Li, J.; Tsaava, T.; Shah, V.; et al. HMGB1 released from nociceptors mediates inflammation. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Cheng, X.; Yang, Y.-L.; Yang, H.; Wang, Y.-H.; Du, G.-H. Kaempferol alleviates LPS-induced neuroinflammation and BBB dysfunction in mice via inhibiting HMGB1 release and down-regulating TLR4/MyD88 pathway. Int. Immunopharmacol. 2018, 56, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Shi, Y.; Du, P.; Wang, J.; Han, Y.; Sun, B.; Feng, J. HMGB1/TLR4 promotes apoptosis and reduces autophagy of hippocampal neurons in diabetes combined with OSA. Life Sci. 2019, 239, 117020. [Google Scholar] [CrossRef] [PubMed]
- Laird, M.D.; Shields, J.S.; Sukumari-Ramesh, S.; Kimbler, D.E.; Fessler, R.D.; Shakir, B.; Youssef, P.; Yanasak, N.; Vender, J.R.; Dhandapani, K.M. High mobility group box protein-1 promotes cerebral edema after traumatic brain injury via activation of toll-like receptor 4. Glia 2013, 62, 26–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Zhang, L.; Tang, J.; Yang, X.; Huang, J.; Zhu, T.; Zhao, F.; Li, S.; Li, X.; Qu, Y.; et al. Role of toll-like receptor 4 in the regulation of the cell death pathway and neuroinflammation. Brain Res. Bull. 2019, 148, 79–90. [Google Scholar] [CrossRef]
- Oladiran, O.; Shi, X.Q.; Yang, M.; Fournier, S.; Zhang, J. Inhibition of TLR4 signaling protects mice from sensory and motor dysfunction in an animal model of autoimmune peripheral neuropathy. J. Neuroinflamm. 2021, 18, 1–17. [Google Scholar] [CrossRef]
- Paudel, Y.N.; Khan, S.U.; Othman, I.; Shaikh, M.F. Naturally Occurring HMGB1 Inhibitor, Glycyrrhizin, Modulates Chronic Seizures-Induced Memory Dysfunction in Zebrafish Model. ACS Chem. Neurosci. 2021, 12, 3288–3302. [Google Scholar] [CrossRef]
- Paudel, Y.N.; Othman, I.; Shaikh, M.F. Anti-High Mobility Group Box-1 Monoclonal Antibody Attenuates Seizure-Induced Cognitive Decline by Suppressing Neuroinflammation in an Adult Zebrafish Model. Front. Pharmacol. 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- Paudel, Y.N.; Shaikh, M.F.; Chakraborti, A.; Kumari, Y.; Aledo-Serrano, A.; Aleksovska, K.; Alvim, M.K.M.; Othman, I. HMGB1: A Common Biomarker and Potential Target for TBI, Neuroinflammation, Epilepsy, and Cognitive Dysfunction. Front. Neurosci. 2018, 12, 628. [Google Scholar] [CrossRef] [Green Version]
- Su, W.; Cui, H.; Wu, D.; Yu, J.; Ma, L.; Zhang, X.; Huang, Y.; Ma, C. Suppression of TLR4-MyD88 signaling pathway attenuated chronic mechanical pain in a rat model of endometriosis. J. Neuroinflamm. 2021, 18, 1–17. [Google Scholar] [CrossRef]
- Sun, X.; Zeng, H.; Wang, Q.; Yu, Q.; Wu, J.; Feng, Y.; Deng, P.; Zhang, H. Glycyrrhizin ameliorates inflammatory pain by inhibiting microglial activation-mediated inflammatory response via blockage of the HMGB1-TLR4-NF-kB pathway. Exp. Cell Res. 2018, 369, 112–119. [Google Scholar] [CrossRef]
- Fujita, K.; Motoki, K.; Tagawa, K.; Chen, X.; Hama, H.; Nakajima, K.; Homma, H.; Tamura, T.; Watanabe, H.; Katsuno, M.; et al. HMGB1, a pathogenic molecule that induces neurite degeneration via TLR4-MARCKS, is a potential therapeutic target for Alzheimer’s disease. Sci. Rep. 2016, 6, 31895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, Z.-H.; Chen, X.; Hua, H.-P.; Liang, L.; Liu, L.-J. The Oral Pretreatment of Glycyrrhizin Prevents Surgery-Induced Cognitive Impairment in Aged Mice by Reducing Neuroinflammation and Alzheimer’s-Related Pathology via HMGB1 Inhibition. J. Mol. Neurosci. 2017, 63, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, H.; Ju, Z.; Ragab, A.A.; Lundbäck, P.; Long, W.; Valdés-Ferrer, S.I.; He, M.; Pribis, J.P.; Li, J.; et al. MD-2 is required for disulfide HMGB1-dependent TLR4 signaling. J. Exp. Med. 2015, 212, 5–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiraldi, M.; Raucci, A.; Muñoz, L.M.; Livoti, E.; Celona, B.; Venereau, E.; Apuzzo, T.; De Marchis, F.; Pedotti, M.; Bachi, A.; et al. HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J. Exp. Med. 2012, 209, 551–563. [Google Scholar] [CrossRef] [Green Version]
- Venereau, E.; Casalgrandi, M.; Schiraldi, M.; Antoine, D.J.; Cattaneo, A.; De Marchis, F.; Liu, J.; Antonelli, A.; Preti, A.; Raeli, L.; et al. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J. Exp. Med. 2012, 209, 1519–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchi, M.E.; Crippa, M.P.; Manfredi, A.A.; Mezzapelle, R.; Querini, P.R.; Venereau, E. High-mobility group box 1 protein orchestrates responses to tissue damage via inflammation, innate and adaptive immunity, and tissue repair. Immunol. Rev. 2017, 280, 74–82. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Wang, H.; Chavan, S.S.; Andersson, U. High Mobility Group Box Protein 1 (HMGB1): The Prototypical Endogenous Danger Molecule. Mol. Med. 2015, 21, S6–S12. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Hreggvidsdottir, H.S.; Palmblad, K.; Wang, H.; Ochani, M.; Li, J.; Lu, B.; Chavan, S.; Rosas-Ballina, M.; Al-Abed, Y.; et al. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc. Natl. Acad. Sci. USA 2010, 107, 11942–11947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bierhaus, A.; Humpert, P.M.; Morcos, M.; Wendt, T.; Chavakis, T.; Arnold, B.; Stern, D.M.; Nawroth, P.P. Understanding RAGE, the receptor for advanced glycation end products. J. Mol. Med. 2005, 83, 876–886. [Google Scholar] [CrossRef]
- Kierdorf, K.; Fritz, G. RAGE regulation and signaling in inflammation and beyond. J. Leukoc. Biol. 2013, 94, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Merenmies, J.; Pihlaskari, R.; Laitinen, J.; Wartiovaara, J.; Rauvala, H. 30-kDa heparin-binding protein of brain (amphoterin) involved in neurite outgrowth. Amino acid sequence and localization in the filopodia of the advancing plasma membrane. J. Biol. Chem. 1991, 266, 16722–16729. [Google Scholar] [CrossRef]
- LeBlanc, P.M.; Doggett, T.A.; Choi, J.; Hancock, M.A.; Durocher, Y.; Frank, F.; Nagar, B.; Ferguson, T.A.; Saleh, M. An Immunogenic Peptide in the A-box of HMGB1 Protein Reverses Apoptosis-induced Tolerance through RAGE Receptor. J. Biol. Chem. 2014, 289, 7777–7786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchi, M.E. HMGB1 loves company. J. Leukoc. Biol. 2009, 86, 573–576. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Jiang, Y.; Wang, J.; Shi, X.; Liu, Q.; Liu, Z.; Li, Y.; Scott, M.J.; Xiao, G.; Li, S.; et al. Macrophage endocytosis of high-mobility group box 1 triggers pyroptosis. Cell Death Differ. 2014, 21, 1229–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Liu, H.; Zeng, Q.; Imperato, G.H.; Addorisio, M.E.; Li, J.; He, M.; Cheng, K.F.; Al-Abed, Y.; Harris, H.E.; et al. Inhibition of HMGB1/RAGE-mediated endocytosis by HMGB1 antagonist box A, anti-HMGB1 antibodies, and cholinergic agonists suppresses inflammation. Mol. Med. 2019, 25, 1–13. [Google Scholar] [CrossRef]
- Ling, Y.; Yang, Z.-Y.; Yin, T.; Li, L.; Yuan, W.-W.; Wu, H.-S.; Wang, C.-Y. Heparin changes the conformation of high-mobility group protein 1 and decreases its affinity toward receptor for advanced glycation endproducts in vitro. Int. Immunopharmacol. 2011, 11, 187–193. [Google Scholar] [CrossRef]
- Porat, A.; Giat, E.; Kowal, C.; He, M.; Son, M.; Latz, E.; Ben-Zvi, I.; Al-Abed, Y.; Diamond, B. DNA-Mediated Interferon Signature Induction by SLE Serum Occurs in Monocytes Through Two Pathways: A Mechanism to Inhibit Both Pathways. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.-J.; Jiang, Z.-P.; Lo, H.-R.; Feng, C.-L.; Chen, C.-J.; Yang, C.-Y.; Huang, M.-Z.; Wu, H.-Y.; Chen, Y.-A.; Chiu, C.-H.; et al. Coalescence of RAGE in Lipid Rafts in Response to Cytolethal Distending Toxin-Induced Inflammation. Front. Immunol. 2019, 10, 109. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.; Zhang, J.; Chen, H.; Zhuge, Y.; Chen, H.; Qian, F.; Zhou, K.; Niu, C.; Wang, F.; Qiu, H.; et al. Endothelial cell pyroptosis plays an important role in Kawasaki disease via HMGB1/RAGE/cathespin B signaling pathway and NLRP3 inflammasome activation. Cell Death Dis. 2019, 10, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Mingo Pulido, Á.; Hänggi, K.; Celias, D.P.; Gardner, A.; Li, J.; Batista-Bittencourt, B.; Mohamed, E.; Trillo-Tinoco, J.; Osunmakinde, O.; Pena, R.; et al. The inhibitory receptor TIM-3 limits activation of the cGAS-STING pathway in intra-tumoral dendritic cells by suppressing extracellular DNA uptake. Immunity 2021, 54, 1154–1167.e7. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Yue, Y.; Xiong, S. Extracellular HMGB1 augments macrophage inflammation by facilitating the endosomal accumulation of ALD-DNA via TLR2/4-mediated endocytosis. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166184. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Fernández-Hernando, C. Endothelial HMGB1 (High-Mobility Group Box 1) Regulation of LDL (Low-Density Lipoprotein) Transcytosis: A Novel Mechanism of Intracellular HMGB1 in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2020, 41, 217–219. [Google Scholar] [PubMed]
- Lan, J.; Luo, H.; Wu, R.; Wang, J.; Zhou, B.; Zhang, Y.; Jiang, Y.; Xu, J. Internalization of HMGB1 (High Mobility Group Box 1) Promotes Angiogenesis in Endothelial Cells. Arter. Thromb. Vasc. Biol. 2020, 40, 2922–2940. [Google Scholar] [CrossRef]
- Liu, L.; Yang, M.; Kang, R.; Dai, Y.; Yu, Y.; Gao, F.; Wang, H.; Sun, X.; Li, X.; Li, J.; et al. HMGB1–DNA complex-induced autophagy limits AIM2 inflammasome activation through RAGE. Biochem. Biophys. Res. Commun. 2014, 450, 851–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouhiainen, A.; Nykänen, N.-P.; Kuja-Panula, J.; Vanttola, P.; Huttunen, H.J.; Rauvala, H. Inhibition of Homophilic Interactions and Ligand Binding of the Receptor for Advanced Glycation End Products by Heparin and Heparin-Related Carbohydrate Structures. Medicines 2018, 5, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, S.-W.; Zhao, Y.; Li, P.; Ning, Y.-L.; Huang, Z.-Z.; Yang, N.; Liu, D.; Zhou, Y.-G. HMGB1 mediates cognitive impairment caused by the NLRP3 inflammasome in the late stage of traumatic brain injury. J. Neuroinflamm. 2021, 18, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Cheng, X.; Tang, Y.; Qiu, X.; Wang, Z.; Fu, G.; Wu, J.; Kang, H.; Wang, J.; Wang, H.; et al. The role of type 1 interferons in coagulation induced by gram-negative bacteria. Blood 2020, 135, 1087–1100. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Wang, X.; Li, Z.; He, Z.; Yang, X.; Cheng, X.; Peng, Y.; Xue, Q.; Bai, Y.; Zhang, R.; et al. Heparin prevents caspase-11-dependent septic lethality independent of anticoagulant properties. Immunity 2021, 54, 454–467.e6. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Cheng, X.; Tang, Y.; Qiu, X.; Wang, Y.; Kang, H.; Wu, J.; Wang, Z.; Liu, Y.; Chen, F.; et al. Bacterial Endotoxin Activates the Coagulation Cascade through Gasdermin D-Dependent Phosphatidylserine Exposure. Immunity 2019, 51, 983–996.e6. [Google Scholar] [CrossRef] [PubMed]
- Hagar, J.A.; Powell, D.A.; Aachoui, Y.; Ernst, R.K.; Miao, E.A. Cytoplasmic LPS Activates Caspase-11: Implications in TLR4-Independent Endotoxic Shock. Science 2013, 341, 1250–1253. [Google Scholar] [CrossRef] [Green Version]
- Kayagaki, N.; Wong, M.T.; Stowe, I.B.; Ramani, S.R.; Gonzalez, L.C.; Akashi-Takamura, S.; Miyake, K.; Zhang, J.; Lee, W.P.; Muszyński, A.; et al. Noncanonical Inflammasome Activation by Intracellular LPS Independent of TLR4. Science 2013, 341, 1246–1249. [Google Scholar] [CrossRef] [PubMed]
- Hubert, P.; Roncarati, P.; Demoulin, S.; Pilard, C.; Ancion, M.; Reynders, C.; Lerho, T.; Bruyere, D.; Lebeau, A.; Radermecker, C.; et al. Extracellular HMGB1 blockade inhibits tumor growth through profoundly remodeling immune microenvironment and enhances checkpoint inhibitor-based immunotherapy. J. Immunother. Cancer 2021, 9, e001966. [Google Scholar] [CrossRef]
- Chen, G.-Y.; Tang, J.; Zheng, P.; Liu, Y. CD24 and Siglec-10 Selectively Repress Tissue Damage–Induced Immune Responses. Science 2009, 323, 1722–1725. [Google Scholar] [CrossRef] [Green Version]
- Tian, R.-R.; Zhang, M.-X.; Liu, M.; Fang, X.; Li, D.; Zhang, L.; Zheng, P.; Zheng, Y.-T.; Liu, Y. CD24Fc protects against viral pneumonia in simian immunodeficiency virus-infected Chinese rhesus monkeys. Cell. Mol. Immunol. 2020, 17, 887–888. [Google Scholar] [CrossRef]
- Tian, R.-R.; Zhang, M.-X.; Zhang, L.-T.; Zhang, P.; Ma, J.-P.; Liu, M.; Devenport, M.; Zheng, P.; Zhang, X.-L.; Lian, X.-D.; et al. CD24 and Fc fusion protein protects SIVmac239-infected Chinese rhesus macaque against progression to AIDS. Antivir. Res. 2018, 157, 9–17. [Google Scholar] [CrossRef]
- Toubai, T.; Rossi, C.; Oravecz-Wilson, K.; Zajac, C.; Liu, C.; Braun, T.; Fujiwara, H.; Wu, J.; Sun, Y.; Brabbs, S.; et al. Siglec-G represses DAMP-mediated effects on T cells. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [Green Version]
- Song, N.J.; Allen, C.; Vilgelm, A.E.; Riesenberg, B.P.; Weller, K.P.; Reynolds, K.; Chakravarthy, K.B.; Kumar, A.; Khatiwada, A.; Sun, Z.; et al. Immunological Insights into the Therapeutic Roles of CD24Fc Against Severe COVID-19. medRxiv 2021, 8, 21262258. [Google Scholar]
- Agalave, N.M.; Larsson, M.; Abdelmoaty, S.; Su, J.; Baharpoor, A.; Lundbäck, P.; Palmblad, K.; Andersson, U.; Harris, H.; Svensson, C.I. Spinal HMGB1 induces TLR4-mediated long-lasting hypersensitivity and glial activation and regulates pain-like behavior in experimental arthritis. Pain 2014, 155, 1802–1813. [Google Scholar] [CrossRef]
- Rudjito, R.; Agalave, N.M.; Farinotti, A.B.; Lundbäck, P.; Szabo-Pardi, T.A.; Price, T.J.; Harris, H.E.; Burton, M.D.; Svensson, C.I. Sex- and cell-dependent contribution of peripheral high mobility group box 1 and TLR4 in arthritis-induced pain. Pain 2020, 162, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Frank, M.G.; Weber, M.D.; Watkins, L.R.; Maier, S.F. Stress sounds the alarmin: The role of the danger-associated molecular pattern HMGB1 in stress-induced neuroinflammatory priming. Brain Behav. Immun. 2015, 48, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grace, P.M.; Strand, K.A.; Galer, E.L.; Rice, K.C.; Maier, S.F.; Watkins, L.R. Protraction of neuropathic pain by morphine is mediated by spinal damage associated molecular patterns (DAMPs) in male rats. Brain Behav. Immun. 2017, 72, 45–50. [Google Scholar] [CrossRef]
- Yang, H.; Andersson, U.; Brines, M. Neurons Are a Primary Driver of Inflammation via Release of HMGB1. Cells 2021, 10, 2791. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Chen, H.; Dai, J.; Wan, Z.; Xiong, P.; Xu, Y.; Han, Z.; Chai, W.; Gong, F.; Zheng, F. Glycyrrhizin Protects Mice Against Experimental Autoimmune Encephalomyelitis by Inhibiting High-Mobility Group Box 1 (HMGB1) Expression and Neuronal HMGB1 Release. Front. Immunol. 2018, 9, 1518. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Liu, K.; Agari, T.; Yasuhara, T.; Morimoto, J.; Okazaki, M.; Takeuchi, H.; Toyoshima, A.; Sasada, S.; Shinko, A.; et al. Anti-high mobility group box 1 antibody exerts neuroprotection in a rat model of Parkinson’s disease. Exp. Neurol. 2016, 275, 220–231. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, A.; Ito, T.; Kibata, K.; Inagaki-Katashiba, N.; Amuro, H.; Nishizawa, T.; Son, Y.; Ozaki, Y.; Nomura, S. Serum high-mobility group box 1 is correlated with interferon-α and may predict disease activity in patients with systemic lupus erythematosus. Lupus 2019, 28, 1120–1127. [Google Scholar] [CrossRef]
- Tanaka, H.; Kondo, K.; Fujita, K.; Homma, H.; Tagawa, K.; Jin, X.; Jin, M.; Yoshioka, Y.; Takayama, S.; Masuda, H.; et al. HMGB1 signaling phosphorylates Ku70 and impairs DNA damage repair in Alzheimer’s disease pathology. Commun. Biol. 2021, 4, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Bolay, H.; Karadas, O.; Oztürk, B.; Sonkaya, R.; Tasdelen, B.; Bulut, T.D.S.; Gülbahar, O.; Özge, A.; Baykan, B. HMGB1, NLRP3, IL-6 and ACE2 levels are elevated in COVID-19 with headache: A window to the infection-related headache mechanism. J. Headache Pain 2021, 22, 1–12. [Google Scholar] [CrossRef]
- Chen, L.; Long, X.; Xu, Q.; Tan, J.; Wang, G.; Cao, Y.; Wei, J.; Luo, H.; Zhu, H.; Huang, L.; et al. Elevated serum levels of S100A8/A9 and HMGB1 at hospital admission are correlated with inferior clinical outcomes in COVID-19 patients. Cell. Mol. Immunol. 2020, 17, 992–994. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Huang, Y.; Quan, J.; Liu, J.; Wang, H.; Billiar, T.R.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. HMGB1 as a potential biomarker and therapeutic target for severe COVID-19. Heliyon 2020, 6, e05672. [Google Scholar] [CrossRef] [PubMed]
- Sivakorn, C.; Dechsanga, J.; Jamjumrus, L.; Boonnak, K.; Schultz, M.J.; Dorndorp, A.M.; Phumratanaprapin, W.; Ratanarat, R.; Naorungroj, T.; Wattanawinitchai, P.; et al. High Mobility Group Box 1 and Interleukin 6 at Intensive Care Unit Admission as Biomarkers in Critically Ill COVID-19 Patients. Am. J. Trop. Med. Hyg. 2021, 105, 73–80. [Google Scholar] [CrossRef]
- Barnay-Verdier, S.; Gaillard, C.; Messmer, M.; Borde, C.; Gibot, S.; Maréchal, V. PCA-ELISA: A sensitive method to quantify free and masked forms of HMGB1. Cytokine 2011, 55, 4–7. [Google Scholar] [CrossRef] [PubMed]
- Abdulahad, D.A.; Westra, J.; Bijzet, J.; Limburg, P.C.; Kallenberg, C.G.; Bijl, M. High mobility group box 1 (HMGB1) and anti-HMGB1 antibodies and their relation to disease characteristics in systemic lupus erythematosus. Arthritis Res. Ther. 2011, 13, R71–R79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, B.; Chen, F.; Ji, Y.; Kiss, L.; de Jonge, W.J.; Conejero-Goldberg, C.; Szabo, C.; Deitch, E.A.; Ulloa, L. Alpha7 cholinergic-agonist prevents systemic inflammation and improves survival during resuscitation. J. Cell. Mol. Med. 2008, 13, 3774–3785. [Google Scholar] [CrossRef] [PubMed]
- Crews, F.T.; Fisher, R.; Deason, C.; Vetreno, R.P. Loss of Basal Forebrain Cholinergic Neurons Following Adolescent Binge Ethanol Exposure: Recovery with the Cholinesterase Inhibitor Galantamine. Front. Behav. Neurosci. 2021, 15, 652494. [Google Scholar] [CrossRef]
- Hu, J.; Vacas, S.; Feng, X.; Lutrin, D.; Uchida, Y.; Lai, I.K.; Maze, M. Dexmedetomidine Prevents Cognitive Decline by Enhancing Resolution of High Mobility Group Box 1 Protein–induced Inflammation through a Vagomimetic Action in Mice. Anesthesiology 2018, 128, 921–931. [Google Scholar] [CrossRef] [Green Version]
- Huston, J.M.; Gallowitsch-Puerta, M.; Ochani, M.; Ochani, K.; Yuan, R.; Rosas-Ballina, M.; Ashok, M.; Goldstein, R.S.; Chavan, S.; Pavlov, V.A.; et al. Transcutaneous vagus nerve stimulation reduces serum high mobility group box 1 levels and improves survival in murine sepsis. Crit. Care Med. 2007, 35, 2762–2768. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Chen, Z.; Pan, Q.; Fu, S.; Lin, F.; Ren, H.; Han, H.; Billiar, T.R.; Sun, F.; Li, Q. The Protective Effect of PNU-282987, a Selective α7 Nicotinic Acetylcholine Receptor Agonist, on the Hepatic Ischemia-Reperfusion Injury Is Associated with the Inhibition of High-Mobility Group Box 1 Protein Expression and Nuclear Factor κB Activation in Mice. Shock 2013, 39, 197–203. [Google Scholar] [CrossRef]
- Pavlov, V.A.; Ochani, M.; Yang, L.-H.; Gallowitsch-Puerta, M.; Ochani, K.; Lin, X.; Levi, J.; Parrish, W.R.; Rosas-Ballina, M.; Czura, C.J.; et al. Selective α7-nicotinic acetylcholine receptor agonist GTS-21 improves survival in murine endotoxemia and severe sepsis. Crit. Care Med. 2007, 35, 1139–1144. [Google Scholar] [CrossRef]
- Sitapara, R.A.; Gauthier, A.G.; Valdés-Ferrer, S.I.; Lin, M.; Patel, V.; Wang, M.; Martino, A.T.; Perron, J.C.; Ashby, C.R., Jr.; Tracey, K.J.; et al. The α7 nicotinic acetylcholine receptor agonist, GTS-21, attenuates hyperoxia-induced acute inflammatory lung injury by alleviating the accumulation of HMGB1 in the airways and the circulation. Mol. Med. 2020, 26, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liao, H.; Ochani, M.; Justiniani, M.; Lin, X.; Yang, L.; Al-Abed, Y.; Wang, H.; Metz, C.; Miller, E.J.; et al. Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat. Med. 2004, 10, 1216–1221. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, F.; Li, X.; Yang, Q.; Li, X.; Xu, N.; Huang, Y.; Zhang, Q.; Gou, X.; Chen, S.; et al. Electroacupuncture pretreatment attenuates cerebral ischemic injury through α7 nicotinic acetylcholine receptor-mediated inhibition of high-mobility group box 1 release in rats. J. Neuroinflamm. 2012, 9, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Hou, L.; Yang, H.; Ge, J.; Wang, S.; Tian, W.; Wang, X.; Yang, Z. Electroacupuncture Pretreatment Attenuates Acute Lung Injury Through α7 Nicotinic Acetylcholine Receptor-Mediated Inhibition of HMGB1 Release in Rats After Cardiopulmonary Bypass. Shock 2018, 50, 351–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wazea, S.A.; Wadie, W.; Bahgat, A.K.; El-Abhar, H.S. Galantamine anti-colitic effect: Role of alpha-7 nicotinic acetylcholine receptor in modulating Jak/STAT3, NF-κB/HMGB1/RAGE and p-AKT/Bcl-2 pathways. Sci. Rep. 2018, 8, 5110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Xia, F.; Zhao, H.; Peng, K.; Liu, H.; Meng, X.; Chen, C.; Ji, F. Dexmedetomidine-induced cardioprotection is mediated by inhibition of high mobility group box-1 and the cholinergic anti-inflammatory pathway in myocardial ischemia-reperfusion injury. PLoS ONE 2019, 14, e0218726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Yong, Y.; Li, X.; Hu, Y.; Wang, J.; Wang, Y.-Q.; Song, W.; Chen, W.-T.; Xie, J.; Chen, X.-M.; et al. Vagal modulation of high mobility group box-1 protein mediates electroacupuncture-induced cardioprotection in ischemia-reperfusion injury. Sci. Rep. 2015, 5, 15503. [Google Scholar] [CrossRef] [Green Version]
- Andersson, U.; Tracey, K.J. Neural reflexes in inflammation and immunity. J. Exp. Med. 2012, 209, 1057–1068. [Google Scholar] [CrossRef]
- Andersson, U.; Tracey, K.J. Reflex Principles of Immunological Homeostasis. Annu. Rev. Immunol. 2012, 30, 313–335. [Google Scholar] [CrossRef] [Green Version]
- Pavlov, V.A.; Chavan, S.S.; Tracey, K.J. Molecular and Functional Neuroscience in Immunity. Annu. Rev. Immunol. 2018, 36, 783–812. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, V.; Chavan, S.S.; Tracey, K.J. Bioelectronic Medicine: From Preclinical Studies on the Inflammatory Reflex to New Approaches in Disease Diagnosis and Treatment. Cold Spring Harb. Perspect. Med. 2019, 10, a034140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, Y.-Y.; Xue, M.; Wang, Y.; Huang, Z.-H.; Huang, C. Electroacupuncture Alleviates Spared Nerve Injury-Induced Neuropathic Pain and Modulates HMGB1/NF-κB Signaling Pathway In The Spinal Cord. J. Pain Res. 2019, 12, 2851–2863. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Wang, Z.; Su, Y.; Qi, L.; Yang, W.; Fu, M.; Jing, X.; Wang, Y.; Ma, Q. A neuroanatomical basis for electroacupuncture to drive the vagal–adrenal axis. Nature 2021, 598, 641–645. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, T.; Yin, C.; Li, Y.; Gao, F.; Yu, L.; Wang, Q. Electroacupuncture Pretreatment Ameliorates Anesthesia and Surgery-Induced Cognitive Dysfunction via Activation of an α7-nAChR Signal in Aged Rats. Neuropsychiatr. Dis. Treat. 2021, 17, 2599–2611. [Google Scholar] [CrossRef] [PubMed]
- Zi, S.-F.; Li, J.-H.; Liu, L.; Deng, C.; Ao, X.; Chen, D.-D.; Wu, S.-Z. Dexmedetomidine-mediated protection against septic liver injury depends on TLR4/MyD88/NF-κB signaling downregulation partly via cholinergic anti-inflammatory mechanisms. Int. Immunopharmacol. 2019, 76, 105898. [Google Scholar] [CrossRef]
- Li, D.-J.; Huang, F.; Ni, M.; Fu, H.; Zhang, L.-S.; Shen, F.-M. α7 Nicotinic Acetylcholine Receptor Relieves Angiotensin II–Induced Senescence in Vascular Smooth Muscle Cells by Raising Nicotinamide Adenine Dinucleotide–Dependent SIRT1 Activity. Arter. Thromb. Vasc. Biol. 2016, 36, 1566–1576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aranow, C.; Atish-Fregoso, Y.; Lesser, M.; Mackay, M.; Anderson, E.; Chavan, S.; Zanos, T.P.; Datta-Chaudhuri, T.; Bouton, C.; Tracey, K.J.; et al. Transcutaneous auricular vagus nerve stimulation reduces pain and fatigue in patients with systemic lupus erythematosus: A randomised, double-blind, sham-controlled pilot trial. Ann. Rheum. Dis. 2020, 80, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Nishibori, M.; Mori, S.; Takahashi, H.K. Anti-HMGB1 monoclonal antibody therapy for a wide range of CNS and PNS diseases. J. Pharmacol. Sci. 2019, 140, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Liu, K.; Wake, H.; Teshigawara, K.; Yoshino, T.; Takahashi, H.; Mori, S.; Nishibori, M. Therapeutic effects of anti-HMGB1 monoclonal antibody on pilocarpine-induced status epilepticus in mice. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haruma, J.; Teshigawara, K.; Hishikawa, T.; Wang, D.; Liu, K.; Wake, H.; Mori, S.; Takahashi, H.; Sugiu, K.; Date, I.; et al. Anti-high mobility group box-1 (HMGB1) antibody attenuates delayed cerebral vasospasm and brain injury after subarachnoid hemorrhage in rats. Sci. Rep. 2016, 6, 37755. [Google Scholar] [CrossRef] [PubMed]
- Masai, K.; Kuroda, K.; Isooka, N.; Kikuoka, R.; Murakami, S.; Kamimai, S.; Wang, D.; Liu, K.; Miyazaki, I.; Nishibori, M.; et al. Neuroprotective Effects of Anti-high Mobility Group Box-1 Monoclonal Antibody Against Methamphetamine-Induced Dopaminergic Neurotoxicity. Neurotox. Res. 2021, 39, 1511–1523. [Google Scholar] [CrossRef]
- Nakajo, M.; Uezono, N.; Nakashima, H.; Wake, H.; Komiya, S.; Nishibori, M.; Nakashima, K. Therapeutic time window of anti-high mobility group box-1 antibody administration in mouse model of spinal cord injury. Neurosci. Res. 2018, 141, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Morioka, N.; Abe, H.; Zhang, F.F.; Hisaoka-Nakashima, K.; Liu, K.; Nishibori, M.; Nakata, Y. Neuropathic Pain in Rats with a Partial Sciatic Nerve Ligation Is Alleviated by Intravenous Injection of Monoclonal Antibody to High Mobility Group Box-1. PLoS ONE 2013, 8, e73640. [Google Scholar] [CrossRef] [Green Version]
- Nosaka, N.; Hatayama, K.; Yamada, M.; Fujii, Y.; Yashiro, M.; Wake, H.; Tsukahara, H.; Nishibori, M.; Morishima, T. Anti-high mobility group box-1 monoclonal antibody treatment of brain edema induced by influenza infection and lipopolysaccharide. J. Med Virol. 2018, 90, 1192–1198. [Google Scholar] [CrossRef] [PubMed]
- Okuma, Y.; Liu, K.; Wake, H.; Zhang, J.; Maruo, T.; Date, I.; Yoshino, T.; Ohtsuka, A.; Otani, N.; Tomura, S.; et al. Anti-high mobility group box-1 antibody therapy for traumatic brain injury. Ann. Neurol. 2012, 72, 373–384. [Google Scholar] [CrossRef] [Green Version]
- Okuma, Y.; Wake, H.; Teshigawara, K.; Takahashi, Y.; Hishikawa, T.; Yasuhara, T.; Mori, S.; Takahashi, H.K.; Date, I.; Nishibori, M. Anti–High Mobility Group Box 1 Antibody Therapy May Prevent Cognitive Dysfunction After Traumatic Brain Injury. World Neurosurg. 2019, 122, e864–e871. [Google Scholar] [CrossRef]
- Uezono, N.; Zhu, Y.; Fujimoto, Y.; Yasui, T.; Matsuda, T.; Nakajo, M.; Abematsu, M.; Setoguchi, T.; Mori, S.; Takahashi, H.K.; et al. Prior Treatment with Anti-High Mobility Group Box-1 Antibody Boosts Human Neural Stem Cell Transplantation-Mediated Functional Recovery After Spinal Cord Injury. Stem Cells 2018, 36, 737–750. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Liu, K.; Wake, H.; Teshigawara, K.; Mori, S.; Nishibori, M. Anti-high mobility group box-1 (HMGB1) antibody inhibits hemorrhage-induced brain injury and improved neurological deficits in rats. Sci. Rep. 2017, 7, 46243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Uezono, N.; Yasui, T.; Nakajo, M.; Nagai, T.; Wang, D.; Nishibori, M.; Nakashima, K. Combinatrial treatment of anti-High Mobility Group Box-1 monoclonal antibody and epothilone B improves functional recovery after spinal cord contusion injury. Neurosci. Res. 2021, 172, 13–25. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HMGB1 Redox Form | Partner Molecule | Receptor | Biological Response | Reference |
|---|---|---|---|---|
| All-thiol | CXCL12 | CXCR4 | Chemotaxis | [50] |
| Disulfide | None | TLR4 | Cytokines | [53] |
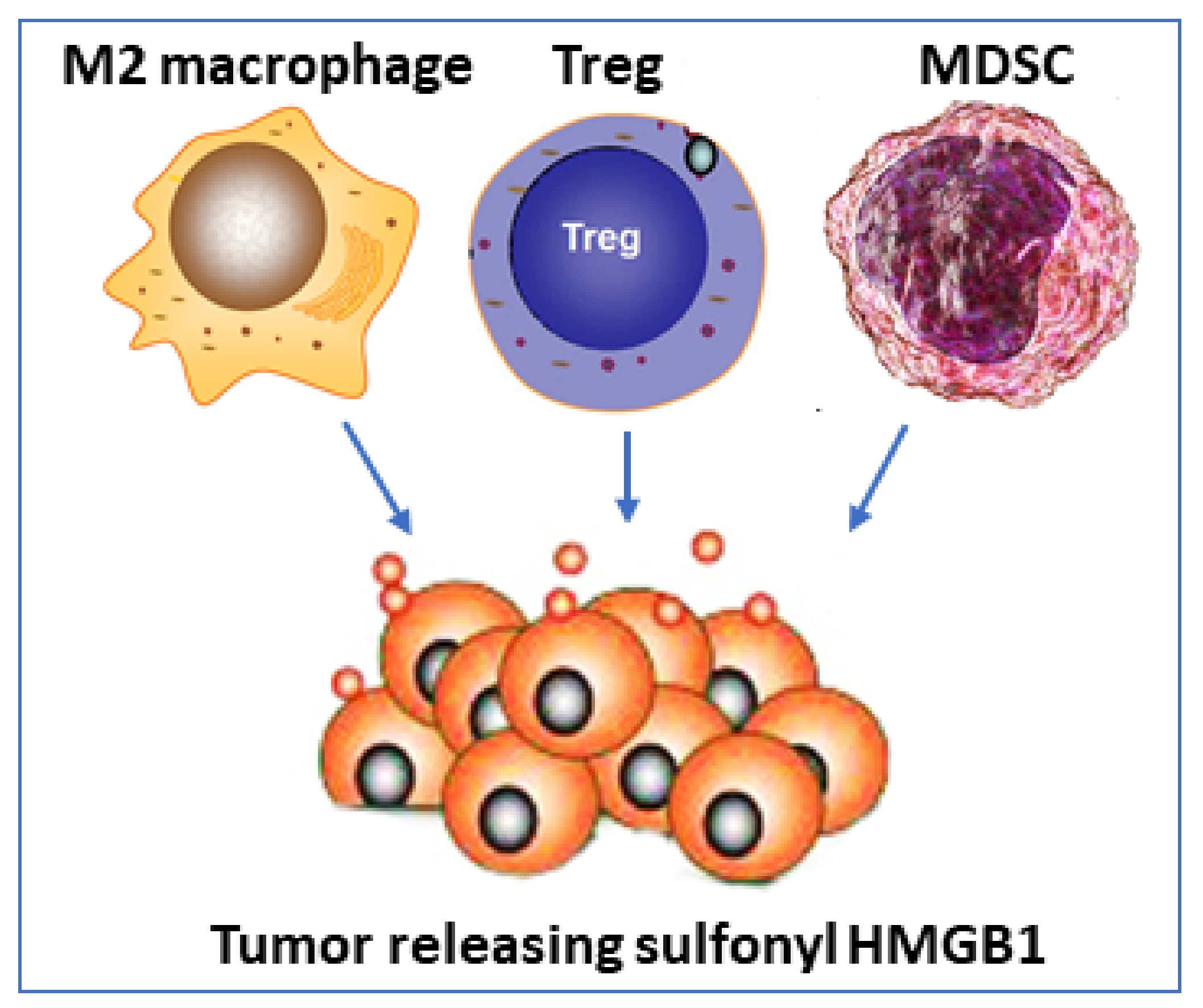
| Sulfonyl | Unknown | RAGE | Accumulation of Tregs and MDSCs, enhanced M2/M1 macrophage ratio and dendritic cell tolerogenicity | [77] |
| Undetermined | Many PAMPs and DAMPs | RAGE | Inflammasome activation, hyperinflammation, coagulation, pyroptosis | [34] |
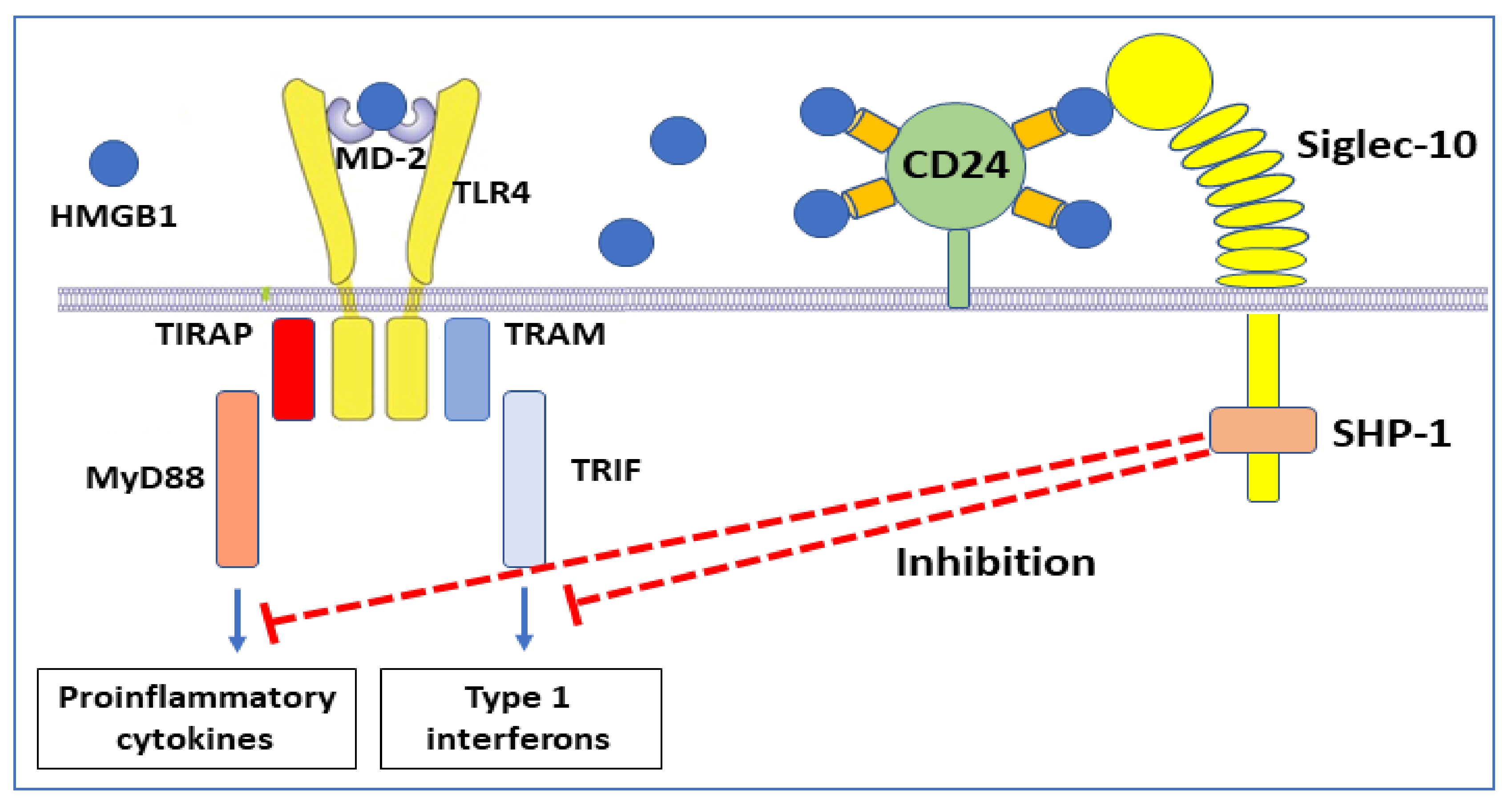
| Undetermined | None | CD24+ Siglec-10 | NF-κΒ inhibition | [78] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andersson, U.; Tracey, K.J.; Yang, H. Post-Translational Modification of HMGB1 Disulfide Bonds in Stimulating and Inhibiting Inflammation. Cells 2021, 10, 3323. https://doi.org/10.3390/cells10123323
Andersson U, Tracey KJ, Yang H. Post-Translational Modification of HMGB1 Disulfide Bonds in Stimulating and Inhibiting Inflammation. Cells. 2021; 10(12):3323. https://doi.org/10.3390/cells10123323
Chicago/Turabian StyleAndersson, Ulf, Kevin J. Tracey, and Huan Yang. 2021. "Post-Translational Modification of HMGB1 Disulfide Bonds in Stimulating and Inhibiting Inflammation" Cells 10, no. 12: 3323. https://doi.org/10.3390/cells10123323
APA StyleAndersson, U., Tracey, K. J., & Yang, H. (2021). Post-Translational Modification of HMGB1 Disulfide Bonds in Stimulating and Inhibiting Inflammation. Cells, 10(12), 3323. https://doi.org/10.3390/cells10123323