
Pathophysiological Mechanisms in Neurodevelopmental Disorders Caused by Rac GTPases Dysregulation: What’s behind Neuro-RACopathies



Abstract
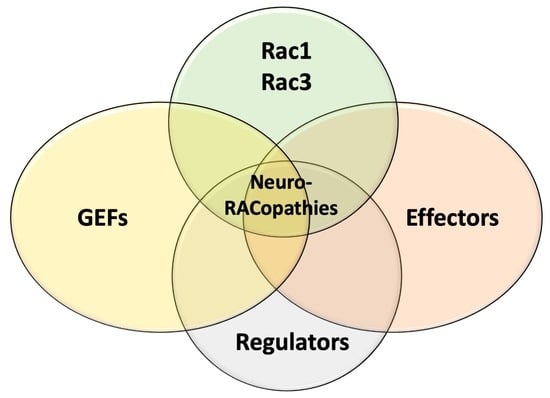
:
1. Introduction
2. RAC1
2.1. RAC1 Structure and Function
2.2. Rac1-Related Disorders
2.3. Underlying Pathogenic Mechanisms
3. RAC3
3.1. RAC3 Structure, Expression, and Function
3.2. Rac3-Related Disorders
3.3. Underlying Pathogenic Mechanisms
4. Implications of Rac Proteins Effectors and Regulators in NDDs
4.1. Activators: Unbalanced RAC Proteins Activation by Guanine Nucleotide-Exchange Factors (GEFs), Such as TRIO, DOCK3, and DOCK4, Is Implicated in NDDs Pathophysiology
4.1.1. Triple Functional Domain Protein (TRIO)
4.1.2. Dedicator of Cytokinesis 3 and 4 (DOCK3 and DOCK4)
4.2. Regulators: Dysfunctional RAC Acivity Modulation by Regulatory Proteins, Such as HACE1, ELMO2, and ELMO3, Contributes to NDDs Pathogenesis
4.2.1. HECT Domain- and Ankyrin Repeat-Containing E3 Ubiquitin Ligase 1 (HACE1)
4.2.2. ELMO/CED12 Domain-Containing Protein 2 and 3 (ELMO2 and ELMO3)
4.3. Effectors: The Emerging Role of RAC Proteins Effectors, Such as PAK1 and PAK3, in Human NDDs
p21 Protein-Activated Kinase 1 and 3 (PAK1 and PAK3)
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ahmad Mokhtar, A.M.; Hashim, I.F.; Mohd Zaini Makhtar, M.; Salikin, N.H.; Amin- Nordin, S. The role of RhoH in TCR signalling and its involvement in diseases. Cells 2021, 10, 950. [Google Scholar] [CrossRef] [PubMed]
- Ahmad Mokhtar, A.; Mbinti Ahmed, S.B.M.; Darling, N.J.; Harris, M.; Mott, H.R.; Owen, D. A complete survey of RhoGDI targets reveals novel interactions with atypical small GTPases. Biochemistry 2021, 60, 1533–1551. [Google Scholar] [CrossRef] [PubMed]
- Stankiewicz, T.R.; Linseman, D.A. Rho family GTPases: Key players in neuronal development, neuronal survival, and neurodegeneration. Front. Cell. Neurosci. 2014, 8, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magalhaes, Y.T.; Farias, J.O.; Silva, L.E.; Forti, F.L. GTPases, genome, actin: A hidden story in DNA damage response and repair mechanisms. DNA Repair 2021, 100, 103070. [Google Scholar] [CrossRef]
- Aspenstrom, P. Fast-cycling rho GTPases. Small GTPases 2020, 11, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Hashim, I.F.; Ahmad Mokhtar, A.M. Small Rho GTPases and their associated RhoGEFs mutations promote immunological defects in primary immunodeficiencies. Int. J. Biochem. Cell Biol. 2021, 137, 106034. [Google Scholar] [CrossRef]
- Mosaddeghzadeh, N.; Ahmadian, M.R. The RHO Family GTPases: Mechanisms of Regulation and Signaling. Cells 2021, 10, 1831. [Google Scholar] [CrossRef]
- Wittinghofer, A.; Vetter, I.R. Structure-function relationships of the G domain, a canonical switch motif. Annu. Rev. Biochem. 2011, 80, 943–971. [Google Scholar] [CrossRef]
- Dvorsky, R.; Ahmadian, M.R. Always look on the bright site of Rho: Structural implications for a conserved intermolecular interface. EMBO Rep. 2004, 5, 1130–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bishop, A.L.; Hall, A. Rho GTPases and their effector proteins. Biochem. J. 2000, 348, 241–255. [Google Scholar] [CrossRef] [PubMed]
- Burridge, K.; Wennerberg, K. Rho and Rac Take Center Stage. Cell 2004, 116, 167–179. [Google Scholar] [CrossRef] [Green Version]
- Roberts, P.J.; Mitin, N.; Keller, P.J.; Chenette, E.J.; Madigan, J.P.; Currin, R.O.; Cox, A.D.; Wilson, O.; Kirschmeier, P.; Der, C.J. Rho Family GTPase modification and dependence on CAAX motif-signaled posttranslational modification. J. Biol. Chem. 2008, 283, 25150–25163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bos, J.L.J.; Rehmann, H.; Wittinghofer, A. GEFs and GAPs: Critical elements in the control of small g proteins. Cell 2007, 129, 865–877. [Google Scholar] [CrossRef] [Green Version]
- Cherfils, J.; Zeghouf, M. Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol. Rev. 2013, 93, 269–309. [Google Scholar] [CrossRef] [Green Version]
- Jaffe, A.B.; Hall, A. RHO GTPASES: Biochemistry and biology. Annu. Rev. Cell Dev. Biol. 2005, 21, 247–269. [Google Scholar] [CrossRef] [Green Version]
- Moll, J.; Sansig, G.; Fattori, E.; Van Der Putten, H. The murine rac1 gene: cDNA cloning, tissue distribution and regulated expression of rac1 mRNA by disassembly of actin microfilaments. Oncogene 1991, 6, 863–866. [Google Scholar]
- Shirsat, N.V.; Pignolo, R.J.; Kreider, B.L.; Rovera, G. A member of the ras gene superfamily is expressed specifically in T, B and myeloid hemopoietic cells. Oncogene 1990, 5, 769–772. [Google Scholar]
- Haataja, L.; Groffen, J.; Heisterkamp, N. Characterization of RAC3, a novel member of the Rho family. J. Biol. Chem. 1997, 272, 20384–20388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malosio, M.L.; Gilardelli, D.; Paris, S.; Albertinazzi, C.; De Curtis, I. Differential expression of distinct members of rho family GTP-binding proteins during neuronal development: Identification of RAC1B, a new neural- specific member of the family. J. Neurosci. 1997, 17, 6717–6728. [Google Scholar] [CrossRef] [Green Version]
- Azzarelli, R.; Kerloch, T.; Pacary, E. Regulation of cerebral cortex development by Rho GTPases: Insights from in vivo studies. Front. Cell. Neurosci. 2015, 8, 445. [Google Scholar] [CrossRef] [Green Version]
- De Curtis, I. The Rac3 GTPase in Neuronal Development, Neurodevelopmental Disorders, and Cancer. Cells 2019, 8, 1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamboni, V.; Jones, R.; Umbach, A.; Ammoni, A.; Passafaro, M.; Hirsch, E.; Merlo, G.R. Rho GTPases in Intellectual Disability: From Genetics to Therapeutic Opportunities. Int. J. Mol. Sci. 2018, 19, 1821. [Google Scholar] [CrossRef] [Green Version]
- Guo, D.; Yang, X.; Shi, L. Rho GTPase Regulators and Effectors in Autism Spectrum Disorders: Animal Models and Insights for Therapeutics. Cells 2020, 9, 835. [Google Scholar] [CrossRef] [Green Version]
- Didsbury, J.; Weber, R.F.; Bokoch, G.M.; Evans, T.; Snyderman, R. Rac, a novel ras-related family of proteins that are botulinum toxin substrates. J. Biol. Chem. 1989, 264, 16378–16382. [Google Scholar] [CrossRef]
- Matos, P.; Skaug, J.; Marques, B.; Beck, S.; Veríssimo, F.; Gespach, C.; Boavida, M.G.; Scherer, S.W.; Jordan, P. Small GTPase Rac1: Structure, localization, and expression of the human gene. Biochem. Biophys. Res. Commun. 2000, 277, 741–751. [Google Scholar] [CrossRef]
- Reijnders, M.R.F.; Ansor, N.M.; Kousi, M.; Yue, W.W.; Tan, P.L.; Clarkson, K.; Clayton-Smith, J.; Corning, K.; Jones, J.R.; Lam, W.W.K.; et al. RAC1 Missense Mutations in Developmental Disorders with Diverse Phenotypes. Am. J. Hum. Genet. 2017, 101, 466–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridley, A.J. Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell Biol. 2006, 16, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Sayyad, W.A.; Fabris, P.; Torre, V. The Role of Rac1 in the Growth Cone Dynamics and Force Generation of DRG Neurons. PLoS ONE 2016, 11, e0146842. [Google Scholar] [CrossRef] [Green Version]
- Pak, C.W.; Flynn, K.C.; Bamburg, J.R. Actin-binding proteins take the reins in growth cones. Nat. Rev. Neurosci. 2008, 9, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Zhang, X.F.; Pollard, T.D.; Forscher, P. Arp2/3 complex-dependent actin networks constrain myosin II function in driving retrograde actin flow. J. Cell Biol. 2012, 197, 939–956. [Google Scholar] [CrossRef] [Green Version]
- Korobova, F.; Svitkina, T. Arp2/3 complex is important for filopodia formation, growth cone motility, and neuritogenesis in neuronal cells. Mol. Biol. Cell 2008, 19, 1561–1574. [Google Scholar] [CrossRef] [Green Version]
- Campellone, K.G.; Welch, M.D. A nucleator arms race: Cellular control of actin assembly. Nat. Rev. Mol. Cell. Biol. 2010, 11, 237–251. [Google Scholar] [CrossRef] [Green Version]
- Millard, T.H.; Sharp, S.J.; Machesky, L.M. Signalling to actin assembly via the WASP (Wiskott-Aldrich syndrome protein)-family proteins and the Arp2/3 complex. Biochem. J. 2004, 380, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steffen, A.; Ladwein, M.; Dimchev, G.A.; Hein, A.; Schwenkmezger, L.; Arens, S.; Ladwein, K.I.; Margit Holleboom, J.; Schur, F.; Victor Small, J.; et al. Rac function is crucial for cell migration but is not required for spreading and focal adhesion formation. J. Cell Sci. 2013, 126, 4572–4588. [Google Scholar] [PubMed] [Green Version]
- Bayer, S.A.; Altman, J.; Dai, X.F.; Humphreys, L. Planar differences in nuclear area and orientation in the subventricular and intermediate zones of the rat embryonic neocortex. J. Comp. Neurol. 1991, 307, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Shah, B.; Püschel, A.W. In vivo functions of small GTPases in neocortical development. Biol. Chem. 2014, 395, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Ohtaka-Maruyama, C.; Okado, H. Molecular Pathways Underlying Projection Neuron Production and Migration during Cerebral Cortical Development. Front. Neurosci. 2015, 9, 447. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, L.; Calcagnotto, M.E.; Paredes, M.F. Cortical Malformations: Lessons in Human Brain Development. Front Cell. Neurosci. 2020, 13, 576. [Google Scholar] [CrossRef]
- Leone, D.P.; Srinivasan, K.; Brakebusch, C.; McConnell, S.K. The rho GTPase Rac1 is required for proliferation and survival of progenitors in the developing forebrain. Dev. Neurobiol. 2010, 70, 659–678. [Google Scholar] [CrossRef] [Green Version]
- Sugihara, K.; Nakatsuji, N.; Nakamura, K.; Nakao, K.; Hashimoto, R.; Otani, H.; Sakagami, H.; Kondo, H.; Nozawa, S.; Aiba, A.; et al. Rac1 is required for the formation of three germ layers during gastrulation. Oncogene 1998, 17, 3427–3433. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Melendez, J.; Campbell, K.; Kuan, C.Y.; Zheng, Y. Rac1 deficiency in the forebrain results in neural progenitor reduction and microcephaly. Dev. Biol. 2009, 325, 162–170. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Liao, G.; Waclaw, R.R.; Burns, K.A.; Linquist, D.; Campbell, K.; Zheng, Y.; Kuan, C.Y. Rac1 controls the formation of midline commissures and the competency of tangential migration in ventral telencephalic neurons. J. Neurosci. 2007, 27, 3884–3893. [Google Scholar] [CrossRef]
- Gahankari, A.; Dong, C.; Bartoletti, G.; Galazo, M.; He, F. Deregulated Rac1 Activity in Neural Crest Controls Cell Proliferation, Migration and Differentiation During Midbrain Development. Front. Cell Dev. Biol. 2021, 9, 704769. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, D.; Wei, F.; Li, Y.; Wang, X.; Li, L.; Wang, G.; Zhang, S.; Zhang, L. Stress-Sensitive Protein Rac1 and Its Involvement in Neurodevelopmental Disorders. Neural Plast. 2020, 2020, 8894372. [Google Scholar] [CrossRef]
- Hori, K.; Nagai, T.; Shan, W.; Sakamoto, A.; Taya, S.; Hashimoto, R.; Hayashi, T.; Abe, M.; Yamazaki, M.; Nakao, K.; et al. Cytoskeletal regulation by AUTS2 in neuronal migration and neuritogenesis. Cell Rep. 2014, 9, 2166–2179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffney, L.J.; Wei, J.; Cheng, J.; Liu, W.; Smith, K.R.; Kittler, J.T.; Yan, Z. Shank3 deficiency induces NMDA receptor hypofunction via an actin-dependent mechanism. J. Neurosci. 2013, 33, 15767–15778. [Google Scholar] [CrossRef] [Green Version]
- Dong, T.; He, J.; Wang, S.; Wang, L.; Cheng, Y.; Zhong, Y. Inability to activate Rac1-dependent forgetting contributes to behavioral inflexibility in mutants of multiple autism-risk genes. Proc. Natl. Acad. Sci. USA 2016, 113, 7644–7649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi-Takagi, A.; Takaki, M.; Graziane, N.; Seshadri, S.; Murdoch, H.; Dunlop, A.J.; Makino, Y.; Seshadri, A.J.; Ishizuka, K.; Srivastava, D.P.; et al. Disrupted-in-Schizophrenia 1 (DISC1) regulates spines of the glutamate synapse via Rac1. Nat. Neurosci. 2010, 13, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Kiraly, D.D.; Lemtiri-Chlieh, F.; Levine, E.S.; Mains, R.E.; Eipper, B.A. Kalirin binds the NR2B subunit of the NMDA receptor, altering its synaptic localization and function. J. Neurosci. 2011, 31, 12554–12565. [Google Scholar] [CrossRef] [Green Version]
- Hwang, S.L.; Hong, Y.R.; Sy, W.D.; Lieu, A.S.; Lin, C.L.; Lee, K.S.; Howng, S.L. Rac1 gene mutations in human brain tumours. Eur. J. Surg. Oncol. 2004, 30, 68–72. [Google Scholar] [CrossRef]
- Khalil, B.D.; El-Sibai, M. Rho GTPases in primary brain tumor malignancy and invasion. J. Neurooncol. 2012, 108, 333–339. [Google Scholar] [CrossRef]
- Jung, I.H.; Leem, G.L.; Jung, D.E.; Kim, M.H.; Kim, E.Y.; Kim, S.H.; Park, H.C.; Park, S.W. Glioma is formed by active Akt1 alone and promoted by active Rac1 in transgenic zebrafish. NeuroOncology 2013, 15, 290–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karpel-Massler, G.; Westhoff, M.A.; Zhou, S.; Nonnenmacher, L.; Dwucet, A.; Kast, R.E.; Bachem, M.G.; Wirtz, C.R.; Debatin, K.M.; Halatsch, M.E. Combined inhibition of HER1/EGFR and RAC1 results in a synergistic antiproliferative effect on established and primary cultured human glioblastoma cells. Mol. Cancer Ther. 2013, 12, 1783–1795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De, P.; Aske, J.C.; Dey, N. RAC1 Takes the Lead in Solid Tumors. Cells 2019, 8, 382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannon, A.C.; Uribe-Alvarez, C.; Chernoff, J. RAC1 as a Therapeutic Target in Malignant Melanoma. Trends Cancer 2020, 6, 478–488. [Google Scholar] [CrossRef]
- Rivière, J.B.; Mirzaa, G.M.; O’Roak, B.J.; Beddaoui, M.; Alcantara, D.; Conway, R.L.; St-Onge, J.; Schwartzentruber, J.A.; Gripp, K.W.; Nikkel, S.M.; et al. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat. Genet. 2012, 44, 934–940. [Google Scholar] [CrossRef] [PubMed]
- Hetmanski, J.H.R.; Schwartz, J.M.; Caswell, P.T. Rationalizing Rac1 and RhoA GTPase signaling: A mathematical approach. Small GTPases 2018, 9, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Hori, K.; Hoshino, M. Neuronal Migration and AUTS2 Syndrome. Brain Sci. 2017, 7, 54. [Google Scholar] [CrossRef] [Green Version]
- Duffney, L.J.; Zhong, P.; Wei, J.; Matas, E.; Cheng, J.; Qin, L.; Ma, K.; Dietz, D.M.; Kajiwara, Y.; Buxbaum, J.D.; et al. Autism-like Deficits in Shank3-Deficient Mice Are Rescued by Targeting Actin Regulators. Cell Rep. 2015, 11, 1400–1413. [Google Scholar] [CrossRef] [Green Version]
- Weaving, L.S.; Ellaway, C.J.; Gécz, J.; Christodoulou, J. Rett syndrome: Clinical review and genetic update. J. Med. Genet. 2005, 42, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Sonoyama, T.; Stadler, L.K.J.; Zhu, M.; Keogh, J.M.; Henning, E.; Hisama, F.; Kirwan, P.; Jura, M.; Blaszczyk, B.K.; DeWitt, D.C.; et al. Human BDNF/TrkB variants impair hippocampal synaptogenesis and associate with neurobehavioural abnormalities. Sci. Rep. 2020, 10, 9028. [Google Scholar] [CrossRef]
- Lu, B.; Pang, P.T.; Woo, N.H. The yin and yang of neurotrophin action. Nat. Rev. Neurosci. 2005, 6, 603–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedrick, N.G.; Harward, S.C.; Hall, C.E.; Murakoshi, H.; McNamara, J.O.; Yasuda, R. Rho GTPase complementation underlies BDNF-dependent homo- and heterosynaptic plasticity. Nature 2016, 538, 104–108. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Pozzo-Miller, L. BDNF deregulation in Rett syndrome. Neuropharmacology 2014, 76, 737–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Zhu, Y.C.; Yu, J.; Miao, S.; Zheng, J.; Xu, L.; Zhou, Y.; Li, D.; Zhang, C.; Tao, J.; et al. CDKL5, a protein associated with rett syndrome, regulates neuronal morphogenesis via Rac1 signaling. J. Neurosci. 2010, 30, 12777–12786. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, C.; Gennaccaro, L.; Trazzi, S.; Bastianini, S.; Bettini, S.; Lo Martire, V.; Ren, E.; Medici, G.; Zoccoli, G.; Rimondini, R.; et al. Heterozygous CDKL5 Knockout Female Mice Are a Valuable Animal Model for CDKL5 Disorder. Neural Plast. 2018, 2018, 9726950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glantz, L.A.; Lewis, D.A. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch. Gen. Psychiatry 2000, 57, 65–73. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z.; Srivastava, D.P.; Photowala, H.; Kai, L.; Cahill, M.E.; Woolfrey, K.M.; Shum, C.Y.; Surmeier, D.J.; Penzes, P. Kalirin-7 controls activity-dependent structural and functional plasticity of dendritic spines. Neuron 2007, 56, 640–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolias, K.F.; Bikoff, J.B.; Burette, A.; Paradis, S.; Harrar, D.; Tavazoie, S.; Weinberg, R.J.; Greenberg, M.E. The Rac1-GEF Tiam1 couples the NMDA receptor to the activity-dependent development of dendritic arbors and spines. Neuron 2005, 45, 525–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazanietz, M.G.; Caloca, M.J. The Rac GTPase in Cancer: From Old Concepts to New Paradigms. Cancer Res. 2017, 77, 5445–5451. [Google Scholar] [CrossRef] [Green Version]
- De, P.; Carlson, J.H.; Jepperson, T.; Willis, S.; Leyland-Jones, B.; Dey, N. RAC1 GTP-ase signals Wnt-beta-catenin pathway mediated integrin-directed metastasis-associated tumor cell phenotypes in triple negative breast cancers. Oncotarget 2017, 8, 3072–3103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozano, E.; Betson, M.; Braga, V.M. Tumor progression: Small GTPases and loss of cell-cell adhesion. Bioessays 2003, 25, 452–463. [Google Scholar] [CrossRef]
- Nohata, N.; Uchida, Y.; Stratman, A.N.; Adams, R.H.; Zheng, Y.; Weinstein, B.M.; Mukouyama, Y.S.; Gutkind, J.S. Temporal-specific roles of Rac1 during vascular development and retinal angiogenesis. Dev. Biol. 2016, 411, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Hofbauer, S.W.; Krenn, P.W.; Ganghammer, S.; Asslaber, D.; Pichler, U.; Oberascher, K.; Henschler, R.; Wallner, M.; Kerschbaum, H.; Greil, R.; et al. Tiam1/Rac1 signals contribute to the proliferation and chemoresistance, but not motility, of chronic lymphocytic leukemia cells. Blood 2014, 123, 2181–2188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corbetta, S.; Gualdoni, S.; Albertinazzi, C.; Paris, S.; Croci, L.; Consalez, G.G.; de Curtis, I. Generation and characterization of Rac3 knockout mice. Mol. Cell. Biol. 2005, 25, 5763–5776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishikawa, M.; Ito, H.; Noda, M.; Hamada, N.; Tabata, H.; Nagata, K.I. Expression analyses of Rac3, a Rho family small GTPase, during mouse brain development. Dev. Neurosci. 2021. Epub ahead of print. [Google Scholar] [CrossRef]
- Aspenström, P.; Fransson, A.; Saras, J. Rho GTPases have diverse effects on the organization of the actin filament system. Biochem. J. 2004, 377, 327–337. [Google Scholar] [CrossRef]
- Corbetta, S.; Gualdoni, S.; Ciceri, G.; Monari, M.; Zuccaro, E.; Tybulewicz, V.L.J.; de Curtis, I. Essential role of Rac1 and Rac3 GTPases in neuronal development. FASEB J. 2009, 23, 1347–1357. [Google Scholar] [CrossRef] [PubMed]
- Pennucci, R.; Gucciardi, I.; de Curtis, I. Rac1 and Rac3 GTPases differently influence the morphological maturation of dendritic spines in hippocampal neurons. PLoS ONE 2019, 14, e0220496. [Google Scholar] [CrossRef] [Green Version]
- Honkura, N.; Matsuzaki, M.; Noguchi, J.; Ellis-Davies, G.C.; Kasai, H. The subspine organization of actin fibers regulates the structure and plasticity of dendritic spines. Neuron 2008, 57, 719–729. [Google Scholar] [CrossRef] [Green Version]
- de Curtis, I. Functions of Rac GTPases during neuronal development. Dev. Neurosci. 2008, 30, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Koizumi, H.; Gleeson, J.G. The doublecortin and doublecortin-like kinase 1 genes cooperate in murine hippocampal development. Cereb. Cortex 2006, 16, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Wonders, C.P.; Anderson, S.A. The origin and specification of cor- tical interneurons. Nat. Rev. Neurosci. 2006, 7, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Bolis, A.; Corbetta, S.; Cioce, A.; De Curtis, I. Differential distribution of Rac1 and Rac3 GTPases in the developing mouse brain: Implications for a role of Rac3 in Purkinje cell differentiation. Eur. J. Neurosci. 2003, 18, 2417–2424. [Google Scholar] [CrossRef] [PubMed]
- Vidaki, M.; Tivodar, S.; Doulgeraki, K.; Tybulewicz, V.; Kessaris, N.; Pachnis, V.; Karagogeos, D. Rac1-dependent cell cycle exit of MGE precursors and GABAergic interneuron migration to the cortex. Cereb. Cortex 2012, 22, 680–692. [Google Scholar] [CrossRef] [Green Version]
- Albertinazzi, C.; Za, L.; Paris, S.; de Curtis, I. ADP-ribosylation factor 6 and a functional PIX/p95-APP1 complex are required for Rac1B-mediated neurite outgrowth. Mol. Biol. Cell 2003, 14, 1295–1307. [Google Scholar] [CrossRef] [PubMed]
- Vaghi, V.; Pennucci, R.; Talpo, F.; Corbetta, S.; Montinaro, V.; Barone, C.; Croci, L.; Spaiardi, P.; Consalez, G.G.; Biella, G.; et al. Rac1 and Rac3 GTPases control synergistically the development of cortical and hippocampal GABAergic interneurons. Cereb. Cortex 2014, 24, 1247–1258. [Google Scholar] [CrossRef] [Green Version]
- Kelsom, C.; Lu, W. Development and specification of GABAergic cortical interneurons. Cell Biosci. 2013, 3, 19. [Google Scholar] [CrossRef] [Green Version]
- Tremblay, R.; Lee, S.; Rudy, B. GABAergic Interneurons in the Neocortex: From Cellular Properties to Circuits. Neuron 2016, 91, 260–292. [Google Scholar] [CrossRef] [Green Version]
- Costain, G.; Callewaert, B.; Gabriel, H.; Tan, T.Y.; Walker, S.; Christodoulou, J.; Lazar, T.; Menten, B.; Orkin, J.; Sadedin, S.; et al. De novo missense variants in RAC3 cause a novel neurodevelopmental syndrome. Genet. Med. 2019, 21, 1021–1026. [Google Scholar] [CrossRef] [PubMed]
- Hiraide, T.; Kaba Yasui, H.; Kato, M.; Nakashima, M.; Saitsu, H. A de novo variant in RAC3 causes severe global developmental delay and a middle interhemispheric variant of holoprosencephaly. J. Hum. Genet. 2019, 64, 1127–1132. [Google Scholar] [CrossRef]
- Scala, M.; Nishikawa, M.; Ito, H.; Tabata, H.; Khan, T.; Accogli, A.; Davids, L.; Martinez, J.; Ruiz, A.; Chiurazzi, P.; et al. RAC3 Variants Impair Axon Guidance and Disrupt Intracortical Neuronal Migration, Leading to Heterogeneous Neurodevelopmental Phenotypes; Department of Molecular Neurobiology, Institute for Developmental Research, Aichi Human Service Center: Aichi, Japan, 2021; manuscript under review. [Google Scholar]
- Albertinazzi, C.; Gilardelli, D.; Paris, S.; Longhi, R.; De Curtis, I. Overexpression of a neural-specific Rho family GTPase, cRac1B, selectively induces enhanced neuritogenesis and neurite branching in primary neurons. J. Cell. Biol. 1998, 142, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Pennucci, R.; Tavano, S.; Tonoli, D.; Gualdoni, S.; de Curtis, I. Rac1 and Rac3 GTPases regulate the development of hilar mossy cells by affecting the migration of their precursors to the hilus. PLoS ONE 2011, 6, 23–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawazu, M.; Ueno, T.; Kontani, K.; Ogita, Y.; Ando, M.; Fukumura, K.; Yamato, A.; Soda, M.; Takeuchi, K.; Miki, Y.; et al. Transforming mutations of RAC guanosine triphosphatases in human cancers. Proc. Natl. Acad. Sci. USA 2013, 110, 3029–3034. [Google Scholar] [CrossRef] [Green Version]
- Porter, A.P.; Papaioannou, A.; Malliri, A. Deregulation of Rho GTPases in cancer. Small GTPases 2016, 7, 123–138. [Google Scholar] [CrossRef] [PubMed]
- de Curtis, I. Roles of Rac1 and Rac3 GTPases during the development of cortical and hippocampal GABAergic interneurons. Front. Cell. Neurosci. 2014, 8, 307. [Google Scholar] [CrossRef] [Green Version]
- García-Mata, R.; Burridge, K. Catching a GEF by its tail. Trends Cell Biol. 2007, 17, 36–43. [Google Scholar] [CrossRef]
- Schmidt, A.; Hall, A. Guanine nucleotide exchange factors for Rho GTPases: Turning on the switch. Genes Dev. 2002, 16, 1587–1609. [Google Scholar] [CrossRef] [Green Version]
- Rossman, K.L.; Der, C.J.; Sondek, J. GEF means go: Turning on RHO GTPases with guanine nucleotide-exchange factors. Nat. Rev. Mol. Cell. Biol. 2005, 6, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Meller, N.; Merlot, S.; Guda, C. CZH proteins: A new family of Rho-GEFs. J. Cell Sci. 2005, 118, 4937–4946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blangy, A.; Vignal, E.; Schmidt, S.; Debant, A.; Gauthier-Rouvière, C.; Fort, P. TrioGEF1 controls Rac- and Cdc42-dependent cell structures through the direct activation of rhoG. J. Cell Sci. 2000, 113, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Debant, A.; Serra-Pagès, C.; Seipel, K.; O’Brien, S.; Tang, M.; Park, S.H.; Streuli, M. The multidomain protein Trio binds the LAR transmembrane tyrosine phosphatase, contains a protein kinase domain, and has separate rac-specific and rho-specific guanine nucleotide exchange factor domains. Proc. Natl. Acad. Sci. USA 1996, 93, 5466–5471. [Google Scholar] [CrossRef] [Green Version]
- Herring, B.E.; Nicoll, R.A. Kalirin and Trio proteins serve critical roles in excitatory synaptic transmission and LTP. Proc. Natl. Acad. Sci. USA 2016, 113, 2264–2269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, S.C.; Wang, D.; Iyer, E.P.; Trunnell, S.A.; Meduri, R.; Shinwari, R.; Sulkowski, M.J.; Cox, D.N. The RhoGEF trio functions in sculpting class specific dendrite morphogenesis in Drosophila sensory neurons. PLoS ONE 2012, 7, e33634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, S.; Debant, A. Function and regulation of the Rho guanine nucleotide exchange factor Trio. Small GTPases 2014, 5, e29769. [Google Scholar] [CrossRef] [Green Version]
- Katrancha, S.M.; Shaw, J.E.; Zhao, A.Y.; Myers, S.A.; Cocco, A.R.; Jeng, A.T.; Zhu, M.; Pittenger, C.; Greer, C.A.; Carr, S.A.; et al. Trio Haploinsufficiency Causes Neurodevelopmental Disease-Associated Deficits. Cell Rep. 2019, 26, 2805–2817.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.J.; He, W.Q.; Tang, J.; Tao, T.; Chen, C.; Gao, Y.Q.; Zhang, W.C.; He, X.Y.; Dai, Y.Y.; Zhu, N.C.; et al. Trio is a key guanine nucleotide exchange factor coordinating regulation of the migration and morphogenesis of granule cells in the developing cerebellum. J. Biol. Chem. 2010, 285, 24834–24844. [Google Scholar] [CrossRef] [Green Version]
- Ba, W.; Yan, Y.; Reijnders, M.R.; Schuurs-Hoeijmakers, J.H.; Feenstra, I.; Bongers, E.M.; Bosch, D.G.; De Leeuw, N.; Pfundt, R.; Gilissen, C.; et al. TRIO loss of function is associated with mild intellectual disability and affects dendritic branching and synapse function. Hum. Mol. Genet. 2016, 25, 892–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercer, C.L.; Keeton, B.; Dennis, N.R. Familial multiple ventricular extrasystoles, short stature, craniofacial abnormalities and digital hypoplasia: A further case of Stoll syndrome? Clin. Dysmorphol. 2008, 17, 91–93. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, S.; Greville-Heygate, S.; Bonnet, M.; Godwin, A.; Fagotto-Kaufmann, C.; Kajava, A.V.; Laouteouet, D.; Mawby, R.; Wai, H.A.; Dingemans, A.J.M.; et al. Opposite Modulation of RAC1 by Mutations in TRIO Is Associated with Distinct, Domain-Specific Neurodevelopmental Disorders. Am. J. Hum. Genet. 2020, 106, 338–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pengelly, R.J.; Greville-Heygate, S.; Schmidt, S.; Seaby, E.G.; Jabalameli, M.R.; Mehta, S.G.; Parker, M.J.; Goudie, D.; Fagotto-Kaufmann, C.; Mercer, C.; et al. Mutations specific to the Rac-GEF domain of TRIO cause intellectual disability and microcephaly. J. Med. Genet. 2016, 53, 735–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vulto-van Silfhout, A.T.; Hehir-Kwa, J.Y.; van Bon, B.W.; Schuurs-Hoeijmakers, J.H.; Meader, S.; Hellebrekers, C.J.; Thoonen, I.J.; de Brouwer, A.P.; Brunner, H.G.; Webber, C.; et al. Clinical significance of de novo and inherited copy-number variation. Hum. Mutat. 2013, 34, 1679–1687. [Google Scholar] [CrossRef] [PubMed]
- Sadybekov, A.; Tian, C.; Arnesano, C.; Katritch, V.; Herring, B.E. An autism spectrum disorder-related de novo mutation hotspot discovered in the GEF1 domain of Trio. Nat. Commun. 2017, 8, 601. [Google Scholar] [CrossRef] [Green Version]
- Gadea, G.; Blangy, A. Dock-family exchange factors in cell migration and disease. Eur. J. Cell Biol. 2014, 93, 466–477. [Google Scholar] [CrossRef] [PubMed]
- Iwata-Otsubo, A.; Ritter, A.L.; Weckselbatt, B.; Ryan, N.R.; Burgess, D.; Conlin, L.K.; Izumi, K. DOCK3-related neurodevelopmental syndrome: Biallelic intragenic deletion of DOCK3 in a boy with developmental delay and hypotonia. Am. J. Med. Genet. A 2018, 176, 241–245. [Google Scholar] [CrossRef]
- Miyamoto, Y.; Yamauchi, J. Cellular signaling of Dock family proteins in neural function. Cell Signal 2010, 22, 175–182. [Google Scholar] [CrossRef]
- Shi, L. Dock protein family in brain development and neurological disease. Commun. Integr. Biol. 2013, 6, e26839. [Google Scholar] [CrossRef] [Green Version]
- Wiltrout, K.; Ferrer, A.; van de Laar, I.; Namekata, K.; Harada, T.; Klee, E.W.; Zimmerman, M.T.; Cousin, M.A.; Kempainen, J.L.; Babovic-Vuksanovic, D.; et al. Variants in DOCK3 cause developmental delay and hypotonia. Eur. J. Hum. Genet. 2019, 27, 1225–1234. [Google Scholar] [CrossRef]
- Huang, M.; Liang, C.; Li, S.; Zhang, J.; Guo, D.; Zhao, B.; Liu, Y.; Peng, Y.; Xu, J.; Liu, W.; et al. Two Autism/Dyslexia Linked Variations of DOCK4 Disrupt the Gene Function on Rac1/Rap1 Activation, Neurite Outgrowth, and Synapse Development. Front. Cell. Neurosci. 2020, 13, 577. [Google Scholar] [CrossRef] [Green Version]
- Pagnamenta, A.T.; Bacchelli, E.; de Jonge, M.V.; Mirza, G.; Scerri, T.S.; Minopoli, F.; Chiocchetti, A.; Ludwig, K.U.; Hoffmann, P.; Paracchini, S.; et al. International Molecular Genetic Study of Autism Consortium. Characterization of a family with rare deletions in CNTNAP5 and DOCK4 suggests novel risk loci for autism and dyslexia. Biol. Psychiatry 2010, 68, 320–328. [Google Scholar] [CrossRef] [Green Version]
- Shao, S.; Kong, R.; Zou, L.; Zhong, R.; Lou, J.; Zhou, J.; Guo, S.; Wang, J.; Zhang, X.; Zhang, J.; et al. The Roles of Genes in the Neuronal Migration and Neurite Outgrowth Network in Developmental Dyslexia: Single- and Multiple-Risk Genetic Variants. Mol. Neurobiol. 2016, 53, 3967–3975. [Google Scholar] [CrossRef] [PubMed]
- Hollstein, R.; Parry, D.A.; Nalbach, L.; Logan, C.V.; Strom, T.M.; Hartill, V.L.; Carr, I.M.; Korenke, G.C.; Uppal, S.; Ahmed, M.; et al. HACE1 deficiency causes an autosomal recessive neurodevelopmental syndrome. J. Med. Genet. 2015, 52, 797–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anglesio, M.S.; Evdokimova, V.; Melnyk, N.; Zhang, L.; Fernandez, C.V.; Grundy, P.E.; Leach, S.; Marra, M.A.; Brooks-Wilson, A.R.; Penninger, J.; et al. Differential expression of a novel ankyrin containing E3 ubiquitin-protein ligase, Hace1, in sporadic Wilms’ tumor versus normal kidney. Hum. Mol. Genet. 2004, 13, 2061–2074. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Anglesio, M.S.; O’Sullivan, M.; Zhang, F.; Yang, G.; Sarao, R.; Mai, P.N.; Cronin, S.; Hara, H.; Melnyk, N.; et al. The E3 ligase HACE1 is a critical chromosome 6q21 tumor suppressor involved in multiple cancers. Nat. Med. 2007, 13, 1060–1069. [Google Scholar] [CrossRef] [PubMed]
- Sakata, M.; Kitamura, Y.H.; Sakuraba, K.; Goto, T.; Mizukami, H.; Saito, M.; Ishibashi, K.; Kigawa, G.; Nemoto, H.; Sanada, Y.; et al. Methylation of HACE1 in gastric carcinoma. Anticancer Res. 2009, 29, 2231–2233. [Google Scholar]
- Torrino, S.; Visvikis, O.; Doye, A.; Boyer, L.; Stefani, C.; Munro, P.; Bertoglio, J.; Gacon, G.; Mettouchi, A.; Lemichez, E. The E3 ubiquitin-ligase HACE1 catalyzes the ubiquitylation of active Rac1. Dev. Cell 2011, 21, 959–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, D.; Xiang, Y.; De Renzis, S.; Rink, J.; Zheng, G.; Zerial, M.; Wang, Y. The ubiquitin ligase HACE1 regulates Golgi membrane dynamics during the cell cycle. Nat. Commun. 2011, 2, 501. [Google Scholar] [CrossRef] [Green Version]
- Lachance, V.; Degrandmaison, J.; Marois, S.; Robitaille, M.; Génier, S.; Nadeau, S.; Angers, S.; Parent, J.L. Ubiquitylation and activation of a Rab GTPase is promoted by a β2AR-HACE1 complex. J. Cell Sci. 2014, 127, 111–123. [Google Scholar]
- Akawi, N.; McRae, J.; Ansari, M.; Balasubramanian, M.; Blyth, M.; Brady, A.F.; Clayton, S.; Cole, T.; Deshpande, C.; Fitzgerald, T.W.; et al. Discovery of four recessive developmental disorders using probabilistic genotype and phenotype matching among 4125 families. Nat. Genet. 2015, 47, 1363–1369. [Google Scholar] [CrossRef]
- Hariharan, N.; Ravi, S.; Pradeep, B.E.; Subramanyam, K.N.; Choudhary, B.; Srinivasan, S.; Khanchandani, P. A novel loss-of-function mutation in HACE1 is linked to a genetic disorder in a patient from India. Hum. Genome Var. 2018, 5, 17061. [Google Scholar] [CrossRef] [Green Version]
- Nagy, V.; Hollstein, R.; Pai, T.P.; Herde, M.K.; Buphamalai, P.; Moeseneder, P.; Lenartowicz, E.; Kavirayani, A.; Korenke, G.C.; Kozieradzki, I.; et al. HACE1 deficiency leads to structural and functional neurodevelopmental defects. Neurol. Genet. 2019, 5, e330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurin, M.; Côté, J.F. Insights into the biological functions of Dock family guanine nucleotide exchange factors. Genes Dev. 2014, 28, 533–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, M.; Pelletier, A.; Côté, J.F. Opening up on ELMO regulation: New insights into the control of Rac signaling by the DOCK180/ELMO complex. Small GTPases 2011, 2, 268–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, L.; Yang, J.; Jo, C.H.; Boland, A.; Zhang, Z.; McLaughlin, S.H.; Abu-Thuraia, A.; Killoran, R.C.; Smith, M.J.; Côté, J.F.; et al. Structure of the DOCK2-ELMO1 complex provides insights into regulation of the auto-inhibited state. Nat. Commun. 2020, 11, 3464. [Google Scholar] [CrossRef]
- Biersmith, B.; Liu, Z.C.; Bauman, K.; Geisbrecht, E.R. The DOCK protein sponge binds to ELMO and functions in Drosophila embryonic CNS development. PLoS ONE 2011, 6, e16120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueda, S.; Negishi, M.; Katoh, H. Rac GEF Dock4 interacts with cortactin to regulate dendritic spine formation. Mol. Biol. Cell 2013, 24, 1602–1613. [Google Scholar] [CrossRef]
- Tran, V.; Goyette, M.A.; Martínez-García, M.; Jiménez de Domingo, A.; Fernández-Mayoralas, D.M.; Fernández-Perrone, A.L.; Tirado, P.; Calleja-Pérez, B.; Álvarez, S.; Côté, J.F.; et al. Biallelic ELMO3 mutations and loss of function for DOCK-mediated RAC1 activation result in intellectual disability. Small GTPases 2021, 4, 1–8, Epub ahead of print. [Google Scholar] [CrossRef]
- Cetinkaya, A.; Xiong, J.R.; Vargel, İ.; Kösemehmetoğlu, K.; Canter, H.İ.; Gerdan, Ö.F.; Longo, N.; Alzahrani, A.; Camps, M.P.; Taskiran, E.Z.; et al. Loss-of-Function Mutations in ELMO2 Cause Intraosseous Vascular Malformation by Impeding RAC1 Signaling. Am. J. Hum. Genet. 2016, 99, 299–317. [Google Scholar] [CrossRef] [Green Version]
- Mehawej, C.; Hoischen, A.; Farah, R.A.; Marey, I.; David, M.; Stora, S.; Lachlan, K.; Brunner, H.G.; Mégarbané, A. Homozygous mutation in ELMO2 may cause Ramon syndrome. Clin. Genet. 2018, 93, 703–706. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Ren, W.; Côté, J.F.; Hinds, P.W.; Hu, X.; Du, K. ClipR-59 interacts with Elmo2 and modulates myoblast fusion. J. Biol. Chem. 2015, 290, 6130–6140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knaus, U.G.; Morris, S.; Dong, H.J.; Chernoff, J.; Bokoch, G.M. Regulation of human leukocyte p21-activated kinases through G protein—Coupled receptors. Science 1995, 269, 221–223. [Google Scholar] [CrossRef]
- Lei, M.; Lu, W.; Meng, W.; Parrini, M.C.; Eck, M.J.; Mayer, B.J.; Harrison, S.C. Structure of PAK1 in an autoinhibited conformation reveals a multistage activation switch. Cell 2000, 102, 387–397. [Google Scholar] [CrossRef] [Green Version]
- Parrini, M.C.; Lei, M.; Harrison, S.C.; Mayer, B.J. Pak1 kinase homodimers are autoinhibited in trans and dissociated upon activation by Cdc42 and Rac1. Mol. Cell 2002, 9, 73–83. [Google Scholar] [CrossRef]
- Rane, C.K.; Minden, A. P21 activated kinases: Structure, regulation, and functions. Small GTPases 2014, 5, e28003. [Google Scholar] [CrossRef] [PubMed]
- Bokoch, G.M. Biology of the p21-activated kinases. Annu. Rev. Biochem. 2003, 72, 743–781. [Google Scholar] [CrossRef]
- Asrar, S.; Meng, Y.; Zhou, Z.; Todorovski, Z.; Huang, W.W.; Jia, Z. Regulation of hippocampal long-term potentiation by p21-activated protein kinase 1 (PAK1). Neuropharmacology 2009, 56, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Zhou, Z.; Asrar, S.; Henkelman, M.; Xie, W.; Jia, Z. p21-Activated kinases 1 and 3 control brain size through coordinating neuronal complexity and synaptic properties. Mol. Cell. Biol. 2011, 31, 388–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harms, F.L.; Kloth, K.; Bley, A.; Denecke, J.; Santer, R.; Lessel, D.; Hempel, M.; Kutsche, K. Activating Mutations in PAK1, Encoding p21-Activated Kinase 1, Cause a Neurodevelopmental Disorder. Am. J. Hum. Genet. 2018, 103, 579–591. [Google Scholar] [CrossRef] [Green Version]
- des Portes, V.; Soufir, N.; Carrié, A.; Billuart, P.; Bienvenu, T.; Vinet, M.C.; Beldjord, C.; Ponsot, G.; Kahn, A.; Boué, J.; et al. Gene for nonspecific X-linked mental retardation (MRX 47) is located in Xq22.3-q24. Am. J. Med. Genet. 1997, 72, 324–328. [Google Scholar] [CrossRef]
- Gedeon, A.K.; Nelson, J.; Gécz, J.; Mulley, J.C. X-linked mild non-syndromic mental retardation with neuropsychiatric problems and the missense mutation A365E in PAK3. Am. J. Med. Genet. A 2003, 120A, 509–517. [Google Scholar] [CrossRef]
- Peippo, M.; Koivisto, A.M.; Särkämö, T.; Sipponen, M.; von Koskull, H.; Ylisaukko-oja, T.; Rehnström, K.; Froyen, G.; Ignatius, J.; Järvelä, I. PAK3 related mental disability: Further characterization of the phenotype. Am. J. Med. Genet. A 2007, 143A, 2406–2416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rejeb, I.; Saillour, Y.; Castelnau, L.; Julien, C.; Bienvenu, T.; Taga, P.; Chaabouni, H.; Chelly, J.; Ben Jemaa, L.; Bahi-Buisson, N. A novel splice mutation in PAK3 gene underlying mental retardation with neuropsychiatric features. Eur. J. Hum. Genet. 2008, 16, 1358–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duarte, K.; Heide, S.; Poëa-Guyon, S.; Rousseau, V.; Depienne, C.; Rastetter, A.; Nava, C.; Attié-Bitach, T.; Razavi, F.; Martinovic, J.; et al. PAK3 mutations responsible for severe intellectual disability and callosal agenesis inhibit cell migration. Neurobiol. Dis. 2020, 136, 104709. [Google Scholar] [CrossRef]
- Magini, P.; Pippucci, T.; Tsai, I.C.; Coppola, S.; Stellacci, E.; Bartoletti-Stella, A.; Turchetti, D.; Graziano, C.; Cenacchi, G.; Neri, I.; et al. A mutation in PAK3 with a dual molecular effect deregulates the RAS/MAPK pathway and drives an X-linked syndromic phenotype. Hum. Mol. Genet. 2014, 23, 3607–3617. [Google Scholar] [CrossRef] [Green Version]
- Pascolini, G.; Gaudioso, F.; Palougarisssarelli, C.; Novelli, A.; Di Giosaffatte, N.; Majore, S.; Grammatico, P. Clinical and Molecular Aspects of the Neurodevelopmental Disorder Associated with PAK3 Perturbation. J. Mol. Neurosci. 2021, 71, 2474–2481, Epub ahead of print. [Google Scholar] [CrossRef]
- Kreis, P.; Thévenot, E.; Rousseau, V.; Boda, B.; Muller, D.; Barnier, J.V. The p21-activated kinase 3 implicated in mental retardation regulates spine morphogenesis through a Cdc42-dependent pathway. J. Biol. Chem. 2007, 282, 21497–21506. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rac Protein | Tissue Distribution | Human Gene (OMIM) | Associated Medical Conditions (OMIM) |
|---|---|---|---|
| RAS-related c3 botulinum toxin substrate 1; RAC1 | Ubiquitous | RAC1 (* 602048) | Mental retardation, autosomal dominant 48; MRD48 (# 617751) |
| RAS-related c3 botulinum toxin substrate 2; RAC2 | Hematopoietic system | RAC2 (* 602049) | Immunodeficiency 73a with defective neutrophil chemotaxis and leukocytosis; IMD73A (# 608203); |
| Immunodeficiency 73b with defective neutrophil chemotaxis and lymphopenia; IMD73B (# 618986); | |||
| Immunodeficiency 73c with defective neutrophil chemotaxis and hypogammaglobulinemia; IMD73C (# 618987) | |||
| RAS-related c3 botulinum toxin substrate 3; RAC3 | Nervous system | RAC3 (* 602050) | Neurodevelopmental disorder with structural brain anomalies and dysmorphic facies; NEDBAF (# 618577) |
| Rac Proteins Interactor. | Human Gene (OMIM) | Category | Pathophysological Mechanism in Relation to Rac Proteins | Associated Medical Conditions (OMIM) |
|---|---|---|---|---|
| Triple functional domain protein | TRIO (* 601893) | GEF | LoF mechanism: ↓ TRIO-mediated RAC1 activation (MRD44); GoF mechanism: indirect ↑ activation of RAC1 (MRD63) | Intellectual developmental disorder, autosomal dominant 44, with microcephaly (MRD44, # 617061); Intellectual developmental disorder, autosomal dominant 63, with macrocephaly (MRD63, # 618825) |
| Dedicator of cytokinesis 3 | DOCK3 (* 603123) | GEF | Lof mechanism: ↓ GEF-mediated RAC activation | Neurodevelopmental disorder with Impaired intellectual development, hypotonia, and ataxia (NEDIDHA, # 618292) |
| Dedicator of cytokinesis 4 | DOCK4 (* 607679) | GEF | Lof mechanism: ↓ GEF-mediated RAC activation | Dyslexia and ASD with poor reading abilities |
| Hect domain- and ankyrin repeat-containing e3 ubiquitin protein ligase 1 | HACE1 (* 610876) | Regulator | Lof mechanism: ↓ ubiquitination leading to ↑ Rac1 activity | Spastic paraplegia and psychomotor retardation with or without seizures (SPPRS, # 616756) |
| Engulfment and cell motility gene 2 | ELMO2 (* 606421) | Regulator | Possible LoF mechanism: abnormal interaction with DOCK proteins | Ramon syndrome (# 266270) |
| Engulfment and cell motility gene 3 | ELMO3 (* 606422) | Regulator | Lof mechanism: ↓ RAC1-GTP-loading by ELMO3/DOCK1 complex | Psychomotor delay and ASD |
| p21 protein-activated kinase 1 | PAK1 (* 602590) | Effector | GoF mechanism: ↑ activation of Rac signaling pathway | Intellectual developmental disorder with macrocephaly, seizures, and speech delay (IDDMSSD, # 618158) |
| p21 protein-activated kinase 3 | PAK3 (* 300142) | Effector | GoF mechanism: ↑ Rac1 binding and activation | Intellectual developmental disorder, x-linked 30 (XLID30, # 300558) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scala, M.; Nishikawa, M.; Nagata, K.-i.; Striano, P. Pathophysiological Mechanisms in Neurodevelopmental Disorders Caused by Rac GTPases Dysregulation: What’s behind Neuro-RACopathies. Cells 2021, 10, 3395. https://doi.org/10.3390/cells10123395
Scala M, Nishikawa M, Nagata K-i, Striano P. Pathophysiological Mechanisms in Neurodevelopmental Disorders Caused by Rac GTPases Dysregulation: What’s behind Neuro-RACopathies. Cells. 2021; 10(12):3395. https://doi.org/10.3390/cells10123395
Chicago/Turabian StyleScala, Marcello, Masashi Nishikawa, Koh-ichi Nagata, and Pasquale Striano. 2021. "Pathophysiological Mechanisms in Neurodevelopmental Disorders Caused by Rac GTPases Dysregulation: What’s behind Neuro-RACopathies" Cells 10, no. 12: 3395. https://doi.org/10.3390/cells10123395
APA StyleScala, M., Nishikawa, M., Nagata, K. -i., & Striano, P. (2021). Pathophysiological Mechanisms in Neurodevelopmental Disorders Caused by Rac GTPases Dysregulation: What’s behind Neuro-RACopathies. Cells, 10(12), 3395. https://doi.org/10.3390/cells10123395