
Fragment-Sized and Bidentate (Immuno)Proteasome Inhibitors Derived from Cysteine and Threonine Targeting Warheads

 , ,
, ,  , , , , and
, , , , and 
Abstract
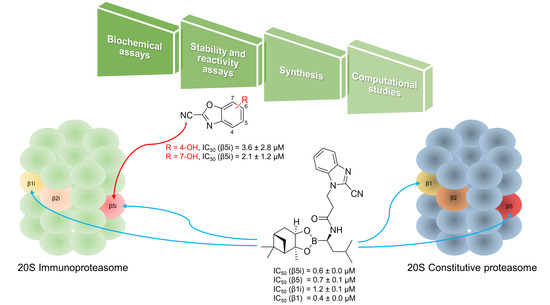
:
1. Introduction
2. Materials and Methods
2.1. General Chemistry Methods
2.1.1. General Methods for the Synthesis of Benzoxazole-2-Carbonitriles Using Appel’s Salt
- Method A.
- To a solution of an appropriate 2-aminophenol derivative (1.0 equiv.) in pyridine (6 mL for 1.0 mmol of the 2-aminophenol derivative), Appel’s salt (1.1 equiv.) was added portion-wise. The reaction mixtures were stirred at different temperatures and the reaction time was also varied (see Supplementary Materials for details). After completion of the reaction, the volatiles were removed under reduced pressure and the crude residue was purified by reversed-phase flash column chromatography using eluents A (0.1% HCOOH in MeCN) and B (0.1% HCOOH in H2O) (gradient from 1:9 to 10:0).
- Method B.
- To a solution of an appropriate 2-aminophenol derivative (1.0 equiv.) in anhydrous THF (6 mL for 1.0 mmol of the 2-aminophenol derivative), Appel’s salt (1.0 equiv.) was added portion-wise. The reaction mixture was stirred at RT for 1 h and then the volatiles were removed under reduced pressure. The residue was re-dissolved in DMSO (4 mL for 1.0 mmol of the 2-aminophenol derivative), the reaction mixture was stirred at 100 °C for 1 h. The volatiles were removed under reduced pressure and the crude residue was purified by reversed-phase flash column chromatography using eluents A (0.1% HCOOH in MeCN) and B (0.1% HCOOH in H2O) (gradient from 1:9 to 10:0).
- Method C.
- To a solution of an appropriate 2-aminophenol derivative (1.0 equiv.) in anhydrous THF (6 mL for 1.0 mmol of the 2-aminophenol derivative), Appel’s salt (1.0 equiv.) was added portion-wise. The reaction mixture was stirred at 140 °C for 20 min in a microwave reactor. After completion of the reaction, the volatiles were removed under reduced pressure and the crude residue was purified by reversed-phase flash column chromatography using eluents A (0.1% HCOOH in MeCN) and B (0.1% HCOOH in H2O) (gradient from 1:9 to 10:0).
2.1.2. Alternative Method for the Synthesis of Benzoxazole-2-Carbonitriles
- Step 1:
- To a solution of an appropriate 2-aminophenol derivative (1.0 equiv.) in EtOH:H2O (5:1, 6 mL for 1.0 mmol of the 2-aminophenol derivative), CS2 (1.0 equiv.) and solid KOH (1.0 equiv.) were added. The reaction mixtures were stirred at different temperatures and the reaction time was also varied (see Supplementary Materials for details). After the reaction was complete, it was cooled to room temperature, diluted with H2O (20 mL for 1.0 mmol of the 2-aminophenol derivative), and the pH was adjusted to 1 with 10% HCl. The precipitate formed was filtered off and washed with H2O (2 × 30 mL).
- Step 2:
- To the obtained 2,3-dihydro-1,3-benzoxazole-2-thiol derivative, SOCl2 (6 mL for 1.0 mmol of the 2,3-dihydro-1,3-benzoxazole-2-thiol derivative) was added drop-wise, followed by the addition of DMF (one drop). The reaction mixture was refluxed for 1 h, the volatiles were then removed under reduced pressure and the crude 2-chloro-2,3-dihydro-1,3-benzoxazole derivative was used in the next step without further purification.
- Step 3:
- To a solution of the obtained 2-chloro-2,3-dihydro-1,3-benzoxazole derivative (1.0 equiv.) in DMF (8 mL for 1.0 mmol of the 2-chloro-2,3-dihydro-1,3-benzoxazole derivative), KCN (1.4 equiv.) was added. The reaction mixtures were stirred at different temperatures and the reaction time was also varied (see Supplementary Materials for details). After the reaction was complete, it was diluted with H2O (30 mL) and extracted with DCM (3 × 30 mL). The volatiles were removed under reduced pressure and the crude residue was purified by reversed-phase flash column chromatography using eluents A (0.1% HCOOH in MeCN) and B (0.1% HCOOH in H2O) (gradient from 1:9 to 10:0).
2.1.3. General O-Acylation Procedure
2.1.4. General Nitro Reduction Procedure
2.1.5. General N-Acylation Procedure
2.1.6. General Synthetic Strategy for the Preparation of Boronic Acid Derivatives
2.2. Residual Activity Measurements
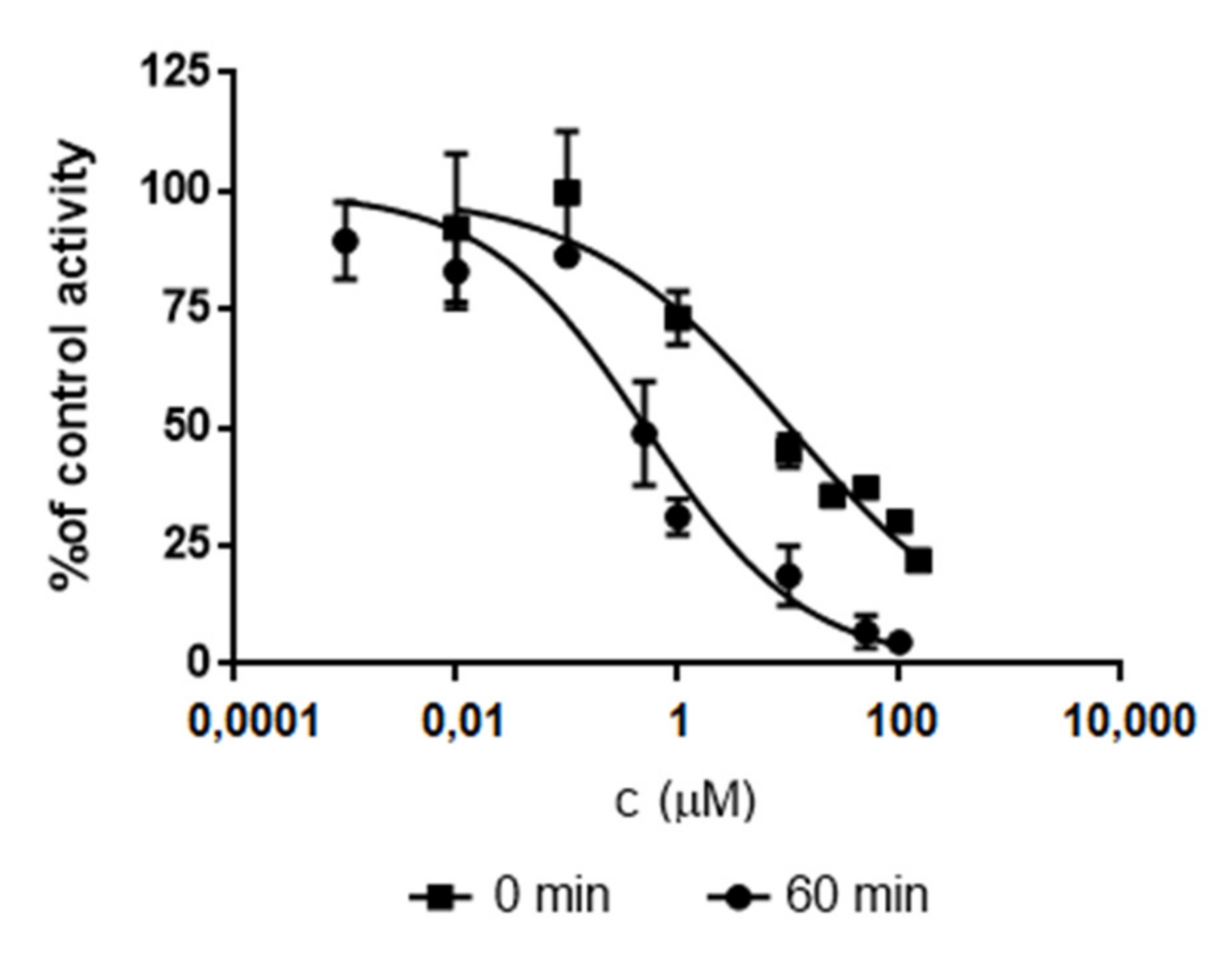
2.3. Determination of IC50 Values
2.4. Screening of the Heterocyclic Electrophilic Compounds
2.5. Reactivity Assays
2.5.1. UV-Vis-Based Stability and Reactivity Assay
2.5.2. HPLC-Based Stability and Reactivity Assay
2.6. LC-MS Measurements
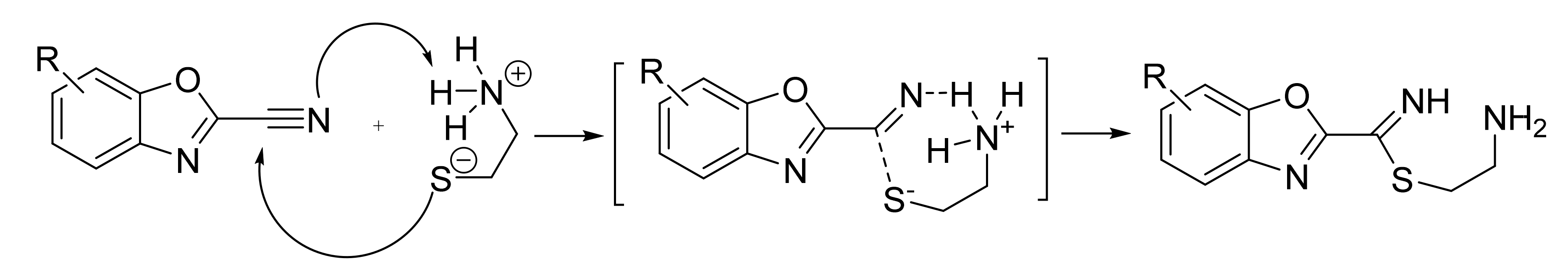
2.7. Reactivity Calculations
2.8. Computational Docking
2.9. Cholinesterase Assay
2.10. Monoamine Oxidase Assay
2.11. Caspase-1 Assay
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef] [PubMed]
- Scherer, D.C.; Brockman, J.A.; Chen, Z.; Maniatis, T.; Ballard, D.W. Signal-induced degradation of IκBα requires site-specific ubiquitination. Proc. Natl. Acad. Sci. USA 1995, 92, 11259–11263. [Google Scholar] [CrossRef] [Green Version]
- Zerfas, B.L.; Maresh, M.E.; Trader, D.J. The Immunoproteasome: An Emerging Target in Cancer and Autoimmune and Neurological Disorders. J. Med. Chem. 2020, 63, 1841–1858. [Google Scholar] [CrossRef]
- Raynes, R.; Pomatto, L.C.D.; Davies, K.J.A. Degradation of oxidized proteins by the proteasome: Distinguishing between the 20S, 26S, and immunoproteasome proteolytic pathways. Mol. Aspects Med. 2016, 50, 41–55. [Google Scholar] [CrossRef] [Green Version]
- Thibaudeau, T.A.; Smith, D.M. A Practical Review of Proteasome Pharmacology. Pharmacol. Rev. 2019, 71, 170–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, Y. Structure, Dynamics and Function of the 26S Proteasome. In Macromolecular Protein Complexes III: Structure and Function; Springer: Cham, Switzerland, 2021; pp. 1–151. [Google Scholar]
- Sahu, I.; Glickman, M.H. Proteasome in action: Substrate degradation by the 26S proteasome. Biochem. Soc. Trans. 2021, 49, 629–644. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.A.; Potter, M.W.; McDade, T.P.; Ricciardi, R.; Perugini, R.A.; Elliott, P.J.; Adams, J.; Callery, M.P. 26S proteasome inhibition induces apoptosis and limits growth of human pancreatic cancer. J. Cell. Biochem. 2001, 82, 110–122. [Google Scholar] [CrossRef]
- Michalek, M.T.; Grant, E.P.; Gramm, C.; Goldberg, A.L.; Rock, K.L. A role for the ubiquitin-dependent proteolytic pathway in MHC class I-restricted antigen presentation. Nature 1993, 363, 552–554. [Google Scholar] [CrossRef] [PubMed]
- Machiels, B.M.; Henfling, M.E.; Gerards, W.L.; Broers, J.L.; Bloemendal, F.C.; Ramaekers, H.; Schutte, B. Detailed analysis of cell cycle kinetics upon proteasome inhibition. Cytometry 1997, 28, 243–252. [Google Scholar] [CrossRef]
- Arendt, C.S.; Hochstrasser, M. Identification of the yeast 20S proteasome catalytic centers and subunit interactions required for active-site formation. Proc. Natl. Acad. Sci. USA 1997, 94, 7156–7161. [Google Scholar] [CrossRef] [Green Version]
- Budenholzer, L.; Cheng, C.L.; Li, Y.; Hochstrasser, M. Proteasome Structure and Assembly. J. Mol. Biol. 2017, 429, 3500–3524. [Google Scholar] [CrossRef] [PubMed]
- Altun, M.; Galardy, P.J.; Shringarpure, R.; Hideshima, T.; Leblanc, R.; Anderson, K.C.; Ploegh, H.L.; Kessler, B. Effects of PS-341 on the activity and composition of proteasomes in multiple myeloma cells. Cancer Res. 2005, 65, 7896–7901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, M.; Busch, M.; Friese-Hamim, M.; Crosignani, S.; Fuchss, T.; Musil, D.; Rohdich, F.; Sanderson, M.P.; Seenisamy, J.; Walter-Bausch, G.; et al. Structure-Based Optimization and Discovery of M3258, a Specific Inhibitor of the Immunoproteasome Subunit LMP7 (β5i). J. Med. Chem. 2021, 64, 10230–10245. [Google Scholar] [CrossRef] [PubMed]
- Noda, C.; Tanahashi, N.; Shimbara, N.; Hendil, K.B.; Tanaka, K. Tissue distribution of constitutive proteasomes, immunoproteasomes, and PA28 in rats. Biochem. Biophys. Res. Commun. 2000, 277, 348–354. [Google Scholar] [CrossRef]
- Haller, K.; Seki, K.; Wei, C.; Castelli, C.; Rivoltini, L.; Kiessling, R.; Levitskaya, J. Tumor necrosis factor-α induces coordinated changes in major histocompatibility class I presentation pathway, resulting in increased stability of class I complexes at the cell surface. Blood 2001, 98, 1108–1115. [Google Scholar] [CrossRef] [PubMed]
- Almond, J.B.; Cohen, G.M. The proteasome: A novel target for cancer chemotherapy. Leukemia 2002, 16, 433–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basler, M.; Mundt, S.; Bitzer, A.; Schmidt, C.; Groettrup, M. The immunoproteasome: A novel drug target for autoimmune diseases. Clin. Exp. Rheumatol. 2015, 33, 74–79. [Google Scholar]
- Limanaqi, F.; Biagioni, F.; Gaglione, A.; Busceti, C.L.; Fornai, F. A Sentinel in the Crosstalk Between the Nervous and Immune System: The (Immuno)-Proteasome. Front. Immunol. 2019, 10, 628. [Google Scholar] [CrossRef] [Green Version]
- Cengiz Seval, G.; Beksac, M. The safety of bortezomib for the treatment of multiple myeloma. Expert Opin. Drug Saf. 2018, 17, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Ettari, R.; Zappalà, M.; Grasso, S.; Musolino, C.; Innao, V.; Allegra, A. Immunoproteasome-selective and non-selective inhibitors: A promising approach for the treatment of multiple myeloma. Pharmacol. Ther. 2018, 182, 176–192. [Google Scholar] [CrossRef] [PubMed]
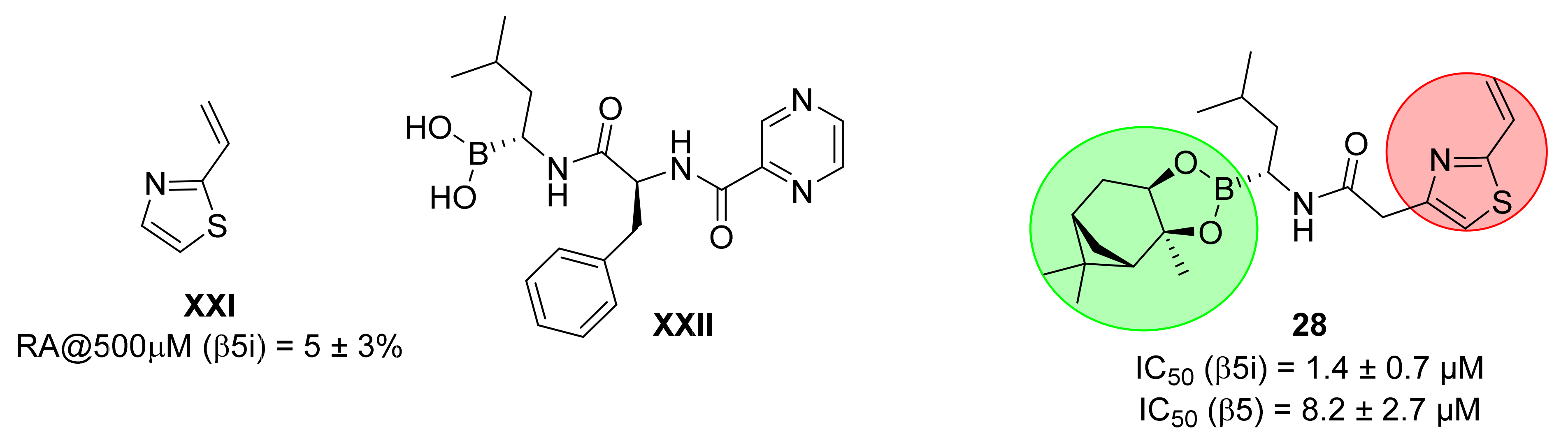
- Kollár, L.; Gobec, M.; Szilágyi, B.; Proj, M.; Knez, D.; Ábrányi-Balogh, P.; Petri, L.; Imre, T.; Bajusz, D.; Ferenczy, G.G.; et al. Discovery of selective fragment-sized immunoproteasome inhibitors. Eur. J. Med. Chem. 2021, 219, 113455. [Google Scholar] [CrossRef]
- Keeley, A.; Ábrányi-Balogh, P.; Keseru, G.M. Design and characterization of a heterocyclic electrophilic fragment library for the discovery of cysteine-targeted covalent inhibitors. MedChemComm 2019, 10, 263–267. [Google Scholar] [CrossRef] [Green Version]
- Petri, L.; Ábrányi-Balogh, P.; Varga, P.R.; Imre, T.; Keserű, G.M. Comparative reactivity analysis of small-molecule thiol surrogates. Bioorg. Med. Chem. 2020, 28, 115357. [Google Scholar] [CrossRef]
- JChem for Office; Version 21.8.0.865; ChemAxon. 2021. Available online: http://www.chemaxon.com (accessed on 10 October 2021).
- Berteotti, A.; Vacondio, F.; Lodola, A.; Bassi, M.; Silva, C.; Mor, M.; Cavalli, A. Predicting the reactivity of nitrile-carrying compounds with cysteine: A combined computational and experimental study. ACS Med. Chem. Lett. 2014, 5, 501–505. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M.; Tomasi, J. Geometry optimization of molecular structures in solution by the polarizable continuum model. J. Comput. Chem. 1998, 19, 404–417. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Hratchian, H.P.; Schlegel, H.B. Accurate reaction paths using a Hessian based predictor–corrector integrator. J. Chem. Phys. 2004, 120, 9918–9924. [Google Scholar] [CrossRef]
- Ladi, E.; Everett, C.; Stivala, C.E.; Daniels, B.E.; Durk, M.R.; Harris, S.F.; Huestis, M.; Purkey, H.; Staben, S.T.; Augustin, M.; et al. Design and evaluation of highly selective human immunoproteasome inhibitors reveal a compensatory process that preserves immune cell viability. J. Med. Chem. 2019, 62, 7032–7041. [Google Scholar] [CrossRef]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Zhu, K.; Borrelli, K.W.; Greenwood, J.R.; Day, T.; Abel, R.; Farid, R.S.; Harder, E. Docking Covalent Inhibitors: A Parameter Free Approach To Pose Prediction and Scoring. J. Chem. Inf. Model. 2014, 54, 1932–1940. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Zhou, M.; Panchuk-Voloshina, N. A one-step fluorometric method for the continuous measurement of monoamine oxidase activity. Anal. Biochem. 1997, 253, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Košak, U.; Knez, D.; Coquelle, N.; Brus, B.; Pišlar, A.; Nachon, F.; Brazzolotto, X.; Kos, J.; Colletier, J.-P.; Gobec, S. N-Propargylpiperidines with naphthalene-2-carboxamide or naphthalene-2-sulfonamide moieties: Potential multifunctional anti-Alzheimer’s agents. Bioorganic Med. Chem. 2017, 25, 633–645. [Google Scholar] [CrossRef]
- Von der Eltz, H.; Guder, H.-J.; Miihlegger, K. New Hydrolase Substrates. U.S. Patent 4,900,822, 13 February 1990. [Google Scholar]
- Verdonk, M.L.; Rees, D.C. Group Efficiency: A Guideline for Hits-to-Leads Chemistry. ChemMedChem 2008, 3, 1179–1180. [Google Scholar] [CrossRef] [PubMed]
- Hung, A.W.; Silvestre, H.L.; Wen, S.; George, G.P.C.; Boland, J.; Blundell, T.L.; Ciulli, A.; Abell, C. Optimization of Inhibitors of Mycobacterium tuberculosis Pantothenate Synthetase Based on Group Efficiency Analysis. ChemMedChem 2016, 11, 38–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groll, M.; Berkers, C.R.; Ploegh, H.L.; Ovaa, H. Crystal structure of the boronic acid-based proteasome inhibitor bortezomib in complex with the yeast 20S proteasome. Structure 2006, 14, 451–456. [Google Scholar] [CrossRef] [Green Version]
- Huber, E.M.; Heinemeyer, W.; Bruin, G.; Overkleeft, H.S.; Groll, M. A umanized yeast proteasome identifies unique binding modes of inhibitors for the immunosubunit β5i. EMBO J. 2016, 35, 2602–2613. [Google Scholar] [CrossRef] [Green Version]
- Verdoes, M.; Florea, B.I.; Menendez-Benito, V.; Maynard, C.J.; Witte, M.D.; van der Linden, W.A.; Nieuwendijk, A.M.V.D.; Hofmann, T.; Berkers, C.R.; van Leeuwen, F.; et al. A Fluorescent Broad-Spectrum Proteasome Inhibitor for Labeling Proteasomes In Vitro and In Vivo. Chem. Biol. 2006, 13, 1217–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Zhao, X.; Zhu, X.; Wu, G.; Li, Y.; Ma, Y.; Yuan, Y.; Yang, J.; Hu, Y.; Ai, L.; et al. Design, synthesis, biological evaluation, and Structure-Activity Relationship (SAR) discussion of dipeptidyl boronate proteasome inhibitors, Part I: Comprehensive understanding of the SAR of α-amino acid boronates. J. Med. Chem. 2009, 52, 4192–4199. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Adwal, A.; Turner, A.G.; Callen, D.F.; Abell, A.D. New Peptidomimetic Boronates for Selective Inhibition of the Chymotrypsin-like Activity of the 26S Proteasome. ACS Med. Chem. Lett. 2016, 7, 1039–1043. [Google Scholar] [CrossRef] [Green Version]
- Berkers, C.R.; Verdoes, M.; Lichtman, E.; Fiebiger, E.; Kessler, B.; Anderson, K.C.; Ploegh, H.L.; Ovaa, H.; Galardy, P.J. Activity probe for in vivo profiling of the specificity of proteasome inhibitor bortezomib. Nat. Methods 2005, 2, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Feaster, S.R.; Quinn, D.M. Mechanism-Based Inhibitors of Mammalian Cholesterol Esterase. Methods Enzymol. 1997, 286, 231–252. [Google Scholar] [CrossRef] [PubMed]
- Kettner, C.A.; Shenvi, A.B. Inhibition of the serine proteases leukocyte elastase, pancreatic elastase, cathepsin G, and chymotrypsin by peptide boronic acids. J. Biol. Chem. 1984, 259, 15106–15114. [Google Scholar] [CrossRef]
- Quach, D.; Tang, G.; Anantharajan, J.; Baburajendran, N.; Poulsen, A.; Wee, J.L.K.; Retna, P.; Li, R.; Liu, B.; Tee, D.H.Y.; et al. Strategic Design of Catalytic Lysine-Targeting Reversible Covalent BCR-ABL Inhibitors. Angew. Chem. Int. Ed. 2021, 60, 17131–17137. [Google Scholar] [CrossRef]
- Alves, L.; Santos, D.A.; Cendron, R.; Rocho, F.R.; Matos, T.K.; Leitão, A.; Montanari, C.A. Nitrile-based peptoids as cysteine protease inhibitors. Bioorganic Med. Chem. 2021, 41, 116211. [Google Scholar] [CrossRef]
- Petri, L.; Ábrányi-Balogh, P.; Tímea, I.; Pálfy, G.; Perczel, A.; Knez, D.; Hrast, M.; Gobec, M.; Sosič, I.; Nyíri, K.; et al. Assessment of Tractable Cysteines for Covalent Targeting by Screening Covalent Fragments. ChemBioChem 2021, 22, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Muchamuel, T.; Basler, M.; Aujay, M.A.; Suzuki, E.; Kalim, K.W.; Lauer, C.; Sylvain, C.; Ring, E.R.; Shields, J.; Jiang, J.; et al. A selective inhibitor of the immunoproteasome subunit LMP7 blocks cytokine production and attenuates progression of experimental arthritis. Nat. Med. 2009, 15, 781–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Britton, M.; Lucas, M.M.; Downey, S.; Screen, M.; Pletnev, A.A.; Verdoes, M.; Tokhunts, R.A.; Amir, O.; Goddard, A.L.; Pelphrey, P.M.; et al. Selective Inhibitor of Proteasome’s Caspase-like Sites Sensitizes Cells to Specific Inhibition of Chymotrypsin-like Sites. Chem. Biol. 2009, 16, 1278–1289. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
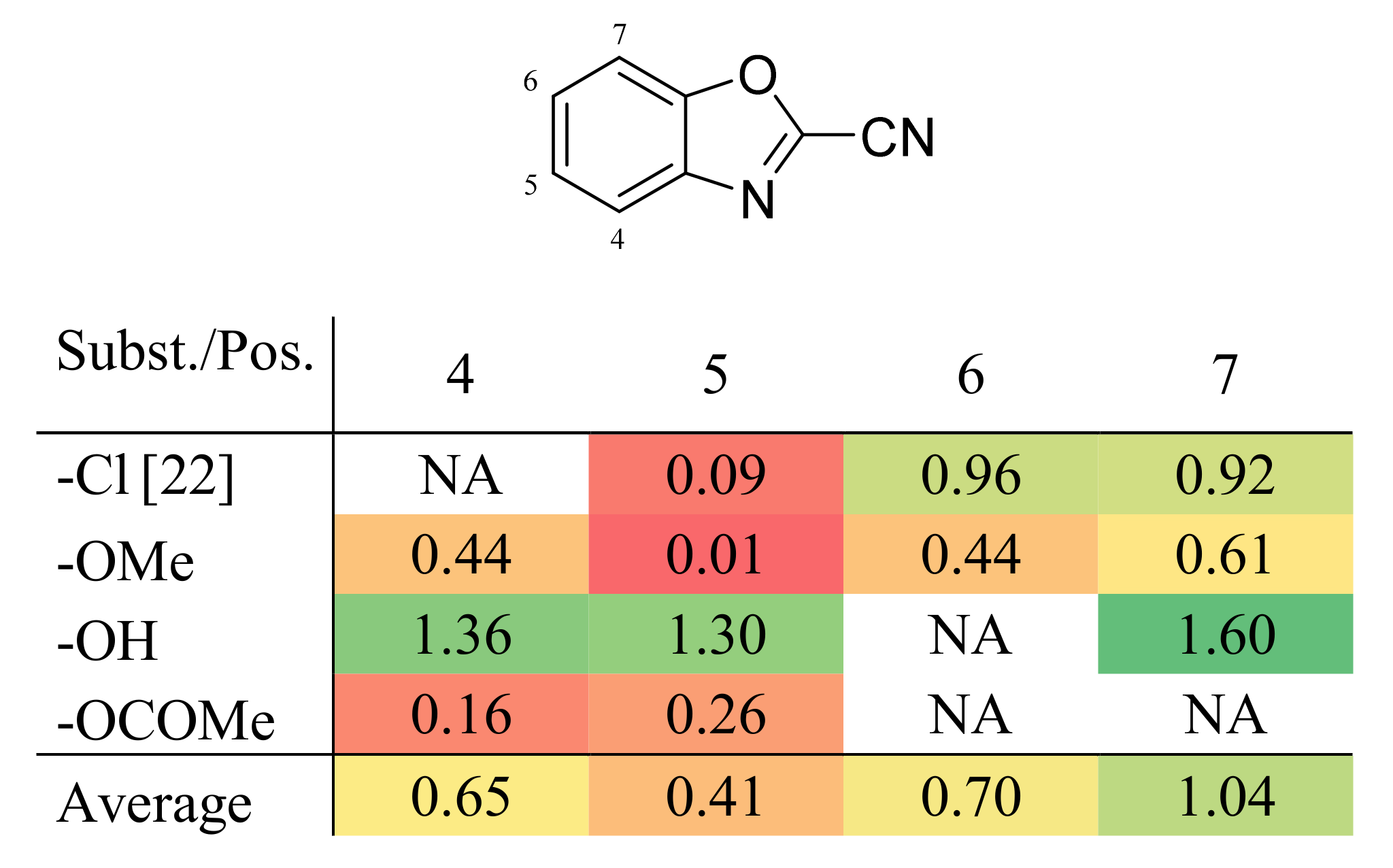
| Substituent | Position | Label | IC50 (µM) a |
|---|---|---|---|
| - | - | 1 | 83 ± 6.0 |
| -Cl | 5 | 2 | 67 ± 11 |
| 6 | 3 | 9.1 ± 4.5 | |
| 7 | 4 | 10 ± 4.6 |
| Substituent | Position | Cpd | Stability in Buffer | RA (%) at 10 µM or IC50 (µM) a |
|---|---|---|---|---|
| -Me b | 6 | 5 | low absorbance c/stable d | 81 ± 4% |
| -OMe | 4 | 6 | stable c,d | 11 ± 7 µM |
| 5 | 7 | stable c,d | 84 ± 11% | |
| 6 | 8 | intermediate c/stable d | 11 ± 6 µM e | |
| 7 | 9 | stable c,d | 20 ± 6% f | |
| -OH | 4 | 10 | stable c | 3.6 ± 2.8 µM |
| 5 | 11 | stable c | 4.2 ± 2.5 µM | |
| 6 | 12 | intermediate c | ND g | |
| 7 | 13 | stable c | 2.1 ± 1.2 µM | |
| -OCOMe | 4 | 14 | stable c | 19 ± 9 µM |
| 5 | 15 | stable c | 7.4 ± 1.7 µM | |
| -NO2 | 4 | 16 | intermediatec/unstable d | NA h |
| 5 | 17 | NA i | NA i | |
| 6 | 18 | unstable c | NA h | |
| 7 | 19 | stable c | 82 ± 2% | |
| -NH2 | 4 | 20 | stable c | 94 ± 7% |
| 5 | 21 | stable c | 81 ± 3% | |
| 6 | 22 | stable c,d | ND g | |
| 7 | 23 | stable c | 98 ± 4% | |
| -NHCOPh | 4 | 24 | stable c | 74 ± 17% |
| 5 | 25 | stable c,d | ND g | |
| 6 | 26 | unstable c/stable d | NA h | |
| 7 | 27 | stable c | 103 ± 8% |
| Cpd | Residual Activity or IC50 (µM) According to CP, Subunit, and Substrate | |||||
|---|---|---|---|---|---|---|
| iCP | cCP | |||||
| β5i (IC50 [μM]) a | β2i (RA [%]) a | β1i (IC50 [μM]) a | β5 (RA [%] or IC50 [μM]) a | β2 (RA [%]) a | β1 (RA [%]) a | |
| Suc-LLVY-AMC | Boc-LRR-AMC | Ac-PAL-AMC | Suc-LLVY-AMC | Boc-LRR-AMC | Ac-nLPnLD-AMC | |
| 6 | 11 ± 7 | NA | 78 | 63% c | NA | 82% |
| 8 | 11 ± 6 b | NA | 69 | 74% b | NA | NA |
| 10 | 3.6 ± 2.8 | NA | NA | 30 ± 12 | NA | NA |
| 11 | 4.2 ± 2.5 | NA | NA | 53% c | NA | NA |
| 13 | 2.1 ± 1.2 | 83% | NA | 11 ± 9 | 71% | 76% |
| 14 | 19 ± 9 | NA | NA | 56% c | NA | NA |
| 15 | 7.4 ± 1.7 | NA | NA | 55% c | NA | NA |
 | |||||||
|---|---|---|---|---|---|---|---|
| n | X | Y | R1, R2 | Cpd | Stability in Buffer | RA (%) at 10 µM or IC50 (µM) a | |
| β5i | β5 | ||||||
| 1 | - | - | pinanediol ester | 29 | unstable b,c | ND d | ND d |
| 2 | - | - | pinanediol ester | 30 | stable b,c | 0.6 ± 0.0 µM | 0.7 ± 0.1 µM |
| 3 | - | - | pinanediol ester | 31 | stable b,c | 5.7 ± 0.9 µM | 13 ± 4 µM |
| 4 | - | - | pinanediol ester | 32 | intermediate b/stable c | 13 ± 2 µM e | 1.6 ± 0.8 µM e |
| 1 | - | - | boronic acid | 33 | unstable b,c | ND d | ND d |
| 2 | - | - | boronic acid | 34 | low absorbance b | 2.2 ± 0.6 µM | 1.8 ± 0.4 µM |
| 3 | - | - | boronic acid | 35 | stable b | 7.9 ± 0.5 µM | 16 ± 8 µM |
| 4 | - | - | boronic acid | 36 | stable b | 59 ± 7 µM | 6.4 ± 2.6 µM |
| 2 | -Cl | - | pinanediol ester | 37 | intermediate b/stable c | 1.6 ± 1.3 µM e | 14 ± 1% e |
| 2 | - | -Cl | pinanediol ester | 38 | stable b,c | 0.6 ± 0.1 µM | 2.0 ± 0.7 µM |
| Structure | Cpd | Stability in Buffer | IC50 (µM) a | |
|---|---|---|---|---|
| β5i | β5 | |||
 | 39 | stable b,c | 3.1 ± 0.3 µM | 3.6 ± 1.1 µM |
 | 40 | stable b | 2.4 ± 1.0 µM | 2.0 ± 0.7 µM |
| Cpd | Residual Activity or IC50 (µM) According to CP, Subunit, and Substrate | |||
|---|---|---|---|---|
| iCP | cCP | |||
| β2i (RA [%]) a | β1i (IC50 [μM]) a | β2 (RA [%]) a | β1 (IC50 [μM]) a | |
| Boc-LRR-AMC | Ac-PAL-AMC | Boc-LRR-AMC | Ac-nLPnLD-AMC | |
| 28 | NA | 2.2 ± 0.1 | NA | 4.8 ± 0.2 |
| 30 | NA | 1.2 ± 0.1 | NA | 0.4 ± 0.0 |
| 31 | NA | 6.0 ± 3.2 | NA | 8.4 ± 1.6 |
| 34 | NA | 2.8 ± 0.5 | NA | 3.2 ± 0.2 |
| 35 | NA | 9.4 ± 1.9 | NA | 13 ± 2 |
| 36 | NA | 4.9 ± 0.4 | NA | 23 ± 6 |
| 38 | NA | 1.9 ± 0.0 | NA | 5.0 ± 0.1 |
| 39 | NA | 1.3 ± 0.0 | NA | 2.9 ± 0.4 |
| 40 | NA | 2.1 ± 0.1 | NA | 3.7 ± 0.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kollár, L.; Gobec, M.; Proj, M.; Smrdel, L.; Knez, D.; Imre, T.; Gömöry, Á.; Petri, L.; Ábrányi-Balogh, P.; Csányi, D.; et al. Fragment-Sized and Bidentate (Immuno)Proteasome Inhibitors Derived from Cysteine and Threonine Targeting Warheads. Cells 2021, 10, 3431. https://doi.org/10.3390/cells10123431
Kollár L, Gobec M, Proj M, Smrdel L, Knez D, Imre T, Gömöry Á, Petri L, Ábrányi-Balogh P, Csányi D, et al. Fragment-Sized and Bidentate (Immuno)Proteasome Inhibitors Derived from Cysteine and Threonine Targeting Warheads. Cells. 2021; 10(12):3431. https://doi.org/10.3390/cells10123431
Chicago/Turabian StyleKollár, Levente, Martina Gobec, Matic Proj, Lara Smrdel, Damijan Knez, Tímea Imre, Ágnes Gömöry, László Petri, Péter Ábrányi-Balogh, Dorottya Csányi, and et al. 2021. "Fragment-Sized and Bidentate (Immuno)Proteasome Inhibitors Derived from Cysteine and Threonine Targeting Warheads" Cells 10, no. 12: 3431. https://doi.org/10.3390/cells10123431
APA StyleKollár, L., Gobec, M., Proj, M., Smrdel, L., Knez, D., Imre, T., Gömöry, Á., Petri, L., Ábrányi-Balogh, P., Csányi, D., Ferenczy, G. G., Gobec, S., Sosič, I., & Keserű, G. M. (2021). Fragment-Sized and Bidentate (Immuno)Proteasome Inhibitors Derived from Cysteine and Threonine Targeting Warheads. Cells, 10(12), 3431. https://doi.org/10.3390/cells10123431