
Ion Channels and Pumps in Autophagy: A Reciprocal Relationship

 , ,
, , 
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Ion Channels and Autophagy
2.1. K+ Channels
2.2. Ca2+ Channels
2.2.1. Sarco/Endoplasmic Ca2+ ATPase
2.2.2. Inositol Triphosphate Receptor Ca2+ Channel
2.2.3. Transient Receptor Potential Mucolipin Channels (TRPMLs)
2.2.4. Two-Pore Channels TPC1 and TPC2
2.2.5. Store-Operated Calcium Channels (SOCCs)
2.3. Vacuolar H+-ATPase
3. Regulation of Lysosomal Ion Channels
3.1. Regulation by Lipids
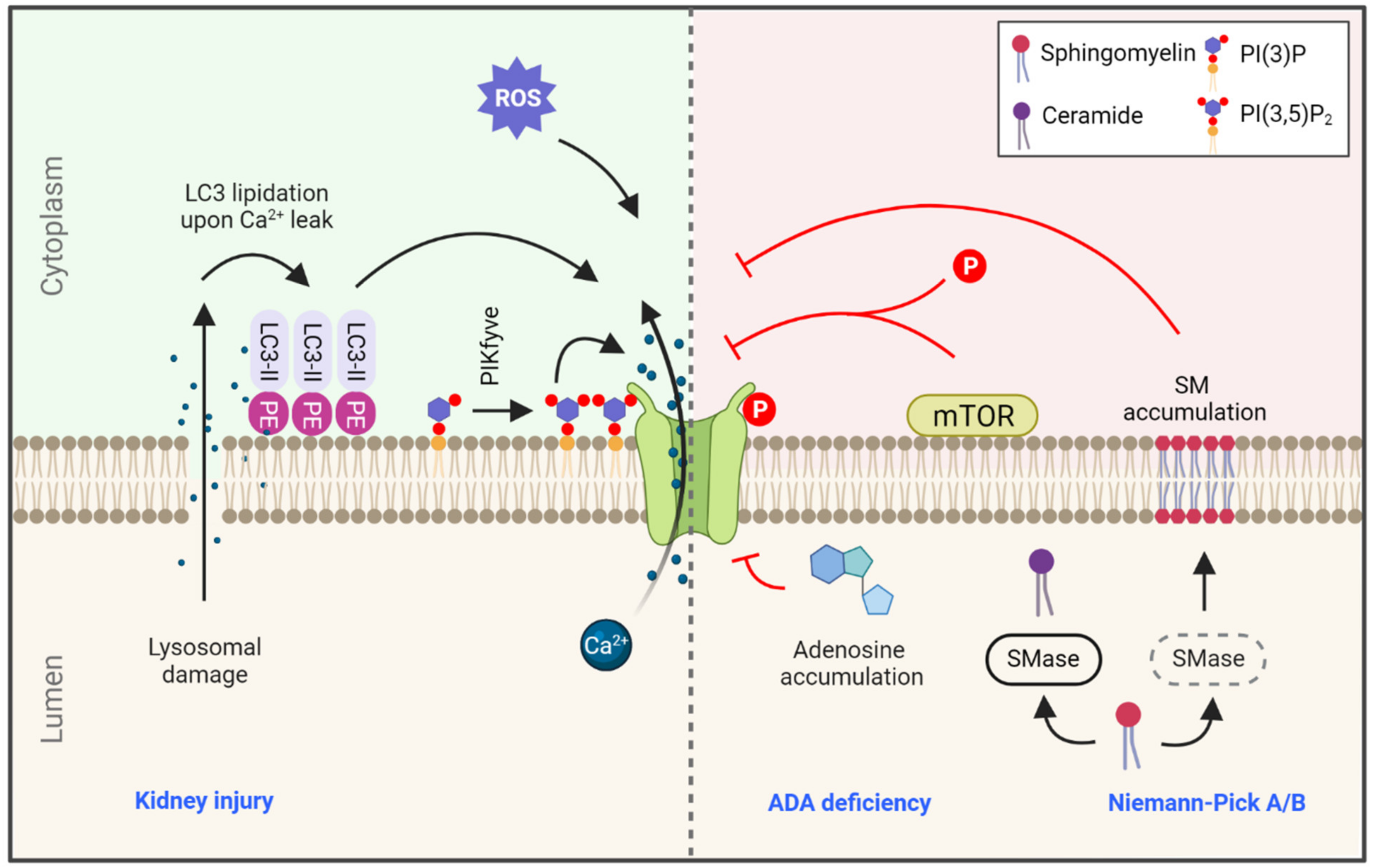
3.1.1. PI(3,5)P2 and Sphingomyelin Regulation of TRPML1 Function
3.1.2. TRPML1 Regulation by Lipidated LC3 upon Injury
3.2. Regulation by mTOR and Calcium
3.3. Regulation by Small Molecules
4. Interrelationship between Calcium and V-ATPase during Autophagy
5. Lysosomal Ion Channels in Health and Disease
6. Conclusions and Perspectives
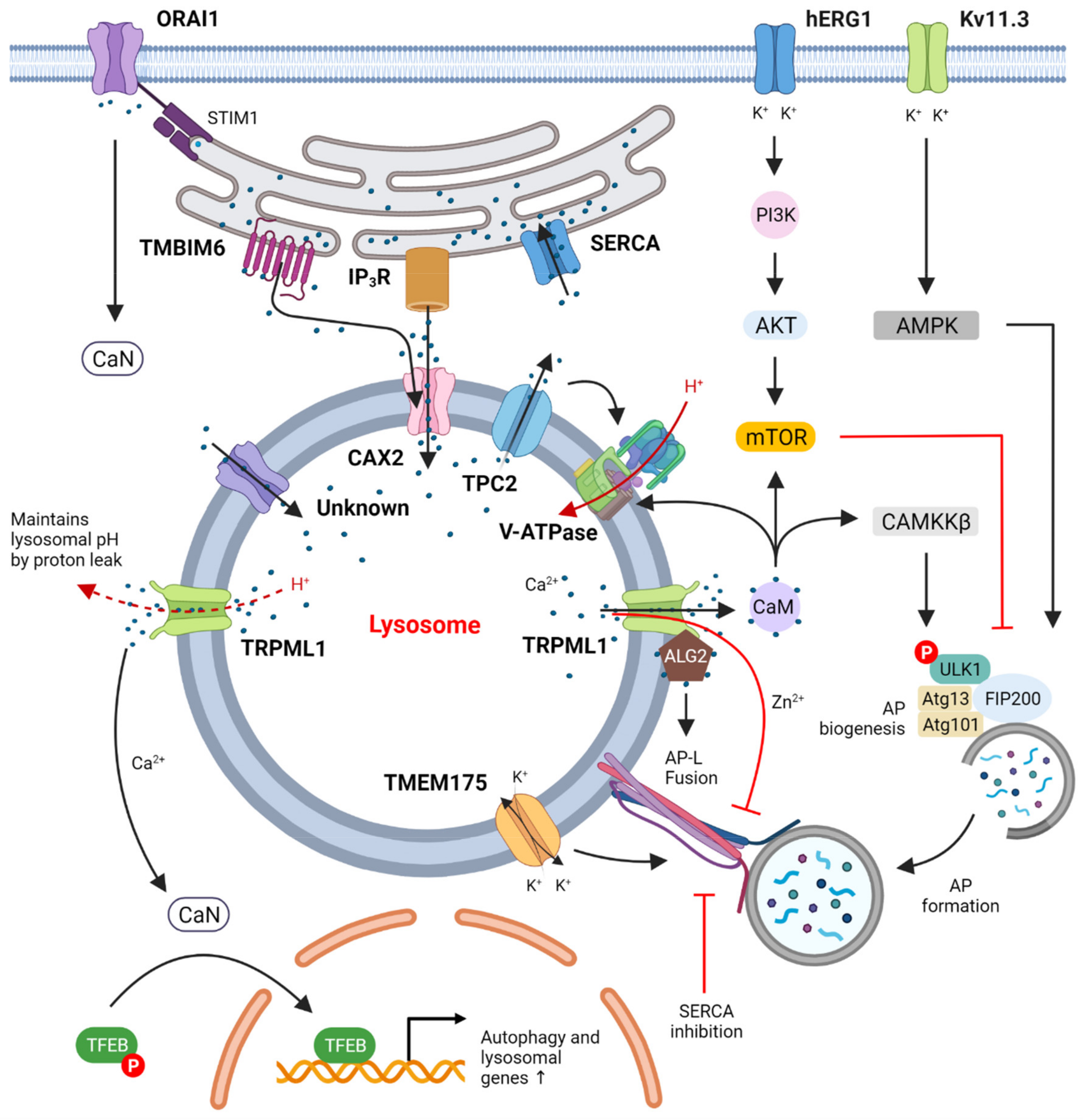
- It appears that defects of ion homeostasis either in the lysosome or in cytosol influence autophagic flux. The multitude of roles of TRPML1 is especially surprising in this regard: Ca2+ and Zn2+ transport by TRPML1 seem to have opposite effects on autophagic progression. This might indicate that Ca2+ and Zn2+ efflux by TRPML1 is context-sensitive—e.g., zinc efflux being prominent in proliferating tumor cells vs. calcium efflux being required for completion of basal autophagy. Additionally, TRPML1 functions as a H+ leaking channel to maintain physiological pH[lys], and absence of this proton permeability function likely contributes to over-acidified autolysosomes containing undegraded cargo in Trpml mutant fly tissue. Whether this proton leakage is coordinated with V-ATPase function during autophagy is an intriguing question, and it could represent a Ca2+ independent layer of V-ATPase regulation by TRPML1.
- ER-lysosome contact sites are especially important for lysosomal calcium replenishment. How dynamic these contact sites are during nutrient replete versus nutrient depleted conditions remains to be established. Additionally, it will be important to study the relationship of ER microdomains and exit sites (which are often sites of autophagosome biogenesis, [161]) during autophagosome formation, ER-lysosome contact site maintenance and autophagosome-lysosome fusion events.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AA | amino acid |
| ADA | Adenosine deaminase |
| AKT | three serine/threonine-specific protein kinases |
| ALG-2 | apoptosis-linked gene-2 protein |
| ALR | aumiddlehagic lysosome reformation |
| ALSAM2 | amyotrophic lateral sclerosisM2 proton channel, influenza virus A |
| AMPK | AMP-activated protein kinase |
| Arl8 | Arf like protein 8 |
| ATF6 | activating transcription factor 6 |
| Atg | aumiddlehagy genes |
| ATP | adenosine triphosphate |
| CaM | calmodulin |
| CAMKKβ | Ca2+/calmodulin dependent protein kinase kinase-beta |
| CaN | calcineurin |
| CAPS1/CADPS | calcium dependent activator protein for secretion |
| Cav2.2 | (N-type) voltage-gated Ca2+ channels |
| CFTR | cystic fibrosis transmembrane conductance regulator |
| ChT | chloramine T |
| CLA | clarithromycin |
| CPP | calcium phosphate precipitate |
| DENV2 | dengue-virus, strain 2 |
| DEPTOR | DEP- domain-containing mTOR- interacting protein |
| DFCP1 | double FYVe domain-containing protein 1 |
| EDME | estradiol methyl ether |
| ENT3 | equilibrative nucleoside transporter 3 |
| ER | endoplasmic reticulum |
| ERK1/2 | extracellular signal-regulated protein kinase |
| F-ATPase | F-type ATPase/ATP synthase |
| FIP200 | FAK family–interacting protein of 200 kD |
| FOXO3 | forkhead box protein O3 |
| GRP75 | glucose-regulated protein 75 |
| GTPase | guanosine triphosphate enzyme |
| H2O2 | hydrogen peroxide |
| hERG1 | Ether-A-Go-Go-Related Gene Potassium Channel 1 |
| HOPS | homotypic fusion and vacuole protein sorting |
| IP3R | inositol trisphosphate receptor |
| IRE1 | inositol-requiring enzyme 1 |
| JNK | c-Jun N-terminal kinases |
| KEL | K+-selective channel |
| KO | knockout |
| Kv11.3 | pore-forming (alpha) subunit of voltage-gated potassium channel |
| L-BMAA | neurotoxin β-N-methylamino-L-alanine |
| LC3 | microtubule-associated light chain-3 |
| LPS | lipopolysaccharide |
| mASMs | mouse aortic smooth muscle cells |
| MCOLN1 | mucolipin-1 |
| ML-SA1 | mucolipin specific agonist, 1 |
| ML2-SA | mucolipin2 specific agonist |
| MLIV | mucolipidosis type IV |
| mLST8 | mammalian lethal with SEC13 protein 8 |
| MTMRs | myotubularin-related proteins |
| mTOR | mechanistic target of rapamycin |
| mtROS | mitochondrial reactive oxygen species |
| NAADP | nicotinic acid adenine dinucleotide phosphate |
| NH4Cl | ammonium chloride |
| ORAI1 | calcium release-activated calcium channel protein 1 |
| p38 | mitogen-activated protein kinase p38 beta |
| p62 | sequestosome-1 |
| PE | phosphatidylethanolamine |
| PERK | protein kinase (PKR)-like ER kinase |
| PI(3,5)P2 | phosphatidylinositol 3,5-bisphosphate |
| PI3K-III | class III phosphatidylinositol 3-kinase complex |
| PI3P | phosphatidylinositol 3-phosphate |
| PIKFYVE | phosphoinositide kinase, FYVE-Type Zinc Finger Containing |
| PRAS40 | raptor recruits proline-rich AKT substrate 40 kDa |
| PROTOR1/2 | protein associated with rictor 1 or 2 |
| PSEN2 | presenilin 2 |
| Rab7 | Ras-related protein7 |
| Raptor | regulatory-associated protein of mTOR |
| RAVE | regulator of ATPase of vacuoles and endosomes |
| Rbcn | rabconnectins |
| ROS | reactive oxygen species |
| RYR | ryanodine receptor |
| SCID | severe combined immunodeficiency |
| SERCA | sarco/endoplasmic reticulum calcium ATPase |
| SM | sphingomyelin |
| SMase | sphingomelinase |
| SNAP25 | synaptosomal-associated protein 25 |
| SNAP29 | synaptosomal-associated protein 29 |
| SNARE | soluble N-ethylmaleimide-sensitive factor attachment protein receptor |
| SOCC | store-operated calcium channels |
| SOCE | store-operated Ca2+ entry |
| STIM1 | stromal interaction molecule 1 precursor |
| TBHP | t-butyl hydroperoxide |
| TFEB | transcription factor EB |
| TG | thapsigargin |
| TMBIM6 | transmembrane Bax inhibitor motif-containing 6 |
| TMEM175 | transmembrane protein 175 |
| TOM70 | component of the TOM (translocase of outer membrane) complex |
| TPC2 | two pore channel 2 |
| CAX2 | vacuolar cation/proton exchanger 2 |
| TRPM2 | transient receptor potential cation channel subfamily M member 2 |
| TRPML | transient receptor potential mucolipin channel |
| TSC2 | tuberous sclerosis complex-2/tuberin |
| ULK1 | unc-51 like aumiddlehagy activating kinase 1 |
| V-ATPase | vacuolar-ATPase |
| VacA | vacuolating toxin a |
| VAMP2 | synaptobrevin-2/ vesicle-associated membrane protein 2 |
| VAMP7 | vesicle-associated membrane protein 7 |
| VAMP8 | vesicle-associated membrane protein 8 |
| VDAC1 | voltage-dependent anion-selective channel protein 1 |
| VhaAC39-1 | Drosophila melanogaster vacuolar H+ ATPase AC39 subunit 1 |
| VMP1 | vacuolar membrane protein-1 |
| VPS3 | vacuolar protein sorting 34 |
| ZIKV | Zika-virus |
References
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef]
- Tekirdag, K.; Cuervo, A.M. Chaperone-mediated autophagy and endosomal microautophagy: Joint by a chaperone. J. Biol. Chem. 2018, 293, 5414–5424. [Google Scholar] [CrossRef] [Green Version]
- Abdrakhmanov, A.; Gogvadze, V.; Zhivotovsky, B. To Eat or to Die: Deciphering Selective Forms of Autophagy. Trends Biochem. Sci. 2020, 45, 347–364. [Google Scholar] [CrossRef]
- Johansen, T.; Lamark, T. Selective Autophagy: ATG8 Family Proteins, LIR Motifs and Cargo Receptors. J. Mol. Biol. 2020, 432, 80–103. [Google Scholar] [CrossRef]
- Itakura, E.; Kishi-Itakura, C.; Mizushima, N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 2012, 151, 1256–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegedus, K.; Takats, S.; Boda, A.; Jipa, A.; Nagy, P.; Varga, K.; Kovacs, A.L.; Juhasz, G. The Ccz1-Mon1-Rab7 module and Rab5 control distinct steps of autophagy. Mol. Biol. Cell 2016, 27, 3132–3142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boda, A.; Lorincz, P.; Takats, S.; Csizmadia, T.; Toth, S.; Kovacs, A.L.; Juhasz, G. Drosophila Arl8 is a general positive regulator of lysosomal fusion events. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Rydzewski, N.; Hider, A.; Zhang, X.; Yang, J.; Wang, W.; Gao, Q.; Cheng, X.; Xu, H. A molecular mechanism to regulate lysosome motility for lysosome positioning and tubulation. Nat. Cell Biol. 2016, 18, 404–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takats, S.; Pircs, K.; Nagy, P.; Varga, A.; Karpati, M.; Hegedus, K.; Kramer, H.; Kovacs, A.L.; Sass, M.; Juhasz, G. Interaction of the HOPS complex with Syntaxin 17 mediates autophagosome clearance in Drosophila. Mol. Biol. Cell 2014, 25, 1338–1354. [Google Scholar] [CrossRef]
- Takats, S.; Nagy, P.; Varga, A.; Pircs, K.; Karpati, M.; Varga, K.; Kovacs, A.L.; Hegedus, K.; Juhasz, G. Autophagosomal Syntaxin17-dependent lysosomal degradation maintains neuronal function in Drosophila. J. Cell Biol. 2013, 201, 531–539. [Google Scholar] [CrossRef] [Green Version]
- Takats, S.; Glatz, G.; Szenci, G.; Boda, A.; Horvath, G.V.; Hegedus, K.; Kovacs, A.L.; Juhasz, G. Non-canonical role of the SNARE protein Ykt6 in autophagosome-lysosome fusion. PLoS Genet. 2018, 14, e1007359. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Xu, T.; Guan, K.L. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003, 17, 1829–1834. [Google Scholar] [CrossRef] [Green Version]
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008, 320, 1496–1501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013, 15, 741–750. [Google Scholar] [CrossRef] [Green Version]
- Polson, H.E.; de Lartigue, J.; Rigden, D.J.; Reedijk, M.; Urbe, S.; Clague, M.J.; Tooze, S.A. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy 2010, 6, 506–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dooley, H.C.; Razi, M.; Polson, H.E.; Girardin, S.E.; Wilson, M.I.; Tooze, S.A. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol. Cell 2014, 55, 238–252. [Google Scholar] [CrossRef] [Green Version]
- Atakpa, P.; Thillaiappan, N.B.; Mataragka, S.; Prole, D.L.; Taylor, C.W. IP3 Receptors Preferentially Associate with ER-Lysosome Contact Sites and Selectively Deliver Ca(2+) to Lysosomes. Cell Rep. 2018, 25, 3180–3193. [Google Scholar] [CrossRef] [Green Version]
- Garrity, A.G.; Wang, W.; Collier, C.M.; Levey, S.A.; Gao, Q.; Xu, H. The endoplasmic reticulum, not the pH gradient, drives calcium refilling of lysosomes. Elife 2016, 5, e15887. [Google Scholar] [CrossRef] [PubMed]
- Scotto Rosato, A.; Montefusco, S.; Soldati, C.; di Paola, S.; Capuozzo, A.; Monfregola, J.; Polishchuk, E.; Amabile, A.; Grimm, C.; Lombardo, A.; et al. TRPML1 links lysosomal calcium to autophagosome biogenesis through the activation of the CaMKKbeta/VPS34 pathway. Nat. Commun. 2019, 10, 5630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, P.H.; Duann, P.; Komazaki, S.; Park, K.H.; Li, H.; Sun, M.; Sermersheim, M.; Gumpper, K.; Parrington, J.; Galione, A.; et al. Lysosomal two-pore channel subtype 2 (TPC2) regulates skeletal muscle autophagic signaling. J. Biol. Chem. 2015, 290, 3377–3389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christensen, K.A.; Myers, J.T.; Swanson, J.A. pH-dependent regulation of lysosomal calcium in macrophages. J. Cell Sci. 2002, 115, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, B.E.; Huynh, K.K.; Brodovitch, A.; Jabs, S.; Stauber, T.; Jentsch, T.J.; Grinstein, S. A cation counterflux supports lysosomal acidification. J. Cell Biol. 2010, 189, 1171–1186. [Google Scholar] [CrossRef] [Green Version]
- Cao, Q.; Zhong, X.Z.; Zou, Y.; Zhang, Z.; Toro, L.; Dong, X.P. BK Channels Alleviate Lysosomal Storage Diseases by Providing Positive Feedback Regulation of Lysosomal Ca2+ Release. Dev. Cell 2015, 33, 427–441. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhang, X.; Dong, X.P.; Samie, M.; Li, X.; Cheng, X.; Goschka, A.; Shen, D.; Zhou, Y.; Harlow, J.; et al. TPC proteins are phosphoinositide- activated sodium-selective ion channels in endosomes and lysosomes. Cell 2012, 151, 372–383. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Zhang, X.; Gao, Q.; Lawas, M.; Yu, L.; Cheng, X.; Gu, M.; Sahoo, N.; Li, X.; Li, P.; et al. A voltage-dependent K(+) channel in the lysosome is required for refilling lysosomal Ca(2+) stores. J. Cell Biol. 2017, 216, 1715–1730. [Google Scholar] [CrossRef] [Green Version]
- Koivusalo, M.; Steinberg, B.E.; Mason, D.; Grinstein, S. In situ measurement of the electrical potential across the lysosomal membrane using FRET. Traffic 2011, 12, 972–982. [Google Scholar] [CrossRef]
- Rangarajan, N.; Kapoor, I.; Li, S.; Drossopoulos, P.; White, K.K.; Madden, V.J.; Dohlman, H.G. Potassium starvation induces autophagy in yeast. J. Biol. Chem. 2020, 295, 14189–14202. [Google Scholar] [CrossRef]
- Allen, D.H.; Lepple-Wienhues, A.; Cahalan, M.D. Ion channel phenotype of melanoma cell lines. J. Membr. Biol. 1997, 155, 27–34. [Google Scholar] [CrossRef]
- Hernandez, L.; Park, K.H.; Cai, S.Q.; Qin, L.; Partridge, N.; Sesti, F. The antiproliferative role of ERG K+ channels in rat osteoblastic cells. Cell Biochem. Biophys. 2007, 47, 199–208. [Google Scholar] [CrossRef]
- Lansu, K.; Gentile, S. Potassium channel activation inhibits proliferation of breast cancer cells by activating a senescence program. Cell Death Dis. 2013, 4, e652. [Google Scholar] [CrossRef]
- Breuer, E.K.; Fukushiro-Lopes, D.; Dalheim, A.; Burnette, M.; Zartman, J.; Kaja, S.; Wells, C.; Campo, L.; Curtis, K.J.; Romero-Moreno, R.; et al. Potassium channel activity controls breast cancer metastasis by affecting beta-catenin signaling. Cell Death Dis. 2019, 10, 180. [Google Scholar] [CrossRef] [PubMed]
- Perez-Neut, M.; Haar, L.; Rao, V.; Santha, S.; Lansu, K.; Rana, B.; Jones, W.K.; Gentile, S. Activation of hERG3 channel stimulates autophagy and promotes cellular senescence in melanoma. Oncotarget 2016, 7, 21991–22004. [Google Scholar] [CrossRef]
- Lastraioli, E.; Lottini, T.; Bencini, L.; Bernini, M.; Arcangeli, A. hERG1 Potassium Channels: Novel Biomarkers in Human Solid Cancers. Biomed. Res. Int. 2015, 2015, 896432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petroni, G.; Bagni, G.; Iorio, J.; Duranti, C.; Lottini, T.; Stefanini, M.; Kragol, G.; Becchetti, A.; Arcangeli, A. Clarithromycin inhibits autophagy in colorectal cancer by regulating the hERG1 potassium channel interaction with PI3K. Cell Death Dis. 2020, 11, 161. [Google Scholar] [CrossRef] [Green Version]
- Cang, C.; Aranda, K.; Seo, Y.J.; Gasnier, B.; Ren, D. TMEM175 Is an Organelle K(+) Channel Regulating Lysosomal Function. Cell 2015, 162, 1101–1112. [Google Scholar] [CrossRef] [Green Version]
- Jinn, S.; Drolet, R.E.; Cramer, P.E.; Wong, A.H.; Toolan, D.M.; Gretzula, C.A.; Voleti, B.; Vassileva, G.; Disa, J.; Tadin-Strapps, M.; et al. TMEM175 deficiency impairs lysosomal and mitochondrial function and increases alpha-synuclein aggregation. Proc. Natl. Acad. Sci. USA 2017, 114, 2389–2394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.H.; Shih, Y.L.; Ko, W.C.; Wei, Y.H.; Shih, C.M. Cadmium-induced autophagy and apoptosis are mediated by a calcium signaling pathway. Cell Mol. Life Sci. 2008, 65, 3640–3652. [Google Scholar] [CrossRef]
- Yorimitsu, T.; Nair, U.; Yang, Z.; Klionsky, D.J. Endoplasmic reticulum stress triggers autophagy. J. Biol. Chem. 2006, 281, 30299–30304. [Google Scholar] [CrossRef] [Green Version]
- Criollo, A.; Maiuri, M.C.; Tasdemir, E.; Vitale, I.; Fiebig, A.A.; Andrews, D.; Molgo, J.; Diaz, J.; Lavandero, S.; Harper, F.; et al. Regulation of autophagy by the inositol trisphosphate receptor. Cell Death Differ. 2007, 14, 1029–1039. [Google Scholar] [CrossRef] [PubMed]
- Talloczy, Z.; Jiang, W.; Virgin, H.W.T.; Leib, D.A.; Scheuner, D.; Kaufman, R.J.; Eskelinen, E.L.; Levine, B. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc. Natl. Acad. Sci. USA 2002, 99, 190–195. [Google Scholar] [CrossRef] [Green Version]
- Hoyer-Hansen, M.; Bastholm, L.; Szyniarowski, P.; Campanella, M.; Szabadkai, G.; Farkas, T.; Bianchi, K.; Fehrenbacher, N.; Elling, F.; Rizzuto, R.; et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol. Cell 2007, 25, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, M.; Chen, D.; Gao, W.; Guan, J.L.; Komatsu, M.; Yin, X.M. Autophagy induced by calcium phosphate precipitates involves endoplasmic reticulum membranes in autophagosome biogenesis. PLoS ONE 2012, 7, e52347. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Liu, H.; Du, X.; Petlulu, P.; Chen, X.; Wang, R.; Zhang, S.; Tian, Z.; Shi, L.; Guo, H.; et al. IRE1 and CaMKKbeta pathways to reveal the mechanism involved in microcystin-LR-induced autophagy in mouse ovarian cells. Food Chem. Toxicol. 2021, 147, 111911. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, S.; Kakizaki, M.; Hirose, Y.; Ishikawa, Y.; Funato, H.; Yanagisawa, M.; Yu, Y.; Liu, Q. Orexin/hypocretin activates mTOR complex 1 (mTORC1) via an Erk/Akt-independent and calcium-stimulated lysosome v-ATPase pathway. J. Biol. Chem. 2014, 289, 31950–31959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parys, J.B.; Decuypere, J.P.; Bultynck, G. Role of the inositol 1,4,5-trisphosphate receptor/Ca2+-release channel in autophagy. Cell Commun. Signal. 2012, 10, 17. [Google Scholar] [CrossRef] [Green Version]
- Engedal, N.; Torgersen, M.L.; Guldvik, I.J.; Barfeld, S.J.; Bakula, D.; Saetre, F.; Hagen, L.K.; Patterson, J.B.; Proikas-Cezanne, T.; Seglen, P.O.; et al. Modulation of intracellular calcium homeostasis blocks autophagosome formation. Autophagy 2013, 9, 1475–1490. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Fang, Y.; Cheng, X.; Lian, Y.; Xu, H.; Zeng, Z.; Zhu, H. TRPML1 Participates in the Progression of Alzheimer’s Disease by Regulating the PPARgamma/AMPK/Mtor Signalling Pathway. Cell Physiol. Biochem. 2017, 43, 2446–2456. [Google Scholar] [CrossRef]
- Senft, D.; Ronai, Z.A. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 2015, 40, 141–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, B.Y.; Wang, M.; Ma, D.L.; Al-Mousa, F.; Michelangeli, F.; Cheng, S.H.; Ng, M.H.; To, K.F.; Mok, A.Y.; Ko, R.Y.; et al. Alisol B, a novel inhibitor of the sarcoplasmic/endoplasmic reticulum Ca(2+) ATPase pump, induces autophagy, endoplasmic reticulum stress, and apoptosis. Mol. Cancer Ther. 2010, 9, 718–730. [Google Scholar] [CrossRef] [Green Version]
- Van Houte, J.; Jordan, H.V.; Laraway, R.; Kent, R.; Soparkar, P.M.; DePaola, P.F. Association of the microbial flora of dental plaque and saliva with human root-surface caries. J. Dent. Res. 1990, 69, 1463–1468. [Google Scholar] [CrossRef] [PubMed]
- Gannage, M.; Dormann, D.; Albrecht, R.; Dengjel, J.; Torossi, T.; Ramer, P.C.; Lee, M.; Strowig, T.; Arrey, F.; Conenello, G.; et al. Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe 2009, 6, 367–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, J.; Ran, Y.; Xie, H.; Deng, L.; Li, C.; Ling, C. Sarco/Endoplasmic Reticulum Ca(2+) Transporting ATPase (SERCA) Modulates Autophagic, Inflammatory, and Mitochondrial Responses during Influenza A Virus Infection in Human Lung Cells. J. Virol. 2021, 95, e00217-21. [Google Scholar] [CrossRef] [PubMed]
- Mauvezin, C.; Nagy, P.; Juhasz, G.; Neufeld, T.P. Autophagosome-lysosome fusion is independent of V-ATPase-mediated acidification. Nat. Commun. 2015, 6, 7007. [Google Scholar] [CrossRef] [Green Version]
- Shen, D.; Wang, X.; Li, X.; Zhang, X.; Yao, Z.; Dibble, S.; Dong, X.P.; Yu, T.; Lieberman, A.P.; Showalter, H.D.; et al. Lipid storage disorders block lysosomal trafficking by inhibiting a TRP channel and lysosomal calcium release. Nat. Commun. 2012, 3, 731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardenas, C.; Juretic, N.; Bevilacqua, J.A.; Garcia, I.E.; Figueroa, R.; Hartley, R.; Taratuto, A.L.; Gejman, R.; Riveros, N.; Molgo, J.; et al. Abnormal distribution of inositol 1,4,5-trisphosphate receptors in human muscle can be related to altered calcium signals and gene expression in Duchenne dystrophy-derived cells. FASEB J. 2010, 24, 3210–3221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valladares, D.; Utreras-Mendoza, Y.; Campos, C.; Morales, C.; Diaz-Vegas, A.; Contreras-Ferrat, A.; Westermeier, F.; Jaimovich, E.; Marchi, S.; Pinton, P.; et al. IP3 receptor blockade restores autophagy and mitochondrial function in skeletal muscle fibers of dystrophic mice. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3685–3695. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, C.; Miller, R.A.; Smith, I.; Bui, T.; Molgo, J.; Muller, M.; Vais, H.; Cheung, K.H.; Yang, J.; Parker, I.; et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 2010, 142, 270–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filadi, R.; Leal, N.S.; Schreiner, B.; Rossi, A.; Dentoni, G.; Pinho, C.M.; Wiehager, B.; Cieri, D.; Cali, T.; Pizzo, P.; et al. TOM70 Sustains Cell Bioenergetics by Promoting IP3R3-Mediated ER to Mitochondria Ca(2+) Transfer. Curr. Biol. 2018, 28, 369–382. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Zhu, Q.; Cao, B.; Cai, Y.; Wen, S.; Bian, J.; Zou, H.; Song, R.; Gu, J.; Liu, X.; et al. Ca(2+) transfer via the ER-mitochondria tethering complex in neuronal cells contribute to cadmium-induced autophagy. Cell Biol. Toxicol. 2021, 1–17. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, Y.; Jiang, Y.; Dou, X.; Li, S.; Chai, H.; Qian, Q.; Wang, M. Upregulated SOCC and IP3R calcium channels and subsequent elevated cytoplasmic calcium signaling promote nonalcoholic fatty liver disease by inhibiting autophagy. Mol. Cell Biochem. 2021, 476, 3163–3175. [Google Scholar] [CrossRef] [PubMed]
- Venkatachalam, K.; Long, A.A.; Elsaesser, R.; Nikolaeva, D.; Broadie, K.; Montell, C. Motor deficit in a Drosophila model of mucolipidosis type IV due to defective clearance of apoptotic cells. Cell 2008, 135, 838–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatachalam, K.; Wong, C.O.; Montell, C. Feast or famine: Role of TRPML in preventing cellular amino acid starvation. Autophagy 2013, 9, 98–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Gao, Q.; Yang, M.; Zhang, X.; Yu, L.; Lawas, M.; Li, X.; Bryant-Genevier, M.; Southall, N.T.; Marugan, J.; et al. Up-regulation of lysosomal TRPML1 channels is essential for lysosomal adaptation to nutrient starvation. Proc. Natl. Acad. Sci. USA 2015, 112, E1373–E1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Yang, Y.; Zhong, X.Z.; Cao, Q.; Zhu, X.H.; Zhu, X.; Dong, X.P. A negative feedback regulation of MTORC1 activity by the lysosomal Ca(2+) channel MCOLN1 (mucolipin 1) using a CALM (calmodulin)-dependent mechanism. Autophagy 2018, 14, 38–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amemiya, Y.; Nakamura, N.; Ikeda, N.; Sugiyama, R.; Ishii, C.; Maki, M.; Shibata, H.; Takahara, T. Amino Acid-Mediated Intracellular Ca(2+) Rise Modulates mTORC1 by Regulating the TSC2-Rheb Axis through Ca(2+)/Calmodulin. Int. J. Mol. Sci. 2021, 22, 6897. [Google Scholar] [CrossRef] [PubMed]
- Decuypere, J.P.; Kindt, D.; Luyten, T.; Welkenhuyzen, K.; Missiaen, L.; de Smedt, H.; Bultynck, G.; Parys, J.B. mTOR-Controlled Autophagy Requires Intracellular Ca(2+) Signaling. PLoS ONE 2013, 8, e61020. [Google Scholar] [CrossRef] [Green Version]
- Li, R.J.; Xu, J.; Fu, C.; Zhang, J.; Zheng, Y.G.; Jia, H.; Liu, J.O. Regulation of mTORC1 by lysosomal calcium and calmodulin. Elife 2016, 5, e19360. [Google Scholar] [CrossRef] [PubMed]
- Medina, D.L.; di Paola, S.; Peluso, I.; Armani, A.; de Stefani, D.; Venditti, R.; Montefusco, S.; Scotto-Rosato, A.; Prezioso, C.; Forrester, A.; et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015, 17, 288–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, C.O.; Li, R.; Montell, C.; Venkatachalam, K. Drosophila TRPML is required for TORC1 activation. Curr. Biol. 2012, 22, 1616–1621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, J.; Xing, Y.; Liu, Y.; Wang, M.M.; Wei, X.; Sui, Z.; Ding, L.; Zhang, Y.; Lu, C.; Fei, Y.H.; et al. MCOLN1/TRPML1 finely controls oncogenic autophagy in cancer by mediating zinc influx. Autophagy 2021, 1–22. [Google Scholar] [CrossRef]
- Curcio-Morelli, C.; Charles, F.A.; Micsenyi, M.C.; Cao, Y.; Venugopal, B.; Browning, M.F.; Dobrenis, K.; Cotman, S.L.; Walkley, S.U.; Slaugenhaupt, S.A. Macroautophagy is defective in mucolipin-1-deficient mouse neurons. Neurobiol. Dis. 2010, 40, 370–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tedeschi, V.; Petrozziello, T.; Sisalli, M.J.; Boscia, F.; Canzoniero, L.M.T.; Secondo, A. The activation of Mucolipin TRP channel 1 (TRPML1) protects motor neurons from L-BMAA neurotoxicity by promoting autophagic clearance. Sci. Rep. 2019, 9, 10743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Cheng, X.; Yu, L.; Yang, J.; Calvo, R.; Patnaik, S.; Hu, X.; Gao, Q.; Yang, M.; Lawas, M.; et al. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat. Commun. 2016, 7, 12109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruhl, P.; Rosato, A.S.; Urban, N.; Gerndt, S.; Tang, R.; Abrahamian, C.; Leser, C.; Sheng, J.; Jha, A.; Vollmer, G.; et al. Estradiol analogs attenuate autophagy, cell migration and invasion by direct and selective inhibition of TRPML1, independent of estrogen receptors. Sci. Rep. 2021, 11, 8313. [Google Scholar] [CrossRef]
- Soyombo, A.A.; Tjon-Kon-Sang, S.; Rbaibi, Y.; Bashllari, E.; Bisceglia, J.; Muallem, S.; Kiselyov, K. TRP-ML1 regulates lysosomal pH and acidic lysosomal lipid hydrolytic activity. J. Biol. Chem. 2006, 281, 7294–7301. [Google Scholar] [CrossRef] [Green Version]
- Isobe, Y.; Nigorikawa, K.; Tsurumi, G.; Takemasu, S.; Takasuga, S.; Kofuji, S.; Hazeki, K. PIKfyve accelerates phagosome acidification through activation of TRPML1 while arrests aberrant vacuolation independent of the Ca2+ channel. J. Biochem. 2019, 165, 75–84. [Google Scholar] [CrossRef]
- Ogunbayo, O.A.; Duan, J.; Xiong, J.; Wang, Q.; Feng, X.; Ma, J.; Zhu, M.X.; Evans, A.M. mTORC1 controls lysosomal Ca(2+) release through the two-pore channel TPC2. Sci. Signal. 2018, 11, eaao5775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, F.S.; Wang, Y.; Dmitriev, P.; Gross, J.; Galione, A.; Pears, C. A two-pore channel protein required for regulating mTORC1 activity on starvation. BMC Biol. 2020, 18, 8. [Google Scholar] [CrossRef]
- Lu, Y.; Hao, B.; Graeff, R.; Yue, J. NAADP/TPC2/Ca(2+) Signaling Inhibits Autophagy. Commun. Integr. Biol. 2013, 6, e27595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakurai, Y.; Kolokoltsov, A.A.; Chen, C.C.; Tidwell, M.W.; Bauta, W.E.; Klugbauer, N.; Grimm, C.; Wahl-Schott, C.; Biel, M.; Davey, R.A. Ebola virus. Two-pore channels control Ebola virus host cell entry and are drug targets for disease treatment. Science 2015, 347, 995–998. [Google Scholar] [CrossRef] [Green Version]
- Ruas, M.; Rietdorf, K.; Arredouani, A.; Davis, L.C.; Lloyd-Evans, E.; Koegel, H.; Funnell, T.M.; Morgan, A.J.; Ward, J.A.; Watanabe, K.; et al. Purified TPC isoforms form NAADP receptors with distinct roles for Ca(2+) signaling and endolysosomal trafficking. Curr. Biol. 2010, 20, 703–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimm, C.; Holdt, L.M.; Chen, C.C.; Hassan, S.; Muller, C.; Jors, S.; Cuny, H.; Kissing, S.; Schroder, B.; Butz, E.; et al. High susceptibility to fatty liver disease in two-pore channel 2-deficient mice. Nat. Commun. 2014, 5, 4699. [Google Scholar] [CrossRef]
- Zhu, Z.D.; Yu, T.; Liu, H.J.; Jin, J.; He, J. SOCE induced calcium overload regulates autophagy in acute pancreatitis via calcineurin activation. Cell Death Dis. 2018, 9, 50. [Google Scholar] [CrossRef]
- Yang, J.; Yu, J.; Li, D.; Yu, S.; Ke, J.; Wang, L.; Wang, Y.; Qiu, Y.; Gao, X.; Zhang, J.; et al. Store-operated calcium entry-activated autophagy protects EPC proliferation via the CAMKK2-MTOR pathway in ox-LDL exposure. Autophagy 2017, 13, 82–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selvaraj, S.; Sun, Y.; Sukumaran, P.; Singh, B.B. Resveratrol activates autophagic cell death in prostate cancer cells via downregulation of STIM1 and the mTOR pathway. Mol. Carcinog. 2016, 55, 818–831. [Google Scholar] [CrossRef]
- Cotter, K.; Stransky, L.; McGuire, C.; Forgac, M. Recent Insights into the Structure, Regulation, and Function of the V-ATPases. Trends Biochem. Sci. 2015, 40, 611–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Benlekbir, S.; Rubinstein, J.L. Electron cryomicroscopy observation of rotational states in a eukaryotic V-ATPase. Nature 2015, 521, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Mijaljica, D.; Prescott, M.; Devenish, R.J. V-ATPase engagement in autophagic processes. Autophagy 2011, 7, 666–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, C.Y.; Shin, H.R.; Berdan, C.A.; Ford, B.; Ward, C.C.; Olzmann, J.A.; Zoncu, R.; Nomura, D.K. Covalent targeting of the vacuolar H(+)-ATPase activates autophagy via mTORC1 inhibition. Nat. Chem. Biol. 2019, 15, 776–785. [Google Scholar] [CrossRef]
- Strasser, B.; Iwaszkiewicz, J.; Michielin, O.; Mayer, A. The V-ATPase proteolipid cylinder promotes the lipid-mixing stage of SNARE-dependent fusion of yeast vacuoles. EMBO J. 2011, 30, 4126–4141. [Google Scholar] [CrossRef] [Green Version]
- Rama, S.; Boumedine-Guignon, N.; Sangiardi, M.; Youssouf, F.; Maulet, Y.; Leveque, C.; Belghazi, M.; Seagar, M.; Debanne, D.; El Far, O. Chromophore-Assisted Light Inactivation of the V-ATPase V0c Subunit Inhibits Neurotransmitter Release Downstream of Synaptic Vesicle Acidification. Mol. Neurobiol. 2019, 56, 3591–3602. [Google Scholar] [CrossRef]
- Kawai, A.; Uchiyama, H.; Takano, S.; Nakamura, N.; Ohkuma, S. Autophagosome-lysosome fusion depends on the pH in acidic compartments in CHO cells. Autophagy 2007, 3, 154–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Yu, W.H.; Kumar, A.; Lee, S.; Mohan, P.S.; Peterhoff, C.M.; Wolfe, D.M.; Martinez-Vicente, M.; Massey, A.C.; Sovak, G.; et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 2010, 141, 1146–1158. [Google Scholar] [CrossRef] [Green Version]
- Brunello, L.; Zampese, E.; Florean, C.; Pozzan, T.; Pizzo, P.; Fasolato, C. Presenilin-2 dampens intracellular Ca2+ stores by increasing Ca2+ leakage and reducing Ca2+ uptake. J. Cell Mol. Med. 2009, 13, 3358–3369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coen, K.; Flannagan, R.S.; Baron, S.; Carraro-Lacroix, L.R.; Wang, D.; Vermeire, W.; Michiels, C.; Munck, S.; Baert, V.; Sugita, S.; et al. Lysosomal calcium homeostasis defects, not proton pump defects, cause endo-lysosomal dysfunction in PSEN-deficient cells. J. Cell Biol. 2012, 198, 23–35. [Google Scholar] [CrossRef]
- Fedeli, C.; Filadi, R.; Rossi, A.; Mammucari, C.; Pizzo, P. PSEN2 (presenilin 2) mutants linked to familial Alzheimer disease impair autophagy by altering Ca(2+) homeostasis. Autophagy 2019, 15, 2044–2062. [Google Scholar] [CrossRef]
- Currinn, H.; Guscott, B.; Balklava, Z.; Rothnie, A.; Wassmer, T. APP controls the formation of PI(3,5)P(2) vesicles through its binding of the PIKfyve complex. Cell Mol. Life Sci. 2016, 73, 393–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, X.P.; Shen, D.; Wang, X.; Dawson, T.; Li, X.; Zhang, Q.; Cheng, X.; Zhang, Y.; Weisman, L.S.; Delling, M.; et al. PI(3,5)P(2) controls membrane trafficking by direct activation of mucolipin Ca(2+) release channels in the endolysosome. Nat. Commun. 2010, 1, 38. [Google Scholar] [CrossRef] [Green Version]
- Jin, N.; Lang, M.J.; Weisman, L.S. Phosphatidylinositol 3,5-bisphosphate: Regulation of cellular events in space and time. Biochem. Soc. Trans. 2016, 44, 177–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devlin, C.; Pipalia, N.H.; Liao, X.; Schuchman, E.H.; Maxfield, F.R.; Tabas, I. Improvement in lipid and protein trafficking in Niemann-Pick C1 cells by correction of a secondary enzyme defect. Traffic 2010, 11, 601–615. [Google Scholar] [CrossRef]
- Nakamura, S.; Shigeyama, S.; Minami, S.; Shima, T.; Akayama, S.; Matsuda, T.; Esposito, A.; Napolitano, G.; Kuma, A.; Namba-Hamano, T.; et al. LC3 lipidation is essential for TFEB activation during the lysosomal damage response to kidney injury. Nat. Cell Biol. 2020, 22, 1252–1263. [Google Scholar] [CrossRef]
- Onyenwoke, R.U.; Sexton, J.Z.; Yan, F.; Diaz, M.C.; Forsberg, L.J.; Major, M.B.; Brenman, J.E. The mucolipidosis IV Ca2+ channel TRPML1 (MCOLN1) is regulated by the TOR kinase. Biochem. J. 2015, 470, 331–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cang, C.; Zhou, Y.; Navarro, B.; Seo, Y.J.; Aranda, K.; Shi, L.; Battaglia-Hsu, S.; Nissim, I.; Clapham, D.E.; Ren, D. mTOR regulates lysosomal ATP-sensitive two-pore Na(+) channels to adapt to metabolic state. Cell 2013, 152, 778–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.K.; Lee, G.H.; Bhattarai, K.R.; Lee, M.S.; Back, S.H.; Kim, H.R.; Chae, H.J. TMBIM6 (transmembrane BAX inhibitor motif containing 6) enhances autophagy through regulation of lysosomal calcium. Autophagy 2021, 17, 761–778. [Google Scholar] [CrossRef] [Green Version]
- Park, H.J.; Gholam-Zadeh, M.; Suh, J.H.; Choi, H.S. Lycorine Attenuates Autophagy in Osteoclasts via an Axis of mROS/TRPML1/TFEB to Reduce LPS-Induced Bone Loss. Oxid. Med. Cell Longev. 2019, 2019, 8982147. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, W.; Gao, Q.; Yang, J.; Yan, X.; Zhao, H.; Su, L.; Yang, M.; Gao, C.; Yao, Y.; et al. Rapamycin directly activates lysosomal mucolipin TRP channels independent of mTOR. PLoS Biol. 2019, 17, e3000252. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.Z.; Zou, Y.; Sun, X.; Dong, G.; Cao, Q.; Pandey, A.; Rainey, J.K.; Zhu, X.; Dong, X.P. Inhibition of Transient Receptor Potential Channel Mucolipin-1 (TRPML1) by Lysosomal Adenosine Involved in Severe Combined Immunodeficiency Diseases. J. Biol. Chem. 2017, 292, 3445–3455. [Google Scholar] [CrossRef] [Green Version]
- Plesch, E.; Chen, C.C.; Butz, E.; Scotto Rosato, A.; Krogsaeter, E.K.; Yinan, H.; Bartel, K.; Keller, M.; Robaa, D.; Teupser, D.; et al. Selective agonist of TRPML2 reveals direct role in chemokine release from innate immune cells. Elife 2018, 7, e39720. [Google Scholar] [CrossRef]
- Grimm, C.; Jors, S.; Saldanha, S.A.; Obukhov, A.G.; Pan, B.; Oshima, K.; Cuajungco, M.P.; Chase, P.; Hodder, P.; Heller, S. Small molecule activators of TRPML3. Chem. Biol. 2010, 17, 135–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Churchill, G.C.; Okada, Y.; Thomas, J.M.; Genazzani, A.A.; Patel, S.; Galione, A. NAADP mobilizes Ca(2+) from reserve granules, lysosome-related organelles, in sea urchin eggs. Cell 2002, 111, 703–708. [Google Scholar] [CrossRef] [Green Version]
- Brailoiu, E.; Churamani, D.; Cai, X.; Schrlau, M.G.; Brailoiu, G.C.; Gao, X.; Hooper, R.; Boulware, M.J.; Dun, N.J.; Marchant, J.S.; et al. Essential requirement for two-pore channel 1 in NAADP-mediated calcium signaling. J. Cell Biol. 2009, 186, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Calcraft, P.J.; Ruas, M.; Pan, Z.; Cheng, X.; Arredouani, A.; Hao, X.; Tang, J.; Rietdorf, K.; Teboul, L.; Chuang, K.T.; et al. NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature 2009, 459, 596–600. [Google Scholar] [CrossRef] [Green Version]
- Lin-Moshier, Y.; Walseth, T.F.; Churamani, D.; Davidson, S.M.; Slama, J.T.; Hooper, R.; Brailoiu, E.; Patel, S.; Marchant, J.S. Photoaffinity labeling of nicotinic acid adenine dinucleotide phosphate (NAADP) targets in mammalian cells. J. Biol. Chem. 2012, 287, 2296–2307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, A.; Ahuja, M.; Patel, S.; Brailoiu, E.; Muallem, S. Convergent regulation of the lysosomal two-pore channel-2 by Mg(2)(+), NAADP, PI(3,5)P(2) and multiple protein kinases. EMBO J. 2014, 33, 501–511. [Google Scholar] [CrossRef]
- Pereira, G.J.; Antonioli, M.; Hirata, H.; Ureshino, R.P.; Nascimento, A.R.; Bincoletto, C.; Vescovo, T.; Piacentini, M.; Fimia, G.M.; Smaili, S.S. Glutamate induces autophagy via the two-pore channels in neural cells. Oncotarget 2017, 8, 12730–12740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasanthakumar, T.; Rubinstein, J.L. Structure and Roles of V-type ATPases. Trends Biochem. Sci. 2020, 45, 295–307. [Google Scholar] [CrossRef]
- Cipriano, D.J.; Wang, Y.; Bond, S.; Hinton, A.; Jefferies, K.C.; Qi, J.; Forgac, M. Structure and regulation of the vacuolar ATPases. Biochim. Biophys. Acta 2008, 1777, 599–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lafourcade, C.; Sobo, K.; Kieffer-Jaquinod, S.; Garin, J.; van der Goot, F.G. Regulation of the V-ATPase along the endocytic pathway occurs through reversible subunit association and membrane localization. PLoS ONE 2008, 3, e2758. [Google Scholar] [CrossRef] [Green Version]
- Paroutis, P.; Touret, N.; Grinstein, S. The pH of the secretory pathway: Measurement, determinants, and regulation. Physiology 2004, 19, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Futai, M.; Nakanishi-Matsui, M.; Okamoto, H.; Sekiya, M.; Nakamoto, R.K. Rotational catalysis in proton pumping ATPases: From E. coli F-ATPase to mammalian V-ATPase. Biochim. Biophys. Acta 2012, 1817, 1711–1721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marshansky, V.; Rubinstein, J.L.; Gruber, G. Eukaryotic V-ATPase: Novel structural findings and functional insights. Biochim. Biophys. Acta 2014, 1837, 857–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toei, M.; Saum, R.; Forgac, M. Regulation and isoform function of the V-ATPases. Biochemistry 2010, 49, 4715–4723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Wang, X.; Gao, T.; Gu, C.; Sun, F.; Yu, L.; Hu, J. Characterization of the complex involved in regulating V-ATPase activity of the vacuolar and endosomal membrane. J. Bioenerg. Biomembr. 2017, 49, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Jaskolka, M.C.; Kane, P.M. Interaction between the yeast RAVE complex and Vph1-containing Vo sectors is a central glucose-sensitive interaction required for V-ATPase reassembly. J. Biol. Chem. 2020, 295, 2259–2269. [Google Scholar] [CrossRef]
- Smardon, A.M.; Diab, H.I.; Tarsio, M.; Diakov, T.T.; Nasab, N.D.; West, R.W.; Kane, P.M. The RAVE complex is an isoform-specific V-ATPase assembly factor in yeast. Mol. Biol. Cell 2014, 25, 356–367. [Google Scholar] [CrossRef]
- Jaskolka, M.C.; Winkley, S.R.; Kane, P.M. RAVE and Rabconnectin-3 Complexes as Signal Dependent Regulators of Organelle Acidification. Front. Cell Dev. Biol. 2021, 9, 698190. [Google Scholar] [CrossRef]
- Hiesinger, P.R.; Fayyazuddin, A.; Mehta, S.Q.; Rosenmund, T.; Schulze, K.L.; Zhai, R.G.; Verstreken, P.; Cao, Y.; Zhou, Y.; Kunz, J.; et al. The v-ATPase V0 subunit a1 is required for a late step in synaptic vesicle exocytosis in Drosophila. Cell 2005, 121, 607–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Wang, D.; Volk, E.; Bellen, H.J.; Hiesinger, P.R.; Quiocho, F.A. V-ATPase V0 sector subunit a1 in neurons is a target of calmodulin. J. Biol. Chem. 2008, 283, 294–300. [Google Scholar] [CrossRef] [Green Version]
- Di Giovanni, J.; Boudkkazi, S.; Mochida, S.; Bialowas, A.; Samari, N.; Leveque, C.; Youssouf, F.; Brechet, A.; Iborra, C.; Maulet, Y.; et al. V-ATPase membrane sector associates with synaptobrevin to modulate neurotransmitter release. Neuron 2010, 67, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Liu, N.; Xie, X.; Bi, G.; Ba, H.; Li, L.; Zhang, J.; Deng, X.; Yao, Y.; Tang, Z.; et al. The macrophage-specific V-ATPase subunit ATP6V0D2 restricts inflammasome activation and bacterial infection by facilitating autophagosome-lysosome fusion. Autophagy 2019, 15, 960–975. [Google Scholar] [CrossRef]
- Wang, D.; Epstein, D.; Khalaf, O.; Srinivasan, S.; Williamson, W.R.; Fayyazuddin, A.; Quiocho, F.A.; Hiesinger, P.R. Ca2+-Calmodulin regulates SNARE assembly and spontaneous neurotransmitter release via v-ATPase subunit V0a1. J. Cell Biol. 2014, 205, 21–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandini, M.A.; Souza, I.A.; Fan, J.; Li, K.; Wang, D.; Zamponi, G.W. Interactions of Rabconnectin-3 with Cav2 calcium channels. Mol. Brain 2019, 12, 62. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, K.; Kamp, M.A.; Siapich, S.A.; Hescheler, J.; Luke, M.; Schneider, T. Ca(v)2.3 Ca2+ channel interacts with the G1-subunit of V-ATPase. Cell Physiol. Biochem. 2011, 27, 421–432. [Google Scholar] [CrossRef]
- Crummy, E.; Mani, M.; Thellman, J.C.; Martin, T.F.J. The priming factor CAPS1 regulates dense-core vesicle acidification by interacting with rabconnectin3beta/WDR7 in neuroendocrine cells. J. Biol. Chem. 2019, 294, 9402–9415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Y.; Denef, N.; Schupbach, T. The vacuolar proton pump, V-ATPase, is required for notch signaling and endosomal trafficking in Drosophila. Dev. Cell 2009, 17, 387–402. [Google Scholar] [CrossRef] [Green Version]
- Sethi, N.; Yan, Y.; Quek, D.; Schupbach, T.; Kang, Y. Rabconnectin-3 is a functional regulator of mammalian Notch signaling. J. Biol. Chem. 2010, 285, 34757–34764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couoh-Cardel, S.; Hsueh, Y.C.; Wilkens, S.; Movileanu, L. Yeast V-ATPase Proteolipid Ring Acts as a Large-conductance Transmembrane Protein Pore. Sci. Rep. 2016, 6, 24774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wada, Y.; Ohsumi, Y.; Tanifuji, M.; Kasai, M.; Anraku, Y. Vacuolar ion channel of the yeast, Saccharomyces cerevisiae. J. Biol. Chem. 1987, 262, 17260–17263. [Google Scholar] [CrossRef]
- Urbani, A.; Giorgio, V.; Carrer, A.; Franchin, C.; Arrigoni, G.; Jiko, C.; Abe, K.; Maeda, S.; Shinzawa-Itoh, K.; Bogers, J.F.M.; et al. Purified F-ATP synthase forms a Ca(2+)-dependent high-conductance channel matching the mitochondrial permeability transition pore. Nat. Commun. 2019, 10, 4341. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.S.; Stone, D.K. Partial resolution and reconstitution of the subunits of the clathrin-coated vesicle proton ATPase responsible for Ca2+-activated ATP hydrolysis. J. Biol. Chem. 1988, 263, 9859–9867. [Google Scholar] [CrossRef]
- Pullman, M.E.; Penefsky, H.S.; Datta, A.; Racker, E. Partial resolution of the enzymes catalyzing oxidative phosphorylation. I. Purification and properties of soluble dinitrophenol-stimulated adenosine triphosphatase. J. Biol. Chem. 1960, 235, 3322–3329. [Google Scholar] [CrossRef]
- Crider, B.P.; Xie, X.S. Characterization of the functional coupling of bovine brain vacuolar-type H(+)-translocating ATPase. Effect of divalent cations, phospholipids, and subunit H (SFD). J. Biol. Chem. 2003, 278, 44281–44288. [Google Scholar] [CrossRef] [Green Version]
- Cang, C.; Bekele, B.; Ren, D. The voltage-gated sodium channel TPC1 confers endolysosomal excitability. Nat. Chem. Biol. 2014, 10, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Matamala, E.; Castillo, C.; Vivar, J.P.; Rojas, P.A.; Brauchi, S.E. Imaging the electrical activity of organelles in living cells. Commun. Biol. 2021, 4, 389. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Wang, L.; Li, S.; Tang, W.; Sun, F.; Wu, Y.; Miao, L.; Cao, Z. ML-SA1, a selective TRPML agonist, inhibits DENV2 and ZIKV by promoting lysosomal acidification and protease activity. Antivir. Res. 2020, 182, 104922. [Google Scholar] [CrossRef]
- Ono, Y.; Mori, Y.; Egashira, Y.; Sumiyama, K.; Takamori, S. Expression of plasma membrane calcium ATPases confers Ca(2+)/H(+) exchange in rodent synaptic vesicles. Sci. Rep. 2019, 9, 4289. [Google Scholar] [CrossRef] [Green Version]
- Wie, J.; Liu, Z.; Song, H.; Tropea, T.F.; Yang, L.; Wang, H.; Liang, Y.; Cang, C.; Aranda, K.; Lohmann, J.; et al. A growth-factor-activated lysosomal K(+) channel regulates Parkinson’s pathology. Nature 2021, 591, 431–437. [Google Scholar] [CrossRef]
- Berman, E.R.; Livni, N.; Shapira, E.; Merin, S.; Levij, I.S. Congenital corneal clouding with abnormal systemic storage bodies: A new variant of mucolipidosis. J. Pediatr. 1974, 84, 519–526. [Google Scholar] [CrossRef]
- Boudewyn, L.C.; Walkley, S.U. Current concepts in the neuropathogenesis of mucolipidosis type IV. J. Neurochem. 2019, 148, 669–689. [Google Scholar] [CrossRef] [Green Version]
- Kasitinon, S.Y.; Eskiocak, U.; Martin, M.; Bezwada, D.; Khivansara, V.; Tasdogan, A.; Zhao, Z.; Mathews, T.; Aurora, A.B.; Morrison, S.J. TRPML1 Promotes Protein Homeostasis in Melanoma Cells by Negatively Regulating MAPK and mTORC1 Signaling. Cell Rep. 2019, 28, 2293–2305. [Google Scholar] [CrossRef] [Green Version]
- Morelli, M.B.; Amantini, C.; Tomassoni, D.; Nabissi, M.; Arcella, A.; Santoni, G. Transient Receptor Potential Mucolipin-1 Channels in Glioblastoma: Role in Patient’s Survival. Cancers 2019, 11, 525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, C.; Zhang, H.; Liu, X.; Zhang, H.; Zhang, Y.; Bai, X.; Wang, L.; Li, H.; Li, X.; Zhang, S.; et al. Downregulated MCOLN1 Attenuates the Progression Of Non-Small-Cell Lung Cancer By Inhibiting Lysosome-Autophagy. Cancer Manag Res. 2019, 11, 8607–8617. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.S.; Muallem, S.; Kim, S.H.; Kwon, K.B.; Kim, M.S. Exosomal release through TRPML1-mediated lysosomal exocytosis is required for adipogenesis. Biochem. Biophys. Res. Commun. 2019, 510, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Chandra, M.; Zhou, H.; Li, Q.; Muallem, S.; Hofmann, S.L.; Soyombo, A.A. A role for the Ca2+ channel TRPML1 in gastric acid secretion, based on analysis of knockout mice. Gastroenterology 2011, 140, 857–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capurro, M.I.; Greenfield, L.K.; Prashar, A.; Xia, S.; Abdullah, M.; Wong, H.; Zhong, X.Z.; Bertaux-Skeirik, N.; Chakrabarti, J.; Siddiqui, I.; et al. VacA generates a protective intracellular reservoir for Helicobacter pylori that is eliminated by activation of the lysosomal calcium channel TRPML1. Nat. Microbiol. 2019, 4, 1411–1423. [Google Scholar] [CrossRef]
- Zhao, Q.; Li, J.; Ko, W.H.; Kwan, Y.W.; Jiang, L.; Sun, L.; Yao, X. TRPM2 promotes autophagic degradation in vascular smooth muscle cells. Sci. Rep. 2020, 10, 20719. [Google Scholar] [CrossRef] [PubMed]
- Arora, K.; Liyanage, P.; Zhong, Q.; Naren, A.P. A SNARE protein Syntaxin 17 captures CFTR to potentiate autophagosomal clearance under stress. FASEB J. 2021, 35, e21185. [Google Scholar] [CrossRef] [PubMed]
- Vervliet, T.; Pintelon, I.; Welkenhuyzen, K.; Bootman, M.D.; Bannai, H.; Mikoshiba, K.; Martinet, W.; Nadif Kasri, N.; Parys, J.B.; Bultynck, G. Basal ryanodine receptor activity suppresses autophagic flux. Biochem. Pharmacol. 2017, 132, 133–142. [Google Scholar] [CrossRef]
- Graef, M.; Friedman, J.R.; Graham, C.; Babu, M.; Nunnari, J. ER exit sites are physical and functional core autophagosome biogenesis components. Mol. Biol. Cell 2013, 24, 2918–2931. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abuammar, H.; Bhattacharjee, A.; Simon-Vecsei, Z.; Blastyák, A.; Csordás, G.; Páli, T.; Juhász, G. Ion Channels and Pumps in Autophagy: A Reciprocal Relationship. Cells 2021, 10, 3537. https://doi.org/10.3390/cells10123537
Abuammar H, Bhattacharjee A, Simon-Vecsei Z, Blastyák A, Csordás G, Páli T, Juhász G. Ion Channels and Pumps in Autophagy: A Reciprocal Relationship. Cells. 2021; 10(12):3537. https://doi.org/10.3390/cells10123537
Chicago/Turabian StyleAbuammar, Hussein, Arindam Bhattacharjee, Zsófia Simon-Vecsei, András Blastyák, Gábor Csordás, Tibor Páli, and Gábor Juhász. 2021. "Ion Channels and Pumps in Autophagy: A Reciprocal Relationship" Cells 10, no. 12: 3537. https://doi.org/10.3390/cells10123537
APA StyleAbuammar, H., Bhattacharjee, A., Simon-Vecsei, Z., Blastyák, A., Csordás, G., Páli, T., & Juhász, G. (2021). Ion Channels and Pumps in Autophagy: A Reciprocal Relationship. Cells, 10(12), 3537. https://doi.org/10.3390/cells10123537