
Immunoglobulin A Mucosal Immunity and Altered Respiratory Epithelium in Cystic Fibrosis



Abstract
:1. Introduction
2. Alterations of the CF Respiratory Epithelium
3. Immunoglobulin A Mucosal Immunity in the CF Respiratory Epithelium
3.1. IgA Structure
3.2. IgA Regulation and Roles
3.3. IgA Immunity in CF
4. Gaps in Knowledge and Perspectives
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Davis, P.B. Cystic fibrosis since 1938. Am. J. Respir. Crit. Care Med. 2006, 173, 475–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elborn, J.S. Cystic fibrosis. Lancet 2016, 388, 2519–2531. [Google Scholar] [CrossRef]
- Ratjen, F.; Bell, S.C.; Rowe, S.M.; Goss, C.H.; Quittner, A.L.; Bush, A. Cystic fibrosis. Nat. Rev. Dis. Primers 2015, 1, 15010. [Google Scholar] [CrossRef]
- De Boeck, K.; Amaral, M.D. Progress in therapies for cystic fibrosis. Lancet Respir. Med. 2016, 4, 662–674. [Google Scholar] [CrossRef]
- Gallati, S. Disease-modifying genes and monogenic disorders: Experience in cystic fibrosis. Appl. Clin. Genet. 2014, 7, 133–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boboli, H.; Boemer, F.; Mastouri, M.; Seghaye, M.C. Neonatal screening for cystic fibrosis: Towards a national implementation in Belgium in 2019. Rev. Med. Liege 2018, 73, 497–501. [Google Scholar] [PubMed]
- Kerem, B.; Rommens, J.M.; Buchanan, J.A.; Markiewicz, D.; Cox, T.K.; Chakravarti, A.; Buchwald, M.; Tsui, L.C. Identification of the cystic fibrosis gene: Genetic analysis. Science 1989, 245, 1073–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riordan, J.R.; Rommens, J.M.; Kerem, B.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.L.; et al. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef]
- Rogan, M.P.; Stoltz, D.A.; Hornick, D.B. Cystic fibrosis transmembrane conductance regulator intracellular processing, trafficking, and opportunities for mutation-specific treatment. Chest 2011, 139, 1480–1490. [Google Scholar] [CrossRef] [PubMed]
- Rommens, J.M.; Iannuzzi, M.C.; Kerem, B.; Drumm, M.L.; Melmer, G.; Dean, M.; Rozmahel, R.; Cole, J.L.; Kennedy, D.; Hidaka, N.; et al. Identification of the cystic fibrosis gene: Chromosome walking and jumping. Science 1989, 245, 1059–1065. [Google Scholar] [CrossRef]
- Brezillon, S.; Dupuit, F.; Hinnrasky, J.; Marchand, V.; Kalin, N.; Tummler, B.; Puchelle, E. Decreased expression of the CFTR protein in remodeled human nasal epithelium from non-cystic fibrosis patients. Lab. Investig. A J. Tech. Methods Pathol. 1995, 72, 191–200. [Google Scholar]
- Cook, D.P.; Rector, M.V.; Bouzek, D.C.; Michalski, A.S.; Gansemer, N.D.; Reznikov, L.R.; Li, X.; Stroik, M.R.; Ostedgaard, L.S.; Abou Alaiwa, M.H.; et al. Cystic Fibrosis Transmembrane Conductance Regulator in Sarcoplasmic Reticulum of Airway Smooth Muscle. Implications for Airway Contractility. Am. J. Respir. Crit. Care Med. 2016, 193, 417–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawford, I.; Maloney, P.C.; Zeitlin, P.L.; Guggino, W.B.; Hyde, S.C.; Turley, H.; Gatter, K.C.; Harris, A.; Higgins, C.F. Immunocytochemical localization of the cystic fibrosis gene product CFTR. Proc. Natl. Acad. Sci. USA 1991, 88, 9262–9266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gifford, A.M.; Chalmers, J.D. The role of neutrophils in cystic fibrosis. Curr. Opin. Hematol. 2014, 21, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Jacquot, J.; Puchelle, E.; Hinnrasky, J.; Fuchey, C.; Bettinger, C.; Spilmont, C.; Bonnet, N.; Dieterle, A.; Dreyer, D.; Pavirani, A.; et al. Localization of the cystic fibrosis transmembrane conductance regulator in airway secretory glands. Eur. Respir. J. 1993, 6, 169–176. [Google Scholar] [PubMed]
- Kartner, N.; Augustinas, O.; Jensen, T.J.; Naismith, A.L.; Riordan, J.R. Mislocalization of delta F508 CFTR in cystic fibrosis sweat gland. Nat. Genet. 1992, 1, 321–327. [Google Scholar] [CrossRef]
- Mall, M.; Bleich, M.; Greger, R.; Schreiber, R.; Kunzelmann, K. The amiloride-inhibitable Na+ conductance is reduced by the cystic fibrosis transmembrane conductance regulator in normal but not in cystic fibrosis airways. J. Clin. Invest. 1998, 102, 15–21. [Google Scholar] [CrossRef]
- Marcorelles, P.; Friocourt, G.; Uguen, A.; Lede, F.; Ferec, C.; Laquerriere, A. Cystic fibrosis transmembrane conductance regulator protein (CFTR) expression in the developing human brain: Comparative immunohistochemical study between patients with normal and mutated CFTR. J. Histochem. Cytochem. 2014, 62, 791–801. [Google Scholar] [CrossRef] [Green Version]
- McGrath, S.A.; Basu, A.; Zeitlin, P.L. Cystic fibrosis gene and protein expression during fetal lung development. Am. J. Respir. Cell Mol. Biol. 1993, 8, 201–208. [Google Scholar] [CrossRef]
- Montoro, D.T.; Haber, A.L.; Biton, M.; Vinarsky, V.; Lin, B.; Birket, S.E.; Yuan, F.; Chen, S.; Leung, H.M.; Villoria, J.; et al. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 2018, 560, 319–324. [Google Scholar] [CrossRef]
- Plasschaert, L.W.; Zilionis, R.; Choo-Wing, R.; Savova, V.; Knehr, J.; Roma, G.; Klein, A.M.; Jaffe, A.B. A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature 2018, 560, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Trezise, A.E.; Buchwald, M. In vivo cell-specific expression of the cystic fibrosis transmembrane conductance regulator. Nature 1991, 353, 434–437. [Google Scholar] [CrossRef]
- Okuda, K.; Dang, H.; Kobayashi, Y.; Carraro, G.; Nakano, S.; Chen, G.; Kato, T.; Asakura, T.; Gilmore, R.C.; Morton, L.C.; et al. Secretory Cells Dominate Airway CFTR Expression and Function in Human Airway Superficial Epithelia. Am. J. Respir. Crit. Care Med. 2021, 203, 1275–1289. [Google Scholar] [CrossRef] [PubMed]
- Devor, D.C.; Singh, A.K.; Lambert, L.C.; DeLuca, A.; Frizzell, R.A.; Bridges, R.J. Bicarbonate and chloride secretion in Calu-3 human airway epithelial cells. J. Gen. Physiol. 1999, 113, 743–760. [Google Scholar] [CrossRef] [Green Version]
- Kartner, N.; Hanrahan, J.W.; Jensen, T.J.; Naismith, A.L.; Sun, S.Z.; Ackerley, C.A.; Reyes, E.F.; Tsui, L.C.; Rommens, J.M.; Bear, C.E.; et al. Expression of the cystic fibrosis gene in non-epithelial invertebrate cells produces a regulated anion conductance. Cell 1991, 64, 681–691. [Google Scholar] [CrossRef]
- Linsdell, P.; Hanrahan, J.W. Glutathione permeability of CFTR. Am. J. Physiol. 1998, 275, C323–C326. [Google Scholar] [CrossRef] [PubMed]
- Moskwa, P.; Lorentzen, D.; Excoffon, K.J.; Zabner, J.; McCray, P.B., Jr.; Nauseef, W.M.; Dupuy, C.; Banfi, B. A novel host defense system of airways is defective in cystic fibrosis. Am. J. Respir. Crit. Care Med. 2007, 175, 174–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cystic Fibrosis Mutation Database (CFTR1). Available online: http://www.genet.sickkids.on.ca/ (accessed on 29 November 2021).
- The Clinical and Functional TRanslation of CFTR (CFTR2). Available online: https://cftr2.org/ (accessed on 29 November 2021).
- ECFS. Annual Data Report (Year 2018) European Cystic Fibrosis Society Patient Registry. 2021. Available online: https://www.ecfs.eu/news/ecfs-patient-registry-annual-data-report-2018 (accessed on 29 November 2021).
- Ramsey, B.W.; Davies, J.; McElvaney, N.G.; Tullis, E.; Bell, S.C.; Drevinek, P.; Griese, M.; McKone, E.F.; Wainwright, C.E.; Konstan, M.W.; et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med. 2011, 365, 1663–1672. [Google Scholar] [CrossRef] [Green Version]
- Boyle, M.P.; Bell, S.C.; Konstan, M.W.; McColley, S.A.; Rowe, S.M.; Rietschel, E.; Huang, X.; Waltz, D.; Patel, N.R.; Rodman, D.; et al. A CFTR corrector (lumacaftor) and a CFTR potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del CFTR mutation: A phase 2 randomised controlled trial. Lancet Respir. Med. 2014, 2, 527–538. [Google Scholar] [CrossRef]
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948. [Google Scholar] [CrossRef]
- Middleton, P.G.; Mall, M.A.; Drevinek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef]
- Ridley, K.; Condren, M. Elexacaftor-Tezacaftor-Ivacaftor: The First Triple-Combination Cystic Fibrosis Transmembrane Conductance Regulator Modulating Therapy. J. Pediatr. Pharmacol. Ther. 2020, 25, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Rowe, S.M.; Daines, C.; Ringshausen, F.C.; Kerem, E.; Wilson, J.; Tullis, E.; Nair, N.; Simard, C.; Han, L.; Ingenito, E.P.; et al. Tezacaftor-Ivacaftor in Residual-Function Heterozygotes with Cystic Fibrosis. N. Engl. J. Med. 2017, 377, 2024–2035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubin, J.L.; O’Callaghan, L.; Pelligra, C.; Konstan, M.W.; Ward, A.; Ishak, J.K.; Chandler, C.; Liou, T.G. Modeling long-term health outcomes of patients with cystic fibrosis homozygous for F508del-CFTR treated with lumacaftor/ivacaftor. Ther. Adv. Respir. Dis. 2019, 13, 1753466618820186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor-Cousar, J.L.; Munck, A.; McKone, E.F.; van der Ent, C.K.; Moeller, A.; Simard, C.; Wang, L.T.; Ingenito, E.P.; McKee, C.; Lu, Y.; et al. Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N. Engl. J. Med. 2017, 377, 2013–2023. [Google Scholar] [CrossRef]
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W.; Marigowda, G.; Huang, X.; Cipolli, M.; Colombo, C.; Davies, J.C.; De Boeck, K.; Flume, P.A.; et al. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 220–231. [Google Scholar] [CrossRef] [Green Version]
- Castellani, C.; Assael, B.M. Cystic fibrosis: A clinical view. Cell. Mol. Life Sci. 2017, 74, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Balazs, A.; Mall, M.A. Mucus obstruction and inflammation in early cystic fibrosis lung disease: Emerging role of the IL-1 signaling pathway. Pediatr. Pulmonol. 2019, 54 (Suppl. 3), S5–S12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boucher, R.C. Muco-Obstructive Lung Diseases. N. Engl. J. Med. 2019, 380, 1941–1953. [Google Scholar] [CrossRef]
- Bergin, D.A.; Hurley, K.; Mehta, A.; Cox, S.; Ryan, D.; O’Neill, S.J.; Reeves, E.P.; McElvaney, N.G. Airway inflammatory markers in individuals with cystic fibrosis and non-cystic fibrosis bronchiectasis. J. Inflamm. Res. 2013, 6, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonfield, T.L.; Panuska, J.R.; Konstan, M.W.; Hilliard, K.A.; Hilliard, J.B.; Ghnaim, H.; Berger, M. Inflammatory cytokines in cystic fibrosis lungs. Am. J. Respir. Crit. Care Med. 1995, 152, 2111–2118. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, S.; Hayes, D., Jr.; Wozniak, D.J. Cystic Fibrosis and Pseudomonas aeruginosa: The Host-Microbe Interface. Clin. Microbiol. Rev. 2019, 32, e00138-18. [Google Scholar] [CrossRef]
- Velsor, L.W.; van Heeckeren, A.; Day, B.J. Antioxidant imbalance in the lungs of cystic fibrosis transmembrane conductance regulator protein mutant mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 281, L31–L38. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.W.; Brownlee, K.G.; Conway, S.P.; Denton, M.; Littlewood, J.M. Evaluation of a new definition for chronic Pseudomonas aeruginosa infection in cystic fibrosis patients. J. Cyst. Fibros. 2003, 2, 29–34. [Google Scholar] [CrossRef] [Green Version]
- Cystic Fibrosis Foundation Patient Registry 2020 Annual Data Report. Available online: https://www.cff.org/medical-professionals/patient-registry (accessed on 29 November 2021).
- Sloane, A.J.; Lindner, R.A.; Prasad, S.S.; Sebastian, L.T.; Pedersen, S.K.; Robinson, M.; Bye, P.T.; Nielson, D.W.; Harry, J.L. Proteomic analysis of sputum from adults and children with cystic fibrosis and from control subjects. Am. J. Respir. Crit. Care Med. 2005, 172, 1416–1426. [Google Scholar] [CrossRef]
- Maury, G.; Pilette, C.; Sibille, Y. Secretory immunity of the airways. Rev. Mal. Respir. 2003, 20, 928–939. [Google Scholar] [PubMed]
- Ganesan, S.; Comstock, A.T.; Sajjan, U.S. Barrier function of airway tract epithelium. Tissue Barriers 2013, 1, e24997. [Google Scholar] [CrossRef] [PubMed]
- Gohy, S.T.; Hupin, C.; Pilette, C.; Ladjemi, M.Z. Chronic inflammatory airway diseases: The central role of the epithelium revisited. Clin. Exp. Allergy 2016, 46, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Lecaille, F.; Lalmanach, G.; Andrault, P.M. Antimicrobial proteins and peptides in human lung diseases: A friend and foe partnership with host proteases. Biochimie 2016, 122, 151–168. [Google Scholar] [CrossRef]
- Whitsett, J.A. Airway Epithelial Differentiation and Mucociliary Clearance. Ann. Am. Thorac. Soc. 2018, 15, S143–S148. [Google Scholar] [CrossRef]
- Carraro, G.; Langerman, J.; Sabri, S.; Lorenzana, Z.; Purkayastha, A.; Zhang, G.; Konda, B.; Aros, C.J.; Calvert, B.A.; Szymaniak, A.; et al. Transcriptional analysis of cystic fibrosis airways at single-cell resolution reveals altered epithelial cell states and composition. Nat. Med. 2021, 27, 806–814. [Google Scholar] [CrossRef]
- Rezaee, F.; Georas, S.N. Breaking barriers. New insights into airway epithelial barrier function in health and disease. Am. J. Respir. Cell. Mol. Biol. 2014, 50, 857–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedrossian, C.W.; Greenberg, S.D.; Singer, D.B.; Hansen, J.J.; Rosenberg, H.S. The lung in cystic fibrosis. A quantitative study including prevalence of pathologic findings among different age groups. Hum. Pathol. 1976, 7, 195–204. [Google Scholar] [CrossRef]
- Sobonya, R.E.; Taussig, L.M. Quantitative aspects of lung pathology in cystic fibrosis. Am. Rev. Respir. Dis. 1986, 134, 290–295. [Google Scholar] [CrossRef]
- Burgel, P.R.; Montani, D.; Danel, C.; Dusser, D.J.; Nadel, J.A. A morphometric study of mucins and small airway plugging in cystic fibrosis. Thorax 2007, 62, 153–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilliard, T.N.; Regamey, N.; Shute, J.K.; Nicholson, A.G.; Alton, E.W.; Bush, A.; Davies, J.C. Airway remodelling in children with cystic fibrosis. Thorax 2007, 62, 1074–1080. [Google Scholar] [CrossRef] [Green Version]
- Regamey, N.; Jeffery, P.K.; Alton, E.W.; Bush, A.; Davies, J.C. Airway remodelling and its relationship to inflammation in cystic fibrosis. Thorax 2011, 66, 624–629. [Google Scholar] [CrossRef] [Green Version]
- Pain, M.; Bermudez, O.; Lacoste, P.; Royer, P.J.; Botturi, K.; Tissot, A.; Brouard, S.; Eickelberg, O.; Magnan, A. Tissue remodelling in chronic bronchial diseases: From the epithelial to mesenchymal phenotype. Eur. Respir. Rev. Off. J. Eur. Respir. Soc. 2014, 23, 118–130. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Invest. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puchelle, E.; Zahm, J.M.; Tournier, J.M.; Coraux, C. Airway epithelial repair, regeneration, and remodeling after injury in chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2006, 3, 726–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adam, D.; Perotin, J.M.; Lebargy, F.; Birembaut, P.; Deslee, G.; Coraux, C. Regeneration of airway epithelium. Rev. Mal. Respir. 2014, 31, 300–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gohy, S.T.; Hupin, C.; Fregimilicka, C.; Detry, B.R.; Bouzin, C.; Gaide Chevronay, H.; Lecocq, M.; Weynand, B.; Ladjemi, M.Z.; Pierreux, C.E.; et al. Imprinting of the COPD airway epithelium for dedifferentiation and mesenchymal transition. Eur. Respir. J. 2015, 45, 1258–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nyabam, S.; Wang, Z.; Thibault, T.; Oluseyi, A.; Basar, R.; Marshall, L.; Griffin, M. A novel regulatory role for tissue transglutaminase in epithelial-mesenchymal transition in cystic fibrosis. Biochim. Biophys. Acta 2016, 1863, 2234–2244. [Google Scholar] [CrossRef] [PubMed]
- Collin, A.M.; Lecocq, M.; Detry, B.; Carlier, F.M.; Bouzin, C.; de Sany, P.; Hoton, D.; Verleden, S.; Froidure, A.; Pilette, C.; et al. Loss of ciliated cells and altered airway epithelial integrity in cystic fibrosis. J. Cyst. Fibros. 2021, 20, e129–e139. [Google Scholar] [CrossRef]
- Quaresma, M.C.; Pankonien, I.; Clarke, L.A.; Sousa, L.S.; Silva, I.A.L.; Railean, V.; Dousova, T.; Fuxe, J.; Amaral, M.D. Mutant CFTR Drives TWIST1 mediated epithelial-mesenchymal transition. Cell Death Dis. 2020, 11, 920. [Google Scholar] [CrossRef] [PubMed]
- Shah, V.S.; Meyerholz, D.K.; Tang, X.X.; Reznikov, L.; Abou Alaiwa, M.; Ernst, S.E.; Karp, P.H.; Wohlford-Lenane, C.L.; Heilmann, K.P.; Leidinger, M.R.; et al. Airway acidification initiates host defense abnormalities in cystic fibrosis mice. Science 2016, 351, 503–507. [Google Scholar] [CrossRef] [Green Version]
- Pezzulo, A.A.; Tang, X.X.; Hoegger, M.J.; Abou Alaiwa, M.H.; Ramachandran, S.; Moninger, T.O.; Karp, P.H.; Wohlford-Lenane, C.L.; Haagsman, H.P.; van Eijk, M.; et al. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature 2012, 487, 109–113. [Google Scholar] [CrossRef]
- Birket, S.E.; Chu, K.K.; Liu, L.; Houser, G.H.; Diephuis, B.J.; Wilsterman, E.J.; Dierksen, G.; Mazur, M.; Shastry, S.; Li, Y.; et al. A functional anatomic defect of the cystic fibrosis airway. Am. J. Respir. Crit. Care Med. 2014, 190, 421–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsui, H.; Grubb, B.R.; Tarran, R.; Randell, S.H.; Gatzy, J.T.; Davis, C.W.; Boucher, R.C. Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell 1998, 95, 1005–1015. [Google Scholar] [CrossRef] [Green Version]
- Balloy, V.; Varet, H.; Dillies, M.A.; Proux, C.; Jagla, B.; Coppee, J.Y.; Tabary, O.; Corvol, H.; Chignard, M.; Guillot, L. Normal and Cystic Fibrosis Human Bronchial Epithelial Cells Infected with Pseudomonas aeruginosa Exhibit Distinct Gene Activation Patterns. PLoS ONE 2015, 10, e0140979. [Google Scholar] [CrossRef]
- Ling, K.M.; Garratt, L.W.; Gill, E.E.; Lee, A.H.Y.; Agudelo-Romero, P.; Sutanto, E.N.; Iosifidis, T.; Rosenow, T.; Turvey, S.E.; Lassmann, T.; et al. Rhinovirus Infection Drives Complex Host Airway Molecular Responses in Children With Cystic Fibrosis. Front. Immunol. 2020, 11, 1327. [Google Scholar] [CrossRef] [PubMed]
- Carrabino, S.; Carpani, D.; Livraghi, A.; Di Cicco, M.; Costantini, D.; Copreni, E.; Colombo, C.; Conese, M. Dysregulated interleukin-8 secretion and NF-kappaB activity in human cystic fibrosis nasal epithelial cells. J. Cyst. Fibros. 2006, 5, 113–119. [Google Scholar] [CrossRef] [Green Version]
- Osika, E.; Cavaillon, J.M.; Chadelat, K.; Boule, M.; Fitting, C.; Tournier, G.; Clement, A. Distinct sputum cytokine profiles in cystic fibrosis and other chronic inflammatory airway disease. Eur. Respir. J. 1999, 14, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Tiringer, K.; Treis, A.; Fucik, P.; Gona, M.; Gruber, S.; Renner, S.; Dehlink, E.; Nachbaur, E.; Horak, F.; Jaksch, P.; et al. A Th17- and Th2-skewed cytokine profile in cystic fibrosis lungs represents a potential risk factor for Pseudomonas aeruginosa infection. Am. J. Respir. Crit. Care Med. 2013, 187, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; De, B.P.; Choudhary, S.; Comhair, S.A.; Goggans, T.; Slee, R.; Williams, B.R.; Pilewski, J.; Haque, S.J.; Erzurum, S.C. Impaired innate host defense causes susceptibility to respiratory virus infections in cystic fibrosis. Immunity 2003, 18, 619–630. [Google Scholar] [CrossRef] [Green Version]
- Sutanto, E.N.; Kicic, A.; Foo, C.J.; Stevens, P.T.; Mullane, D.; Knight, D.A.; Stick, S.M. Australian Respiratory Early Surveillance Team for Cystic Fibrosis. Innate inflammatory responses of pediatric cystic fibrosis airway epithelial cells: Effects of nonviral and viral stimulation. Am. J. Respir. Cell Mol. Biol. 2011, 44, 761–767. [Google Scholar] [CrossRef]
- Kaetzel, C.S. The polymeric immunoglobulin receptor: Bridging innate and adaptive immune responses at mucosal surfaces. Immunol. Rev. 2005, 206, 83–99. [Google Scholar] [CrossRef]
- Woof, J.M.; Kerr, M.A. The function of immunoglobulin A in immunity. J Pathol 2006, 208, 270–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerutti, A. The regulation of IgA class switching. Nat. Rev. Immunol. 2008, 8, 421–434. [Google Scholar] [CrossRef]
- Pilette, C.; Ouadrhiri, Y.; Godding, V.; Vaerman, J.P.; Sibille, Y. Lung mucosal immunity: Immunoglobulin-A revisited. Eur. Respir. J. 2001, 18, 571–588. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.R.; Chaker, L.; Ikram, M.A.; Peeters, R.P.; van Hagen, P.M.; Dalm, V. Determinants and Reference Ranges of Serum Immunoglobulins in Middle-Aged and Elderly Individuals: A Population-Based Study. J. Clin. Immunol. 2021, 41, 1902–1914. [Google Scholar] [CrossRef] [PubMed]
- Apodaca, G.; Katz, L.A.; Mostov, K.E. Receptor-mediated transcytosis of IgA in MDCK cells is via apical recycling endosomes. J. Cell Biol. 1994, 125, 67–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breitfeld, P.P.; Harris, J.M.; Mostov, K.E. Postendocytotic sorting of the ligand for the polymeric immunoglobulin receptor in Madin-Darby canine kidney cells. J. Cell Biol. 1989, 109, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Nagura, H.; Nakane, P.K.; Brown, W.R. Secretory component in immmunoglobulin deficiency: And immunoelectron microscopic study of intestinal epithelium. Scand J. Immunol. 1980, 12, 359–363. [Google Scholar] [CrossRef]
- Johansen, F.E.; Kaetzel, C.S. Regulation of the polymeric immunoglobulin receptor and IgA transport: New advances in environmental factors that stimulate pIgR expression and its role in mucosal immunity. Mucosal Immunol. 2011, 4, 598–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musil, L.S.; Baenziger, J.U. Cleavage of membrane secretory component to soluble secretory component occurs on the cell surface of rat hepatocyte monolayers. J. Cell Biol. 1987, 104, 1725–1733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, R.C.; Gibbons, R.J. Inhibition of bacterial adherence by secretory immunoglobulin A: A mechanism of antigen disposal. Science 1972, 177, 697–699. [Google Scholar] [CrossRef] [PubMed]
- Corthesy, B.; Benureau, Y.; Perrier, C.; Fourgeux, C.; Parez, N.; Greenberg, H.; Schwartz-Cornil, I. Rotavirus anti-VP6 secretory immunoglobulin A contributes to protection via intracellular neutralization but not via immune exclusion. J. Virol. 2006, 80, 10692–10699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazanec, M.B.; Coudret, C.L.; Fletcher, D.R. Intracellular neutralization of influenza virus by immunoglobulin A anti-hemagglutinin monoclonal antibodies. J. Virol. 1995, 69, 1339–1343. [Google Scholar] [CrossRef] [Green Version]
- Wright, A.; Yan, H.; Lamm, M.E.; Huang, Y.T. Immunoglobulin A antibodies against internal HIV-1 proteins neutralize HIV-1 replication inside epithelial cells. Virology 2006, 356, 165–170. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Chen, Z.; Ni, Y.; Li, Z. CFTR mutations in men with congenital bilateral absence of the vas deferens (CBAVD): A systemic review and meta-analysis. Hum. Reprod. 2012, 27, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Norderhaug, I.N.; Johansen, F.E.; Schjerven, H.; Brandtzaeg, P. Regulation of the formation and external transport of secretory immunoglobulins. Crit. Rev. Immunol. 1999, 19, 481–508. [Google Scholar]
- Pilette, C.; Ouadrhiri, Y.; Dimanche, F.; Vaerman, J.P.; Sibille, Y. Secretory component is cleaved by neutrophil serine proteinases but its epithelial production is increased by neutrophils through NF-kappa B- and p38 mitogen-activated protein kinase-dependent mechanisms. Am. J. Respir. Cell Mol. Biol. 2003, 28, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Renegar, K.B.; Jackson, G.D.; Mestecky, J. In vitro comparison of the biologic activities of monoclonal monomeric IgA, polymeric IgA, and secretory IgA. J. Immunol. 1998, 160, 1219–1223. [Google Scholar] [PubMed]
- Phalipon, A.; Cardona, A.; Kraehenbuhl, J.P.; Edelman, L.; Sansonetti, P.J.; Corthesy, B. Secretory component: A new role in secretory IgA-mediated immune exclusion in vivo. Immunity 2002, 17, 107–115. [Google Scholar] [CrossRef] [Green Version]
- Hammerschmidt, S.; Talay, S.R.; Brandtzaeg, P.; Chhatwal, G.S. SpsA, a novel pneumococcal surface protein with specific binding to secretory immunoglobulin A and secretory component. Mol. Microbiol. 1997, 25, 1113–1124. [Google Scholar] [CrossRef] [PubMed]
- Marshall, L.J.; Perks, B.; Ferkol, T.; Shute, J.K. IL-8 released constitutively by primary bronchial epithelial cells in culture forms an inactive complex with secretory component. J. Immunol. 2001, 167, 2816–2823. [Google Scholar] [CrossRef] [Green Version]
- Corthesy, B. Role of secretory immunoglobulin A and secretory component in the protection of mucosal surfaces. Future Microbiol. 2010, 5, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Gohy, S.T.; Detry, B.R.; Lecocq, M.; Bouzin, C.; Weynand, B.A.; Amatngalim, G.D.; Sibille, Y.M.; Pilette, C. Polymeric Immunoglobulin Receptor Down-regulation in Chronic Obstructive Pulmonary Disease. Persistence in the Cultured Epithelium and Role of Transforming Growth Factor-beta. Am. J. Respir. Crit. Care Med. 2014, 190, 509–521. [Google Scholar] [CrossRef]
- Pilette, C.; Godding, V.; Kiss, R.; Delos, M.; Verbeken, E.; Decaestecker, C.; De Paepe, K.; Vaerman, J.P.; Decramer, M.; Sibille, Y. Reduced epithelial expression of secretory component in small airways correlates with airflow obstruction in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2001, 163, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Polosukhin, V.V.; Cates, J.M.; Lawson, W.E.; Zaynagetdinov, R.; Milstone, A.P.; Massion, P.P.; Ocak, S.; Ware, L.B.; Lee, J.W.; Bowler, R.P.; et al. Bronchial secretory immunoglobulin a deficiency correlates with airway inflammation and progression of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2011, 184, 317–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hupin, C.; Rombaux, P.; Bowen, H.; Gould, H.; Lecocq, M.; Pilette, C. Downregulation of polymeric immunoglobulin receptor and secretory IgA antibodies in eosinophilic upper airway diseases. Allergy 2013, 68, 1589–1597. [Google Scholar] [CrossRef] [PubMed]
- Elm, C.; Braathen, R.; Bergmann, S.; Frank, R.; Vaerman, J.P.; Kaetzel, C.S.; Chhatwal, G.S.; Johansen, F.E.; Hammerschmidt, S. Ectodomains 3 and 4 of human polymeric Immunoglobulin receptor (hpIgR) mediate invasion of Streptococcus pneumoniae into the epithelium. J. Biol. Chem. 2004, 279, 6296–6304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodson, M.E.; Morris, L.; Batten, J.C. Serum immunoglobulins and immunoglobulin G subclasses in cystic fibrosis related to the clinical state of the patient. Eur. Respir. J. 1988, 1, 701–705. [Google Scholar] [PubMed]
- Hassan, J.; Feighery, C.; Bresnihan, B.; Keogan, M.; Fitzgerald, M.X.; Whelan, A. Serum IgA and IgG subclasses during treatment for acute respiratory exacerbation in cystic fibrosis: Analysis of patients colonised with mucoid or non-mucoid strains of pseudomonas aeruginosa. Immunol. Invest. 1994, 23, 1–13. [Google Scholar] [CrossRef]
- Van Bever, H.P.; Gigase, P.L.; De Clerck, L.S.; Bridts, C.H.; Franckx, H.; Stevens, W.J. Immune complexes and Pseudomonas aeruginosa antibodies in cystic fibrosis. Arch. Dis. Child. 1988, 63, 1222–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konstan, M.W.; Hilliard, K.A.; Norvell, T.M.; Berger, M. Bronchoalveolar lavage findings in cystic fibrosis patients with stable, clinically mild lung disease suggest ongoing infection and inflammation. Am. J. Respir. Crit. Care Med. 1994, 150, 448–454. [Google Scholar] [CrossRef]
- Marshall, L.J.; Perks, B.; Bodey, K.; Suri, R.; Bush, A.; Shute, J.K. Free secretory component from cystic fibrosis sputa displays the cystic fibrosis glycosylation phenotype. Am. J. Respir. Crit. Care Med. 2004, 169, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Schiotz, P.O.; Hoiby, N.; Permin, H.; Wiik, A. IgA and IgG antibodies against surface antigens of Pseudomonas aeruginosa in sputum and serum from patients with cystic fibrosis. Acta Pathol. Microbiol. Scand. Sect. C Immunol. 1979, 87, 229–233. [Google Scholar]
- Pedersen, S.S.; Espersen, F.; Hoiby, N.; Jensen, T. Immunoglobulin A and immunoglobulin G antibody responses to alginates from Pseudomonas aeruginosa in patients with cystic fibrosis. J. Clin. Microbiol. 1990, 28, 747–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kronborg, G.; Fomsgaard, A.; Galanos, C.; Freudenberg, M.A.; Hoiby, N. Antibody responses to lipid A, core, and O sugars of the Pseudomonas aeruginosa lipopolysaccharide in chronically infected cystic fibrosis patients. J. Clin. Microbiol. 1992, 30, 1848–1855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aanaes, K.; Johansen, H.K.; Poulsen, S.S.; Pressler, T.; Buchwald, C.; Hoiby, N. Secretory IgA as a diagnostic tool for Pseudomonas aeruginosa respiratory colonization. J. Cyst. Fibros. 2013, 12, 81–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, J.; McGarry, D.P.; Joseph, N.; Peppers, B.; Hostoffer, R. Salivary IgA deficiency in a patient with cystic fibrosis (genotype M470V/V520F). Ann. Allergy Asthma Immunol. 2018, 121, 619–620. [Google Scholar] [CrossRef]
- Hallberg, K.; Mattsson-Rydberg, A.; Fandriks, L.; Strandvik, B. Gastric IgA in cystic fibrosis in relation to the migrating motor complex. Scand J. Gastroenterol 2001, 36, 843–848. [Google Scholar] [CrossRef]
- Eckman, E.A.; Mallender, W.D.; Szegletes, T.; Silski, C.L.; Schreiber, J.R.; Davis, P.B.; Ferkol, T.W. In vitro transport of active alpha(1)-antitrypsin to the apical surface of epithelia by targeting the polymeric immunoglobulin receptor. Am. J. Respir. Cell Mol. Biol. 1999, 21, 246–252. [Google Scholar] [CrossRef]
- Collin, A.M.; Lecocq, M.; Noel, S.; Detry, B.; Carlier, F.M.; Aboubakar Nana, F.; Bouzin, C.; Leal, T.; Vermeersch, M.; De Rose, V.; et al. Lung immunoglobulin A immunity dysregulation in cystic fibrosis. EBioMedicine 2020, 60, 102974. [Google Scholar] [CrossRef] [PubMed]
- Frija-Masson, J.; Martin, C.; Regard, L.; Lothe, M.N.; Touqui, L.; Durand, A.; Lucas, B.; Damotte, D.; Alifano, M.; Fajac, I.; et al. Bacteria-driven peribronchial lymphoid neogenesis in bronchiectasis and cystic fibrosis. Eur. Respir. J. 2017, 49, 1601873. [Google Scholar] [CrossRef] [Green Version]
- Grootjans, J.; Krupka, N.; Hosomi, S.; Matute, J.D.; Hanley, T.; Saveljeva, S.; Gensollen, T.; Heijmans, J.; Li, H.; Limenitakis, J.P.; et al. Epithelial endoplasmic reticulum stress orchestrates a protective IgA response. Science 2019, 363, 993–998. [Google Scholar] [CrossRef] [Green Version]
- Tan, H.L.; Regamey, N.; Brown, S.; Bush, A.; Lloyd, C.M.; Davies, J.C. The Th17 pathway in cystic fibrosis lung disease. Am. J. Respir. Crit. Care Med. 2011, 184, 252–258. [Google Scholar] [CrossRef]
- Kushwah, R.; Gagnon, S.; Sweezey, N.B. Intrinsic predisposition of naive cystic fibrosis T cells to differentiate towards a Th17 phenotype. Respir. Res. 2013, 14, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberson, E.C.; Tully, J.E.; Guala, A.S.; Reiss, J.N.; Godburn, K.E.; Pociask, D.A.; Alcorn, J.F.; Riches, D.W.; Dienz, O.; Janssen-Heininger, Y.M.; et al. Influenza induces endoplasmic reticulum stress, caspase-12-dependent apoptosis, and c-Jun N-terminal kinase-mediated transforming growth factor-beta release in lung epithelial cells. Am. J. Respir. Cell Mol. Biol. 2012, 46, 573–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazel-Sanchez, B.; Iwaszkiewicz, J.; Bonifacio, J.P.P.; Silva, F.; Niu, C.; Strohmeier, S.; Eletto, D.; Krammer, F.; Tan, G.; Zoete, V.; et al. Influenza A viruses balance ER stress with host protein synthesis shutoff. Proc. Natl. Acad. Sci. USA 2021, 118, e2024681118. [Google Scholar] [CrossRef]
- Palm, N.W.; de Zoete, M.R.; Cullen, T.W.; Barry, N.A.; Stefanowski, J.; Hao, L.; Degnan, P.H.; Hu, J.; Peter, I.; Zhang, W.; et al. Immunoglobulin A coating identifies colitogenic bacteria in inflammatory bowel disease. Cell 2014, 158, 1000–1010. [Google Scholar] [CrossRef] [Green Version]
- Rochereau, N.; Roblin, X.; Michaud, E.; Gayet, R.; Chanut, B.; Jospin, F.; Corthesy, B.; Paul, S. NOD2 deficiency increases retrograde transport of secretory IgA complexes in Crohn’s disease. Nat. Commun. 2021, 12, 261. [Google Scholar] [CrossRef] [PubMed]
- Bunker, J.J.; Erickson, S.A.; Flynn, T.M.; Henry, C.; Koval, J.C.; Meisel, M.; Jabri, B.; Antonopoulos, D.A.; Wilson, P.C.; Bendelac, A. Natural polyreactive IgA antibodies coat the intestinal microbiota. Science 2017, 358, eaan6619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catanzaro, J.R.; Strauss, J.D.; Bielecka, A.; Porto, A.F.; Lobo, F.M.; Urban, A.; Schofield, W.B.; Palm, N.W. IgA-deficient humans exhibit gut microbiota dysbiosis despite secretion of compensatory IgM. Sci. Rep. 2019, 9, 13574. [Google Scholar] [CrossRef] [PubMed]
- Price, C.E.; O’Toole, G.A. The Gut-Lung Axis in Cystic Fibrosis. J. Bacteriol. 2021, 203, e0031121. [Google Scholar] [CrossRef] [PubMed]
- Hoen, A.G.; Li, J.; Moulton, L.A.; O’Toole, G.A.; Housman, M.L.; Koestler, D.C.; Guill, M.F.; Moore, J.H.; Hibberd, P.L.; Morrison, H.G.; et al. Associations between Gut Microbial Colonization in Early Life and Respiratory Outcomes in Cystic Fibrosis. J. Pediatr. 2015, 167, 138–147.e1-3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruane, D.; Chorny, A.; Lee, H.; Faith, J.; Pandey, G.; Shan, M.; Simchoni, N.; Rahman, A.; Garg, A.; Weinstein, E.G.; et al. Microbiota regulate the ability of lung dendritic cells to induce IgA class-switch recombination and generate protective gastrointestinal immune responses. J. Exp. Med. 2016, 213, 53–73. [Google Scholar] [CrossRef] [PubMed]
- Sagel, S.D.; Kapsner, R.; Osberg, I.; Sontag, M.K.; Accurso, F.J. Airway inflammation in children with cystic fibrosis and healthy children assessed by sputum induction. Am. J. Respir. Crit. Care Med. 2001, 164, 1425–1431. [Google Scholar] [CrossRef]
- Wilke, M.; Buijs-Offerman, R.M.; Aarbiou, J.; Colledge, W.H.; Sheppard, D.N.; Touqui, L.; Bot, A.; Jorna, H.; de Jonge, H.R.; Scholte, B.J. Mouse models of cystic fibrosis: Phenotypic analysis and research applications. J. Cyst. Fibros. 2011, 10 (Suppl. 2), S152–S171. [Google Scholar] [CrossRef] [Green Version]
- Yi, B.; Dalpke, A.H.; Boutin, S. Changes in the Cystic Fibrosis Airway Microbiome in Response to CFTR Modulator Therapy. Front. Cell. Infect. Microbiol. 2021, 11, 548613. [Google Scholar] [CrossRef] [PubMed]



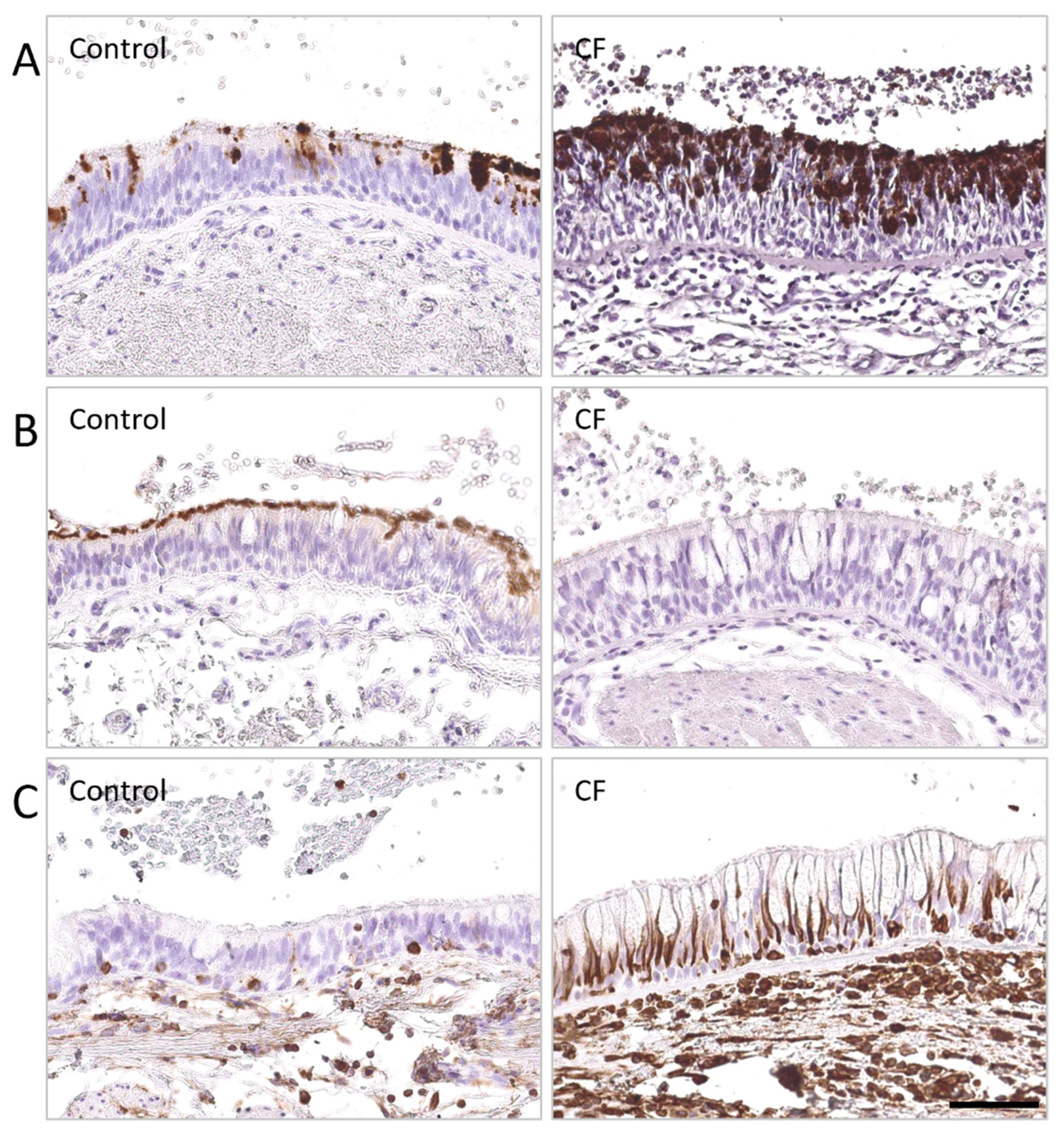{kind=link}
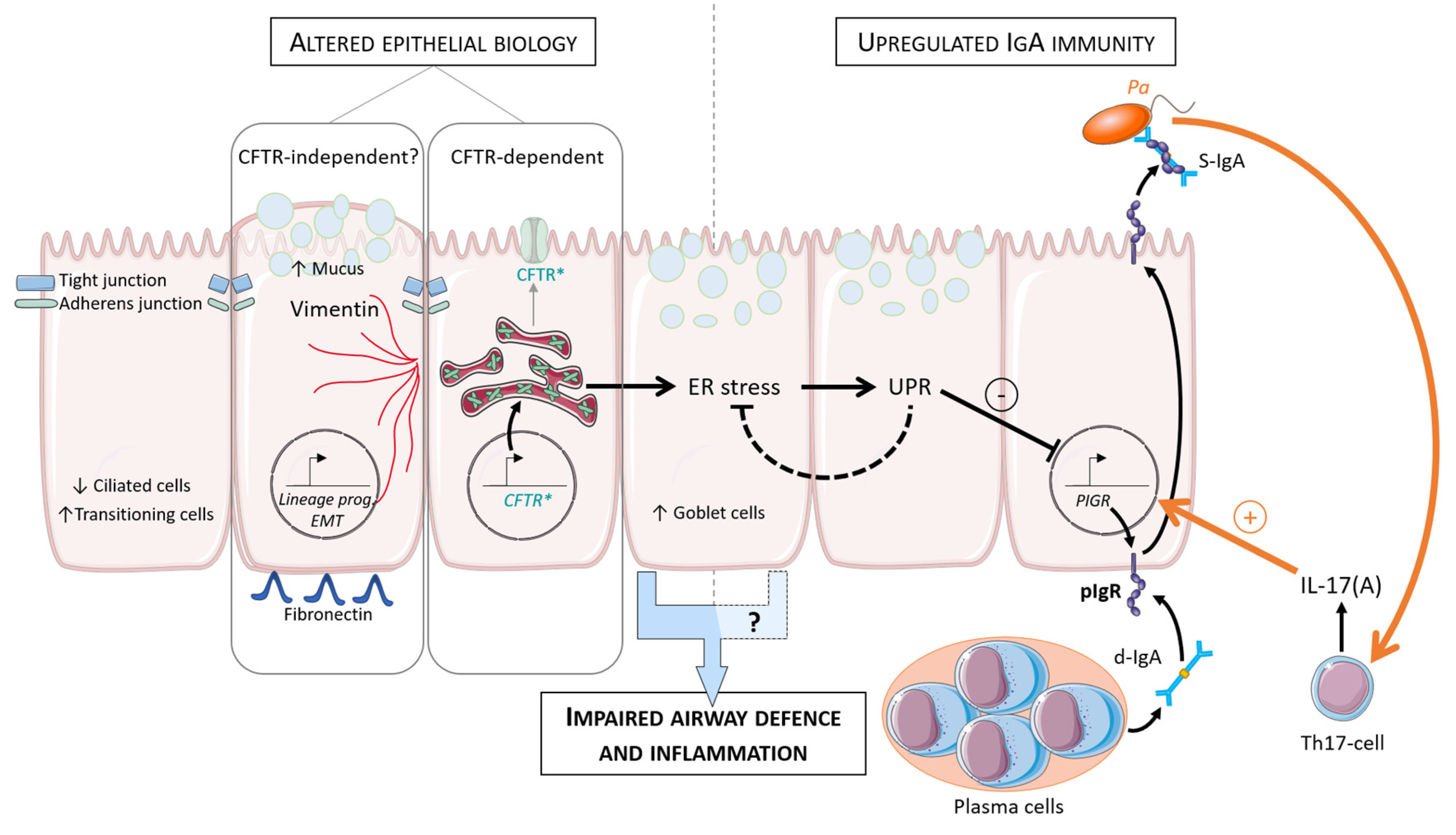{kind=link}
| Findings | Model | Study |
|---|---|---|
| Squamous metaplasia, inflammatory infiltrates, bronchiectasis, mucopurulent plugs | Human autopsies | Bedrossian et al., 1976 [57] Sobonya and Taussig, 1986 [58] |
| Decrease of ciliated cells and increase of goblet cells | Lung explants | Burgel et al., 2007 [59] Collin et al., 2021 [68] |
| Thickening of the reticular basement membrane | Endobronchial biopsies (CF children) | Hilliard et al., 2007 [60] |
| Degradation of structural components of the extracellular matrix | Endobronchial biopsies (CF children) | Regamey et al., 2011 [61] |
| Increased expression of fibronectin and N-cadherin | IB3 cells (CF bronchial cell line) | Nyabam et al., 2016 [67] |
| Decreased ciliated and goblet cells numbers Increased mesenchymal markers expression Increased B-cells, peri-bronchial lymphoid aggregates | Lung explants, air–liquid interface human lower airway culture, C57BL/6N mice and F508del mice | Collin et al., 2021 [68] Frija-Masson et al., 2017 [121] |
| CFTR modulators reverse EMT process | Lung explants, primary human bronchial cells and cell lines (CFBE41o-) | Quaresma et al., 2020 [69] |
| Altered epithelial differentiation (less basal cells, more cells transitioning to ciliated and secretory cells) | Lung explants, air–liquid interface human lower airway culture | Carraro et al., 2021 [55] |
| Exaggerated inflammatory response (basal dysregulated production of IL-8) | Primary nasal epithelial culture | Carrabino et al., 2006 [76] |
| Exaggerated inflammatory response | Sputum and BAL (CF children) | Osika et al., 1999 [77] Tiringer et al., [78] |
| Reduced periciliary liquid volume Decreased pH of airway surface liquid impairing bacterial killing and mucociliary transport | Primary airway epithelial cultures from humans, pigs and mice Pigs trachea | Matsui et al., [73] Shah et al., 2016 [70] Pezzulo et al., 2012 [71] Birket et al., 2014 [72] |
| Innate antiviral defense altered in CF (human parainfluenza 3 virus) | A549 and CV-1 cells, primary bronchial epithelial culture | Zheng et al., 2003 [79] |
| Innate antiviral/bacterial defense altered in CF (rhinovirus, Pseudomonas) | Air–liquid interface human lower airway culture (CF children) | Ling et al., 2020 [75] Balloy et al., 2015 [74] |
| Increased IgA concentration | Serum Sputum and bronchoalveolar lavage | Hodson et al., 1988 [108] Hassan et al., 1994 [109] Van Bever et al., 1988 [110] Konstan et al., 1994 [111] Collin et al., 2020 [120] |
| Increased free SC | Sputum | Marshall et al., 2004 [112] |
| Increased specific anti-Pseudomonas aeruginosa IgA | Serum Sputum Nasal secretion | Schiotz et al., 1979 [113] Kronborg et al., 1992 [115] Pedersen et al., 1990 [114] Aanaes et al., 2013 [116] |
| Decreased IgA secretion | Saliva Gastric luminal perfusate | Oh et al., 2018 [117] Hallberg et al., 2001 [118] |
| Pseudomonas aeruginosa infection overcomes pIgR and IgA down-regulation through an IL-17 inflammatory host response | F508del mice | Collin et al., 2020 [120] |
| Increased Th17 inflammation | Buffy coat, F508del mice. Bronchoalveolar lavage, endobronchial biopsies | Kushwah et al., 2013 [124] Tan et al., 2011 [123] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gohy, S.; Moeremans, A.; Pilette, C.; Collin, A. Immunoglobulin A Mucosal Immunity and Altered Respiratory Epithelium in Cystic Fibrosis. Cells 2021, 10, 3603. https://doi.org/10.3390/cells10123603
Gohy S, Moeremans A, Pilette C, Collin A. Immunoglobulin A Mucosal Immunity and Altered Respiratory Epithelium in Cystic Fibrosis. Cells. 2021; 10(12):3603. https://doi.org/10.3390/cells10123603
Chicago/Turabian StyleGohy, Sophie, Alexandra Moeremans, Charles Pilette, and Amandine Collin. 2021. "Immunoglobulin A Mucosal Immunity and Altered Respiratory Epithelium in Cystic Fibrosis" Cells 10, no. 12: 3603. https://doi.org/10.3390/cells10123603
APA StyleGohy, S., Moeremans, A., Pilette, C., & Collin, A. (2021). Immunoglobulin A Mucosal Immunity and Altered Respiratory Epithelium in Cystic Fibrosis. Cells, 10(12), 3603. https://doi.org/10.3390/cells10123603