
Inhibition of HER Receptors Reveals Distinct Mechanisms of Compensatory Upregulation of Other HER Family Members: Basis for Acquired Resistance and for Combination Therapy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. siRNAs, Plasmids, Tissue Culture, and Animals
2.2. mRNA Preparation, miRNA Preparation and Quantitative RT-PCR
2.3. In Vitro Transfections and Growth Assays
2.4. Apoptosis Assay
2.5. ELISA, FACS Analysis and Immunohistochemistry
2.6. Therapeutic Knockdown in Subcutaneous Tumor Xenografts
3. Results
3.1. The Single Knockdown of HER2 or HER3 Leads to the Rapid Counter-Upregulation of Other HER Family Members
3.2. Combined RNAi-Mediated Knockdown of HER Family Members
3.3. Additive Effects of Double Knockdown of HER Family Members on Cell Proliferation and Apoptosis
3.4. Enhanced Anti-Tumor Effects In Vivo upon Therapeutic HER2/HER3 Double Knockdown through Systemic Treatment of Mice with siRNAs
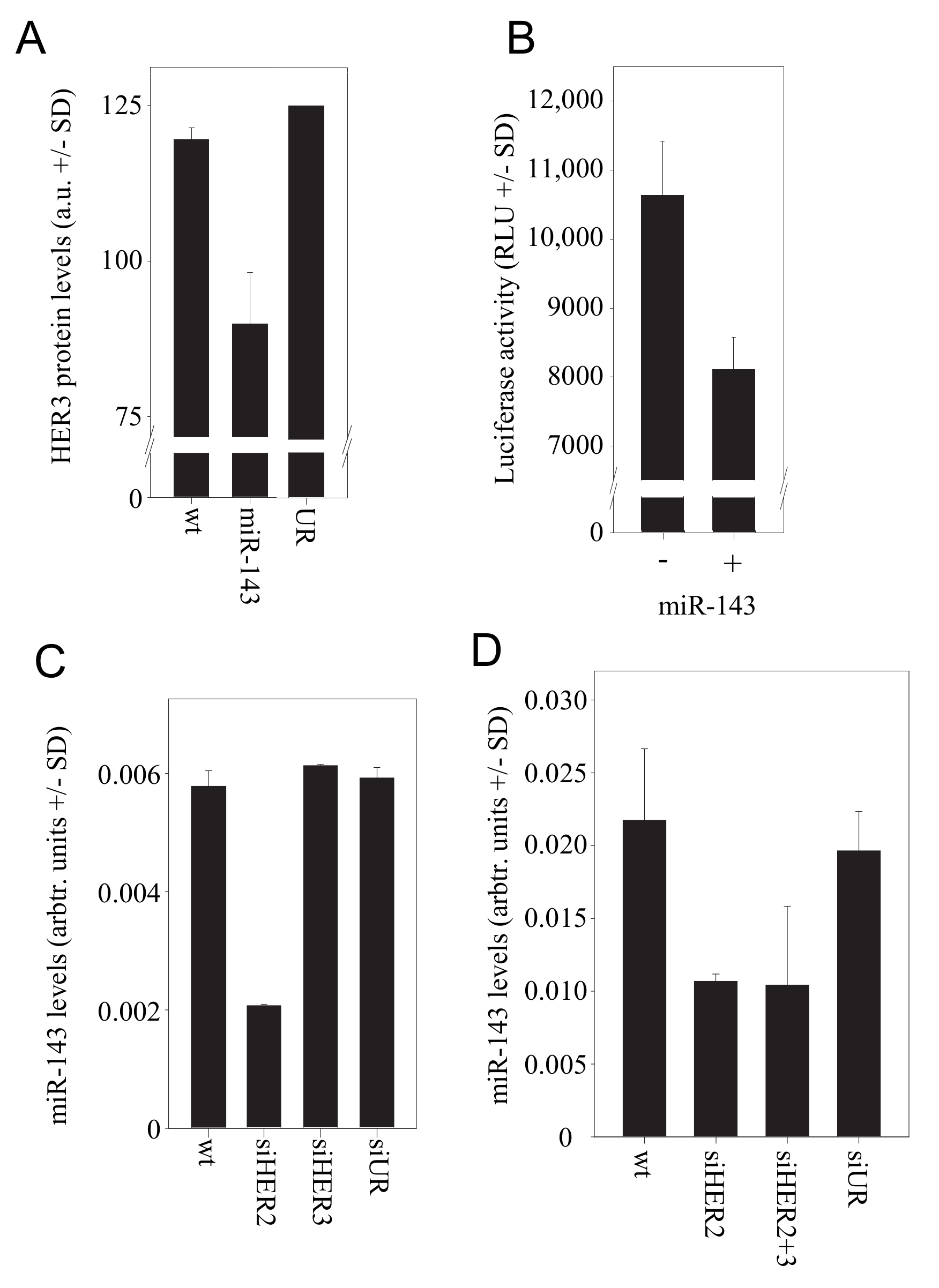
3.5. The microRNA miR-143 Mediates the Counter-Upregulation of HER3 upon HER2 Knockdown
3.6. The Transcriptional Regulator SATB1 Is Responsible for the Inverse Correlation between HER3 and HER2
3.7. The Counter-Regulatory Network of HER Family Members Is also Observed upon Small Molecule Inhibitor- or Antibody-Mediated Inhibition
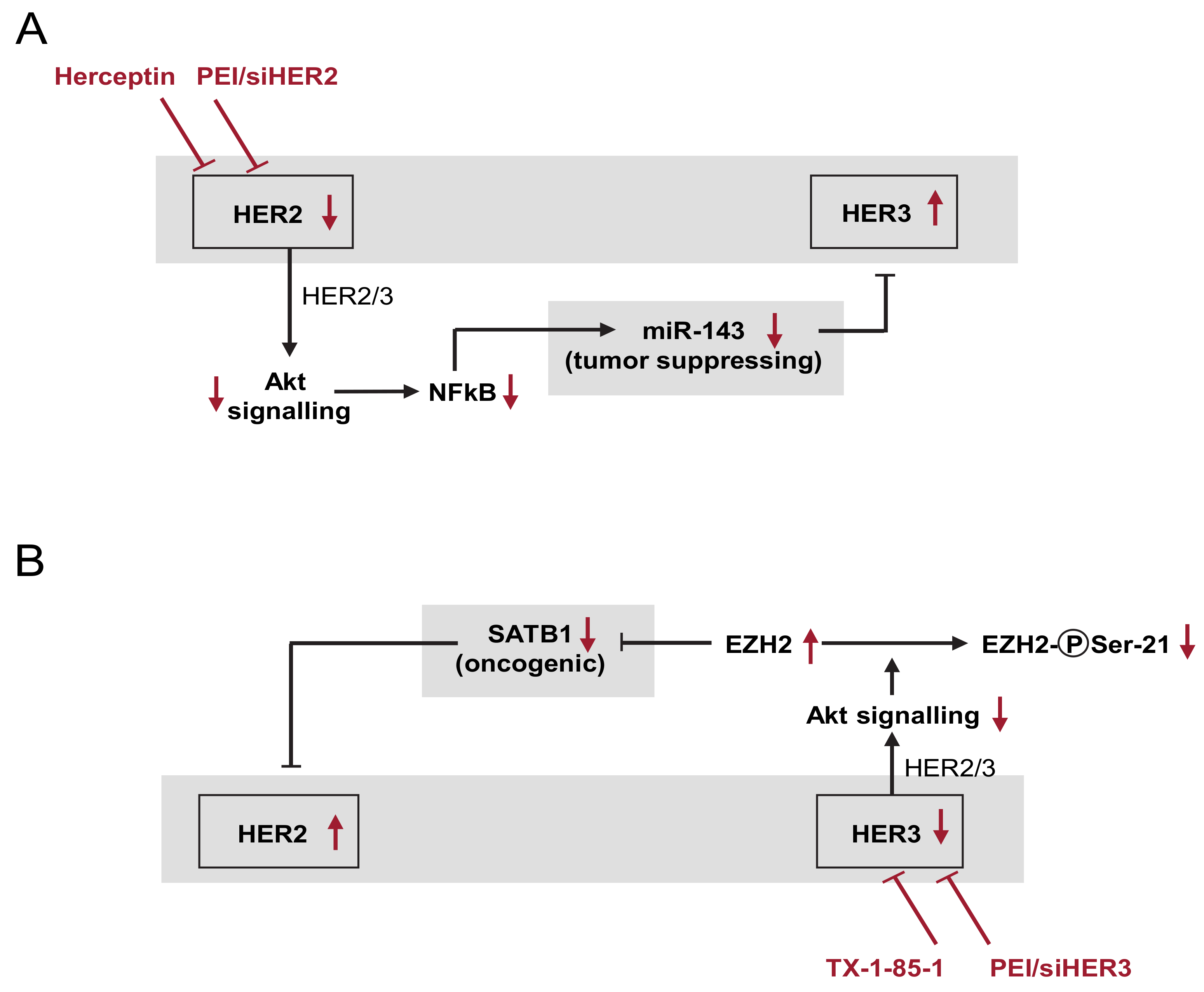
3.8. Model of the Network of Mutual and Compensatory Regulation of HER Expression
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nature reviews. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar]
- Hynes, N.E.; MacDonald, G. ErbB receptors and signaling pathways in cancer. Curr. Opin. Cell Biol. 2009, 21, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.; Ullrich, A.; McGuire, R.L. Human breast cancer: Correlation of relapse and survival with amplification of the HER2/neu oncogene. Science 1987, 235, 177–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, J.S.; Fletcher, J.A. The HER-2/neu oncogene in breast cancer: Prognostic factor, predictive factor, and target for therapy. Stem Cells 1998, 16, 413–428. [Google Scholar] [PubMed]
- Jayson, G.C.; Kohn, E.C.; Kitchener, H.C.; Ledermann, J.A. Ovarian cancer. Lancet 2014, 384, 1376–1388. [Google Scholar] [CrossRef]
- Lurje, G.; Lenz, H.J. EGFR signaling and drug discovery. Oncology 2009, 77, 400–410. [Google Scholar]
- Citri, A.; Yarden, Y. EGF-ERBB signalling: Towards the systems level. Nature reviews. Mol. Cell Biol. 2006, 7, 505–516. [Google Scholar]
- Lyu, H.; Han, A.; Polsdofer, E.; Liu, S.; Liu, B. Understanding the biology of HER3 receptor as a therapeutic target in human cancer. Acta Pharm. Sin. B 2018, 8, 503–510. [Google Scholar] [CrossRef]
- Vogel, C.L.; Cobleigh, M.A.; Tripathy, D.; Gutheil, J.C.; Harris, L.N.; Fehrenbacher, L.; Slamon, D.J.; Murphy, M.; Novotny, W.F.; Burchmore, M.; et al. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J. Clin. Oncol. 2002, 20, 719–726. [Google Scholar] [CrossRef]
- Kaufman, B.; Trudeau, M.; Awada, A.; Blackwell, K.; Bachelot, T.; Salazar, V.; DeSilvio, M.; Westlund, R.; Zaks, T.; Spector, N.; et al. Lapatinib monotherapy in patients with HER2-overexpressing relapsed or refractory inflammatory breast cancer: Final results and survival of the expanded HER2+ cohort in EGF103009, a phase II study. Lancet Oncol. 2009, 10, 581–588. [Google Scholar] [CrossRef]
- Baselga, J.; Albanell, J.; Ruiz, A.; Lluch, A.; Gascon, P.; Guillem, V.; Gonzalez, S.; Sauleda, S.; Marimon, I.; Tabernero, J.M.; et al. Phase II and tumor pharmacodynamic study of gefitinib in patients with advanced breast cancer. J. Clin. Oncol. 2005, 23, 5323–5333. [Google Scholar] [CrossRef] [PubMed]
- Kiavue, N.; Cabel, L.; Melaabi, S.; Bataillon, G.; Callens, C.; Lerebours, F.; Pierga, J.Y.; Bidard, F.C. ERBB3 mutations in cancer: Biological aspects, prevalence and therapeutics. Oncogene 2020, 39, 487–502. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Q.; Liu, X.; Fleming, E.; Yuan, K.; Piao, H.; Chen, J.; Moustafa, Z.; Thomas, R.K.; Greulich, H.; Schinzel, A.; et al. An activated ErbB3/NRG1 autocrine loop supports in vivo proliferation in ovarian cancer cells. Cancer Cell 2010, 17, 298–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holbro, T.; Beerli, R.R.; Maurer, F.; Koziczak, M.; Barbas, C.F., 3rd; Hynes, N.E. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc. Natl. Acad. Sci. USA 2003, 100, 8933–8938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee-Hoeflich, S.T.; Crocker, L.; Yao, E.; Pham, T.; Munroe, X.; Hoeflich, K.P.; Sliwkowski, M.X.; Stern, H.M. A central role for HER3 in HER2-amplified breast cancer: Implications for targeted therapy. Cancer Res. 2008, 68, 5878–5887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacob, W.; James, I.; Hasmann, M.; Weisser, M. Clinical development of HER3-targeting monoclonal antibodies: Perils and progress. Cancer Treat Rev. 2018, 68, 111–123. [Google Scholar] [CrossRef]
- Mishra, R.; Hanker, A.B.; Garrett, J.T. Genomic alterations of ERBB receptors in cancer: Clinical implications. Oncotarget 2017, 8, 114371–114392. [Google Scholar] [CrossRef] [Green Version]
- Sampera, A.; Sanchez-Martin, F.J.; Arpi, O.; Visa, L.; Iglesias, M.; Menendez, S.; Gaye, E.; Dalmases, A.; Clave, S.; Gelabert-Baldrich, M.; et al. HER-Family Ligands Promote Acquired Resistance to Trastuzumab in Gastric Cancer. Mol. Cancer Ther. 2019, 18, 2135–2145. [Google Scholar] [CrossRef] [Green Version]
- Garrett, J.T.; Arteaga, C.L. Resistance to HER2-directed antibodies and tyrosine kinase inhibitors: Mechanisms and clinical implications. Cancer Biol. Ther. 2011, 11, 793–800. [Google Scholar] [CrossRef] [Green Version]
- Garrett, J.T.; Olivares, M.G.; Rinehart, C.; Granja-Ingram, N.D.; Sanchez, V.; Chakrabarty, A.; Dave, B.; Cook, R.S.; Pao, W.; McKinely, E.; et al. Transcriptional and posttranslational up-regulation of HER3 (ErbB3) compensates for inhibition of the HER2 tyrosine kinase. Proc. Natl. Acad. Sci. USA 2011, 108, 5021–5026. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Xu, Y.; Ding, Y.; Li, C.; Zhao, H.; Wang, J.; Meng, S. Posttranscriptional upregulation of HER3 by HER2 mRNA induces trastuzumab resistance in breast cancer. Mol. Cancer 2018, 17, 113. [Google Scholar] [CrossRef] [PubMed]
- Pinkas-Kramarski, R.; Soussan, L.; Waterman, H.; Levkowitz, G.; Alroy, I.; Klapper, L.; Lavi, S.; Seger, R.; Ratzkin, B.J.; Sela, M.; et al. Diversification of Neu differentiation factor and epidermal growth factor signaling by combinatorial receptor interactions. EMBO J. 1996, 15, 2452–2467. [Google Scholar] [CrossRef] [PubMed]
- Graus-Porta, D.; Beerli, R.R.; Daly, J.M.; Hynes, N.E. ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO J. 1997, 16, 1647–1655. [Google Scholar] [CrossRef] [PubMed]
- Wallasch, C.; Weiss, F.U.; Niederfellner, G.; Jallal, B.; Issing, W.; Ullrich, A. Heregulin-dependent regulation of HER2/neu oncogenic signaling by heterodimerization with HER3. EMBO J. 1995, 14, 4267–4275. [Google Scholar] [CrossRef] [PubMed]
- Klapper, L.N.; Glathe, S.; Vaisman, N.; Hynes, N.E.; Andrews, G.C.; Sela, M.; Yarden, Y. The ErbB-2/HER2 oncoprotein of human carcinomas may function solely as a shared coreceptor for multiple stroma-derived growth factors. Proc. Natl. Acad. Sci. USA 1999, 96, 4995–5000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guy, P.M.; Platko, J.V.; Cantley, L.C.; Cerione, R.A.; Carraway, K.L., 3rd. Insect cell-expressed p180erbB3 possesses an impaired tyrosine kinase activity. Proc. Natl. Acad. Sci. USA 1994, 91, 8132–8136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alimandi, M.; Romano, A.; Curia, M.C.; Muraro, R.; Fedi, P.; Aaronson, S.A.; Di Fiore, P.P.; Kraus, M.H. Cooperative signaling of erbB3 and erbB2 in neoplastic transformation and human malignancies. Oncogene 1995, 10, 1813–1821. [Google Scholar] [PubMed]
- Zamore, P.D.; Tuschl, T.; Sharp, P.A.; Bartel, D.P. RNAi: Double-stranded RNA directs the ATP-dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell 2000, 101, 25–33. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.; Park, J.H.; Sailor, M.J. Rekindling RNAi Therapy: Materials Design Requirements for In Vivo siRNA Delivery. Adv. Mater. 2019, 31, 1903637. [Google Scholar] [CrossRef] [Green Version]
- Hampl, V.; Martin, C.; Aigner, A.; Hoebel, S.; Singer, S.; Frank, N.; Sarikas, A.; Ebert, O.; Prywes, R.; Gudermann, T.; et al. Depletion of the transcriptional coactivators Megakaryoblastic Leukemia 1 and 2 abolishes hepatocellular carcinoma xenograft growth by inducing oncogene-induced senescence. EMBO Mol. Med. 2013, 5, 1367–1382. [Google Scholar] [CrossRef]
- Hobel, S.; Koburger, I.; John, M.; Czubayko, F.; Hadwiger, P.; Vornlocher, H.P.; Aigner, A. Polyethylenimine/small interfering RNA-mediated knockdown of vascular endothelial growth factor in vivo exerts anti-tumor effects synergistically with Bevacizumab. J. Gene Med. 2010, 12, 287–300. [Google Scholar]
- Hendruschk, S.; Wiedemuth, R.; Aigner, A.; Topfer, K.; Cartellieri, M.; Martin, D.; Kirsch, M.; Ikonomidou, C.; Schackert, G.; Temme, A. RNA interference targeting survivin exerts antitumoral effects in vitro and in established glioma xenografts in vivo. Neuro Oncol. 2011, 13, 1074–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hobel, S.; Aigner, A. Polyethylenimines for siRNA and miRNA delivery in vivo. Wiley interdisciplinary reviews. Nanomed. Nanobiotechnol. 2013, 5, 485–501. [Google Scholar] [CrossRef] [PubMed]
- Urban-Klein, B.; Werth, S.; Abuharbeid, S.; Czubayko, F.; Aigner, A. RNAi-mediated gene-targeting through systemic application of polyethylenimine (PEI)-complexed siRNA in vivo. Gene Ther. 2005, 12, 461–466. [Google Scholar] [CrossRef]
- Ibrahim, A.F.; Weirauch, U.; Thomas, M.; Grunweller, A.; Hartmann, R.K.; Aigner, A. MicroRNA replacement therapy for miR-145 and miR-33a is efficacious in a model of colon carcinoma. Cancer Res. 2011, 71, 5214–5224. [Google Scholar] [CrossRef] [Green Version]
- Moasser, M.M.; Basso, A.; Averbuch, S.D.; Rosen, N. The tyrosine kinase inhibitor ZD1839 (“Iressa”) inhibits HER2-driven signaling and suppresses the growth of HER2-overexpressing tumor cells. Cancer Res. 2001, 61, 7184–7188. [Google Scholar] [PubMed]
- Aigner, A. Nonviral in vivo delivery of therapeutic small interfering RNAs. Curr. Opin. Mol. Ther. 2007, 9, 345–352. [Google Scholar]
- Mattie, M.D.; Benz, C.C.; Bowers, J.; Sensinger, K.; Wong, L.; Scott, G.K.; Fedele, V.; Ginzinger, D.; Getts, R.; Haqq, C. Optimized high-throughput microRNA expression profiling provides novel biomarker assessment of clinical prostate and breast cancer biopsies. Mol. Cancer 2006, 5, 24. [Google Scholar] [CrossRef] [Green Version]
- Motoyama, K.; Inoue, H.; Takatsuno, Y.; Tanaka, F.; Mimori, K.; Uetake, H.; Sugihara, K.; Mori, M. Over- and under-expressed microRNAs in human colorectal cancer. Int. J. Oncol. 2009, 34, 1069–1075. [Google Scholar]
- Han, H.J.; Russo, J.; Kohwi, Y.; Kohwi-Shigematsu, T. SATB1 reprogrammes gene expression to promote breast tumour growth and metastasis. Nature 2008, 452, 187–193. [Google Scholar] [CrossRef]
- Fromberg, A.; Engeland, K.; Aigner, A. The Special AT-rich Sequence Binding Protein 1 (SATB1) and its role in solid tumors. Cancer Lett. 2018, 417, 96–111. [Google Scholar] [CrossRef] [PubMed]
- Iorns, E.; Hnatyszyn, H.J.; Seo, P.; Clarke, J.; Ward, T.; Lippman, M. The role of SATB1 in breast cancer pathogenesis. J. Natl. Cancer Inst. 2010, 102, 1284–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, D.; Ueno, L.; Vogt, P.K. Akt-mediated regulation of NFkappaB and the essentialness of NFkappaB for the oncogenicity of PI3K and Akt. Int. J. Cancer 2009, 125, 2863–2870. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Liu, S.; Hu, T.; Liu, S.; He, Y.; Sun, S. Up-regulated microRNA-143 transcribed by nuclear factor kappa B enhances hepatocarcinoma metastasis by repressing fibronectin expression. Hepatology 2009, 50, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Ritter, C.A.; Perez-Torres, M.; Rinehart, C.; Guix, M.; Dugger, T.; Engelman, J.A.; Arteaga, C.L. Human breast cancer cells selected for resistance to trastuzumab in vivo overexpress epidermal growth factor receptor and ErbB ligands and remain dependent on the ErbB receptor network. Clin. Cancer Res. 2007, 13, 4909–4919. [Google Scholar] [CrossRef] [Green Version]
- Sergina, N.V.; Rausch, M.; Wang, D.; Blair, J.; Hann, B.; Shokat, K.M.; Moasser, M.M. Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER3. Nature 2007, 445, 437–441. [Google Scholar]
- Wang, Y.; Lee, C.G. MicroRNA and cancer--focus on apoptosis. J. Cell. Mol. Med. 2009, 13, 12–23. [Google Scholar]
- Yan, X.; Chen, X.; Liang, H.; Deng, T.; Chen, W.; Zhang, S.; Liu, M.; Gao, X.; Liu, Y.; Zhao, C.; et al. miR-143 and miR-145 synergistically regulate ERBB3 to suppress cell proliferation and invasion in breast cancer. Mol. Cancer 2014, 13, 220. [Google Scholar] [CrossRef] [Green Version]
- Cha, T.L.; Zhou, B.P.; Xia, W.; Wu, Y.; Yang, C.C.; Chen, C.T.; Ping, B.; Otte, A.P.; Hung, M.C. Akt-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science 2005, 310, 306–310. [Google Scholar] [CrossRef]
- Lei, L.; Lu, L.; Xiang, L.; Xue-song, W.; De-pei, L.; Chih-chuan, L. Epigenetic repression of SATB1 by polycomb group protein EZH2 in epithelial cells. Chin. Med. Sci. J. 2010, 25, 199–205. [Google Scholar] [CrossRef]
- Ghosh, R.; Narasanna, A.; Wang, S.E.; Liu, S.; Chakrabarty, A.; Balko, J.M.; Gonzalez-Angulo, A.M.; Mills, G.B.; Penuel, E.; Winslow, J.; et al. Trastuzumab has preferential activity against breast cancers driven by HER2 homodimers. Cancer Res. 2011, 71, 1871–1882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, H.S.; Mason, K.; Ramyar, K.X.; Stanley, A.M.; Gabelli, S.B.; Denney, D.W., Jr.; Leahy, D.J. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature 2003, 421, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Agus, D.B.; Akita, R.W.; Fox, W.D.; Lewis, G.D.; Higgins, B.; Pisacane, P.I.; Lofgren, J.A.; Tindell, C.; Evans, D.P.; Maiese, K.; et al. Targeting ligand-activated ErbB2 signaling inhibits breast and prostate tumor growth. Cancer Cell 2002, 2, 127–137. [Google Scholar] [CrossRef] [Green Version]
- Xia, W.; Gerard, C.M.; Liu, L.; Baudson, N.M.; Ory, T.L.; Spector, N.L. Combining lapatinib (GW572016), a small molecule inhibitor of ErbB1 and ErbB2 tyrosine kinases, with therapeutic anti-ErbB2 antibodies enhances apoptosis of ErbB2-overexpressing breast cancer cells. Oncogene 2005, 24, 6213–6221. [Google Scholar] [PubMed] [Green Version]
- Nahta, R.; Hung, M.C.; Esteva, F.J. The HER-2-targeting antibodies trastuzumab and pertuzumab synergistically inhibit the survival of breast cancer cells. Cancer Res. 2004, 64, 2343–2346. [Google Scholar] [CrossRef] [Green Version]
- Baselga, J.; Cortes, J.; Im, S.A.; Clark, E.; Ross, G.; Kiermaier, A.; Swain, S.M. Biomarker analyses in CLEOPATRA: A phase III, placebo-controlled study of pertuzumab in human epidermal growth factor receptor 2-positive, first-line metastatic breast cancer. J. Clin. Oncol. 2014, 32, 3753–3761. [Google Scholar] [CrossRef]
- Scheuer, W.; Friess, T.; Burtscher, H.; Bossenmaier, B.; Endl, J.; Hasmann, M. Strongly enhanced antitumor activity of trastuzumab and pertuzumab combination treatment on HER2-positive human xenograft tumor models. Cancer Res. 2009, 69, 9330–9336. [Google Scholar]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gutsch, D.; Jenke, R.; Büch, T.; Aigner, A. Inhibition of HER Receptors Reveals Distinct Mechanisms of Compensatory Upregulation of Other HER Family Members: Basis for Acquired Resistance and for Combination Therapy. Cells 2021, 10, 272. https://doi.org/10.3390/cells10020272
Gutsch D, Jenke R, Büch T, Aigner A. Inhibition of HER Receptors Reveals Distinct Mechanisms of Compensatory Upregulation of Other HER Family Members: Basis for Acquired Resistance and for Combination Therapy. Cells. 2021; 10(2):272. https://doi.org/10.3390/cells10020272
Chicago/Turabian StyleGutsch, Daniela, Robert Jenke, Thomas Büch, and Achim Aigner. 2021. "Inhibition of HER Receptors Reveals Distinct Mechanisms of Compensatory Upregulation of Other HER Family Members: Basis for Acquired Resistance and for Combination Therapy" Cells 10, no. 2: 272. https://doi.org/10.3390/cells10020272
APA StyleGutsch, D., Jenke, R., Büch, T., & Aigner, A. (2021). Inhibition of HER Receptors Reveals Distinct Mechanisms of Compensatory Upregulation of Other HER Family Members: Basis for Acquired Resistance and for Combination Therapy. Cells, 10(2), 272. https://doi.org/10.3390/cells10020272