
NF-κB in Cancer Immunity: Friend or Foe?

Abstract
:1. Introduction: The Growing Role of Immunity in Cancer
2. A Simplified View of the Signaling to NF-κB
3. NF-κB in Innate Immunity and Inflammation
3.1. Pro-Tumoral Roles of NF-κB in Innate Immune Cells: Macrophages and MDSCs
3.1.1. Macrophages
3.1.2. MDSCs
3.2. NF-κB in the Promotion of Anti-Tumor Immunity: DCs and NK Cells
3.2.1. DCs
3.2.2. NK Cells
4. NF-κB in Adaptive Immunity to Cancer
4.1. B Cells
4.2. T Cells
4.2.1. Effector CD4+ and CD8+ T-Cell Subsets
4.2.2. Foxp3+ Treg Cells
5. NF-κB Modulation at the Era of Immunotherapy: Activation or Inhibition?
5.1. NF-κB Activation to Enhance Effector T-Cell Function in Cancer
5.1.1. Putative Functions of NF-κB in T-Cell-Targeting Immunotherapies
5.1.2. Stimulation of NF-κB by TLRs and TNFRSFs Agonistic Reagents as Novel Immunotherapies
5.2. NF-κB Inhibitors as Immunotherapeutic Agents?
5.2.1. Modulation of Tumor Immunity by NF-κB Inhibitors
5.2.2. Combination of NF-κB Inhibitors and Other Immunotherapies
5.3. Limitations of NF-κB-Targeting Therapies and Future Directions
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hayden, M.S.; Ghosh, S. Shared principles in NF-kappaB signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerondakis, S.; Siebenlist, U. Roles of the NF-kappaB pathway in lymphocyte development and function. Cold Spring Harb. Perspect. Biol. 2010, 2, a000182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basseres, D.S.; Ebbs, A.; Levantini, E.; Baldwin, A.S. Requirement of the NF-kappaB subunit p65/RelA for K-Ras-induced lung tumorigenesis. Cancer Res. 2010, 70, 3537–3546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carneiro-Lobo, T.C.; Scalabrini, L.C.; Magalhaes, L.D.S.; Cardeal, L.B.; Rodrigues, F.S.; Dos Santos, E.O.; Baldwin, A.S.; Levantini, E.; Giordano, R.J.; Basseres, D.S. IKKbeta targeting reduces KRAS-induced lung cancer angiogenesis in vitro and in vivo: A potential anti-angiogenic therapeutic target. Lung Cancer 2019, 130, 169–178. [Google Scholar] [CrossRef]
- Meylan, E.; Dooley, A.L.; Feldser, D.M.; Shen, L.; Turk, E.; Ouyang, C.; Jacks, T. Requirement for NF-kappaB signalling in a mouse model of lung adenocarcinoma. Nature 2009, 462, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Splittgerber, R.; Yull, F.E.; Kantrow, S.; Ayers, G.D.; Karin, M.; Richmond, A. Conditional ablation of Ikkb inhibits melanoma tumor development in mice. J. Clin. Investig. 2010, 120, 2563–2574. [Google Scholar] [CrossRef]
- He, G.; Yu, G.Y.; Temkin, V.; Ogata, H.; Kuntzen, C.; Sakurai, T.; Sieghart, W.; Peck-Radosavljevic, M.; Leffert, H.L.; Karin, M. Hepatocyte IKKbeta/NF-kappaB inhibits tumor promotion and progression by preventing oxidative stress-driven STAT3 activation. Cancer Cell 2010, 17, 286–297. [Google Scholar] [CrossRef] [Green Version]
- van Hogerlinden, M.; Rozell, B.L.; Toftgard, R.; Sundberg, J.P. Characterization of the progressive skin disease and inflammatory cell infiltrate in mice with inhibited NF-kappaB signaling. J. Investig. Dermatol. 2004, 123, 101–108. [Google Scholar] [CrossRef] [Green Version]
- Capece, D.; Verzella, D.; Tessitore, A.; Alesse, E.; Capalbo, C.; Zazzeroni, F. Cancer secretome and inflammation: The bright and the dark sides of NF-kappaB. Semin. Cell Dev. Biol. 2018, 78, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.J.; Ratnam, N.M.; Byrd, J.C.; Guttridge, D.C. NF-kappaB functions in tumor initiation by suppressing the surveillance of both innate and adaptive immune cells. Cell Rep. 2014, 9, 90–103. [Google Scholar] [CrossRef] [Green Version]
- Hopewell, E.L.; Zhao, W.; Fulp, W.J.; Bronk, C.C.; Lopez, A.S.; Massengill, M.; Antonia, S.; Celis, E.; Haura, E.B.; Enkemann, S.A.; et al. Lung tumor NF-kappaB signaling promotes T cell-mediated immune surveillance. J. Clin. Investig. 2013, 123, 2509–2522. [Google Scholar] [CrossRef] [Green Version]
- Ji, Z.; He, L.; Regev, A.; Struhl, K. Inflammatory regulatory network mediated by the joint action of NF-kB, STAT3, and AP-1 factors is involved in many human cancers. Proc. Natl. Acad. Sci. USA 2019, 116, 9453–9462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gowrishankar, K.; Gunatilake, D.; Gallagher, S.J.; Tiffen, J.; Rizos, H.; Hersey, P. Inducible but not constitutive expression of PD-L1 in human melanoma cells is dependent on activation of NF-kappaB. PLoS ONE 2015, 10, e0123410. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.O.; Li, C.W.; Xia, W.; Cha, J.H.; Chan, L.C.; Wu, Y.; Chang, S.S.; Lin, W.C.; Hsu, J.M.; Hsu, Y.H.; et al. Deubiquitination and Stabilization of PD-L1 by CSN5. Cancer Cell 2016, 30, 925–939. [Google Scholar] [CrossRef] [Green Version]
- Larionova, I.; Tuguzbaeva, G.; Ponomaryova, A.; Stakheyeva, M.; Cherdyntseva, N.; Pavlov, V.; Choinzonov, E.; Kzhyshkowska, J. Tumor-Associated Macrophages in Human Breast, Colorectal, Lung, Ovarian and Prostate Cancers. Front. Oncol. 2020, 10, 566511. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Liang, H.; Zen, K. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front. Immunol. 2014, 5, 614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Biswas, S.K.; Lewis, C.E. NF-kappaB as a central regulator of macrophage function in tumors. J. Leukoc. Biol. 2010, 88, 877–884. [Google Scholar] [CrossRef]
- Porta, C.; Rimoldi, M.; Raes, G.; Brys, L.; Ghezzi, P.; Di Liberto, D.; Dieli, F.; Ghisletti, S.; Natoli, G.; De Baetselier, P.; et al. Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor kappaB. Proc. Natl. Acad. Sci. USA 2009, 106, 14978–14983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saccani, A.; Schioppa, T.; Porta, C.; Biswas, S.K.; Nebuloni, M.; Vago, L.; Bottazzi, B.; Colombo, M.P.; Mantovani, A.; Sica, A. p50 nuclear factor-kappaB overexpression in tumor-associated macrophages inhibits M1 inflammatory responses and antitumor resistance. Cancer Res. 2006, 66, 11432–11440. [Google Scholar] [CrossRef] [Green Version]
- Caamaño, J.; Alexander, J.; Craig, L.; Bravo, R.; Hunter, C.A. The NF-kappa B family member RelB is required for innate and adaptive immunity to Toxoplasma gondii. J. Immunol. 1999, 163, 4453–4461. [Google Scholar]
- Gasparini, C.; Foxwell, B.M.; Feldmann, M. RelB/p50 regulates TNF production in LPS-stimulated dendritic cells and macrophages. Cytokine 2013, 61, 736–740. [Google Scholar] [CrossRef]
- Li, D.; Beisswenger, C.; Herr, C.; Hellberg, J.; Han, G.; Zakharkina, T.; Voss, M.; Wiewrodt, R.; Bohle, R.M.; Menger, M.D.; et al. Myeloid cell RelA/p65 promotes lung cancer proliferation through Wnt/beta-catenin signaling in murine and human tumor cells. Oncogene 2014, 33, 1239–1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greten, F.R.; Eckmann, L.; Greten, T.F.; Park, J.M.; Li, Z.W.; Egan, L.J.; Kagnoff, M.F.; Karin, M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 2004, 118, 285–296. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Han, L.; Sun, F.; Zhou, J.; Ohaegbulam, K.C.; Tang, X.; Zang, X.; Steinbrecher, K.A.; Qu, Z.; Xiao, G. NF-kappaB RelA renders tumor-associated macrophages resistant to and capable of directly suppressing CD8(+) T cells for tumor promotion. Oncoimmunology 2018, 7, e1435250. [Google Scholar] [CrossRef] [Green Version]
- Hagemann, T.; Wilson, J.; Kulbe, H.; Li, N.F.; Leinster, D.A.; Charles, K.; Klemm, F.; Pukrop, T.; Binder, C.; Balkwill, F.R. Macrophages Induce Invasiveness of Epithelial Cancer Cells Via NF-κB and JNK. J. Immunol. 2005, 175, 1197–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagemann, T.; Lawrence, T.; McNeish, I.; Charles, K.A.; Kulbe, H.; Thompson, R.G.; Robinson, S.C.; Balkwill, F.R. “Re-educating” tumor-associated macrophages by targeting NF-kappaB. J. Exp. Med. 2008, 205, 1261–1268. [Google Scholar] [CrossRef]
- Achyut, B.R.; Angara, K.; Jain, M.; Borin, T.F.; Rashid, M.H.; Iskander, A.S.M.; Ara, R.; Kolhe, R.; Howard, S.; Venugopal, N.; et al. Canonical NFkappaB signaling in myeloid cells is required for the glioblastoma growth. Sci. Rep. 2017, 7, 13754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, T.; Canli, Ö.; Greten, F.R. Canonical NF-κB signaling in myeloid cells promotes lung metastasis in a mouse breast cancer model. Oncotarget 2018, 9, 16775–16791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancino, A.; Lawrence, T. NF-κB and tumor-associated macrophages. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 784–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connelly, L.; Barham, W.; Onishko, H.M.; Chen, L.; Sherrill, T.P.; Zabuawala, T.; Ostrowski, M.C.; Blackwell, T.S.; Yull, F.E. NF-kappaB activation within macrophages leads to an anti-tumor phenotype in a mammary tumor lung metastasis model. Breast Cancer Res. 2011, 13, R83. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Hawkins, O.E.; Barham, W.; Gilchuk, P.; Boothby, M.; Ayers, G.D.; Joyce, S.; Karin, M.; Yull, F.E.; Richmond, A. Myeloid IKKbeta promotes antitumor immunity by modulating CCL11 and the innate immune response. Cancer Res. 2014, 74, 7274–7284. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Kantrow, S.; Sai, J.; Hawkins, O.; Boothby, M.; Ayers, G.D.; Young, E.; Demicco, E.; Lazar, A.; Lev, D.; et al. INK4a/ARF inactivation with activation of the NF-κB/IL-6 pathway is sufficient to drive the development and growth of angiosarcoma. Cancer Res. 2012, 72, 4682–4695. [Google Scholar] [CrossRef] [Green Version]
- Abram, C.L.; Roberge, G.L.; Hu, Y.; Lowell, C.A. Comparative analysis of the efficiency and specificity of myeloid-Cre deleting strains using ROSA-EYFP reporter mice. J. Immunol. Methods 2014, 408, 89–100. [Google Scholar] [CrossRef] [Green Version]
- Suresh, R.; Barakat, D.J.; Barberi, T.; Zheng, L.; Jaffee, E.; Pienta, K.J.; Friedman, A.D. NF-kappaB p50-deficient immature myeloid cell (p50-IMC) adoptive transfer slows the growth of murine prostate and pancreatic ductal carcinoma. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Hu, Z.; Zhu, Z.; Zhang, X.; Wei, Z.; Zhang, Y.; Hu, D.; Cai, Q. The MSHA strain of Pseudomonas aeruginosa (PA-MSHA) inhibits gastric carcinoma progression by inducing M1 macrophage polarization. Tumour Biol. J. Int. Soc. Oncodevelop. Biol. Med. 2016, 37, 6913–6921. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.Y.; Wang, N.; Man, K.; Tsao, S.W.; Che, C.M.; Feng, Y. Autophagy-induced RelB/p52 activation mediates tumour-associated macrophage repolarisation and suppression of hepatocellular carcinoma by natural compound baicalin. Cell Death Dis. 2015, 6, e1942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veglia, F.; Perego, M.; Gabrilovich, D. Myeloid-derived suppressor cells coming of age. Nat. Immunol. 2018, 19, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Law, A.M.K.; Valdes-Mora, F.; Gallego-Ortega, D. Myeloid-Derived Suppressor Cells as a Therapeutic Target for Cancer. Cells 2020, 9, 561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, M.K.; Zhu, L.; Harris-White, M.; Kar, U.K.; Huang, M.; Johnson, M.F.; Lee, J.M.; Elashoff, D.; Strieter, R.; Dubinett, S.; et al. Myeloid suppressor cell depletion augments antitumor activity in lung cancer. PLoS ONE 2012, 7, e40677. [Google Scholar] [CrossRef]
- Stromnes, I.M.; Brockenbrough, J.S.; Izeradjene, K.; Carlson, M.A.; Cuevas, C.; Simmons, R.M.; Greenberg, P.D.; Hingorani, S.R. Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut 2014, 63, 1769–1781. [Google Scholar] [CrossRef] [Green Version]
- Condamine, T.; Gabrilovich, D.I. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. 2011, 32, 19–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flores, R.R.; Clauson, C.L.; Cho, J.; Lee, B.C.; McGowan, S.J.; Baker, D.J.; Niedernhofer, L.J.; Robbins, P.D. Expansion of myeloid-derived suppressor cells with aging in the bone marrow of mice through a NF-kappaB-dependent mechanism. Aging Cell 2017, 16, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Hong, E.H.; Chang, S.Y.; Lee, B.R.; Kim, Y.S.; Lee, J.M.; Kang, C.Y.; Kweon, M.N.; Ko, H.J. Blockade of Myd88 signaling induces antitumor effects by skewing the immunosuppressive function of myeloid-derived suppressor cells. Int. J. Cancer J. Int. Du Cancer 2013, 132, 2839–2848. [Google Scholar] [CrossRef] [PubMed]
- Llitjos, J.F.; Auffray, C.; Alby-Laurent, F.; Rousseau, C.; Merdji, H.; Bonilla, N.; Toubiana, J.; Belaidouni, N.; Mira, J.P.; Lucas, B.; et al. Sepsis-induced expansion of granulocytic myeloid-derived suppressor cells promotes tumour growth through Toll-like receptor 4. J. Pathol. 2016, 239, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, B.; Li, X.; Zhao, X.; Wan, L.; Lin, G.; Yu, M.; Wang, J.; Jiang, X.; Feng, W.; et al. Transmembrane TNF-alpha promotes suppressive activities of myeloid-derived suppressor cells via TNFR2. J. Immunol. 2014, 192, 1320–1331. [Google Scholar] [CrossRef] [Green Version]
- Porta, C.; Consonni, F.M.; Morlacchi, S.; Sangaletti, S.; Bleve, A.; Totaro, M.G.; Larghi, P.; Rimoldi, M.; Tripodo, C.; Strauss, L.; et al. Tumor-Derived Prostaglandin E2 Promotes p50 NF-kappaB-Dependent Differentiation of Monocytic MDSCs. Cancer Res. 2020, 80, 2874–2888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Li, X.; Zamani, A.; Wang, W.; Lee, C.N.; Li, M.; Luo, G.; Eller, E.; Sun, H.; Ghosh, S.; et al. c-Rel is a myeloid checkpoint for cancer immunotherapy. Nat. Cancer 2020, 1, 507–517. [Google Scholar] [CrossRef]
- Yu, J.; Wang, Y.; Yan, F.; Zhang, P.; Li, H.; Zhao, H.; Yan, C.; Yan, F.; Ren, X. Noncanonical NF-κB Activation Mediates STAT3-stimulated IDO Upregulation in Myeloid-Derived Suppressor Cells in Breast Cancer. J. Immunol. 2014, 193, 2574–2586. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, S.A.; Kulshrestha, A.; Katara, G.K.; Riehl, V.; Sahoo, M.; Beaman, K.D. Cancer-associated V-ATPase induces delayed apoptosis of protumorigenic neutrophils. Mol. Oncol. 2020, 14, 590–610. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Shi, H.; Yuan, X.; Jiang, P.; Qian, H.; Xu, W. Tumor-derived exosomes induce N2 polarization of neutrophils to promote gastric cancer cell migration. Mol. Cancer 2018, 17, 146. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Peng, A.; Huang, X.Z.; Shi, D.C.; Wang, J.C.; Zhao, Q.; Lin, H.; Kuang, D.M.; Ke, P.F.; Lao, X.M. Peritumoral stromal neutrophils are essential for c-Met-elicited metastasis in human hepatocellular carcinoma. Oncoimmunology 2016, 5, e1219828. [Google Scholar] [CrossRef] [Green Version]
- Li, X.F.; Chen, D.P.; Ouyang, F.Z.; Chen, M.M.; Wu, Y.; Kuang, D.M.; Zheng, L. Increased autophagy sustains the survival and pro-tumourigenic effects of neutrophils in human hepatocellular carcinoma. J. Hepatol. 2015, 62, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Lecot, P.; Sarabi, M.; Pereira Abrantes, M.; Mussard, J.; Koenderman, L.; Caux, C.; Bendriss-Vermare, N.; Michallet, M.-C. Neutrophil Heterogeneity in Cancer: From Biology to Therapies. Front. Immunol. 2019, 10, 2155. [Google Scholar] [CrossRef] [Green Version]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef]
- Bottcher, J.P.; Reis e Sousa, C. The Role of Type 1 Conventional Dendritic Cells in Cancer Immunity. Trends Cancer 2018, 4, 784–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubert, M.; Gobbini, E.; Couillault, C.; Manh, T.V.; Doffin, A.C.; Berthet, J.; Rodriguez, C.; Ollion, V.; Kielbassa, J.; Sajous, C.; et al. IFN-III is selectively produced by cDC1 and predicts good clinical outcome in breast cancer. Sci. Immunol. 2020, 5. [Google Scholar] [CrossRef] [Green Version]
- Salmon, H.; Idoyaga, J.; Rahman, A.; Leboeuf, M.; Remark, R.; Jordan, S.; Casanova-Acebes, M.; Khudoynazarova, M.; Agudo, J.; Tung, N.; et al. Expansion and Activation of CD103(+) Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 2016, 44, 924–938. [Google Scholar] [CrossRef] [Green Version]
- Binnewies, M.; Mujal, A.M.; Pollack, J.L.; Combes, A.J.; Hardison, E.A.; Barry, K.C.; Tsui, J.; Ruhland, M.K.; Kersten, K.; Abushawish, M.A.; et al. Unleashing Type-2 Dendritic Cells to Drive Protective Antitumor CD4(+) T Cell Immunity. Cell 2019, 177, 556–571.e516. [Google Scholar] [CrossRef]
- Mitchell, D.; Chintala, S.; Dey, M. Plasmacytoid dendritic cell in immunity and cancer. J. Neuroimmunol. 2018, 322, 63–73. [Google Scholar] [CrossRef]
- Sisirak, V.; Faget, J.; Gobert, M.; Goutagny, N.; Vey, N.; Treilleux, I.; Renaudineau, S.; Poyet, G.; Labidi-Galy, S.I.; Goddard-Leon, S.; et al. Impaired IFN-alpha production by plasmacytoid dendritic cells favors regulatory T-cell expansion that may contribute to breast cancer progression. Cancer Res. 2012, 72, 5188–5197. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Lin, L.; Zhang, Z.; Zhang, H.; Hu, H. Targeting NF-kappaB pathway for the therapy of diseases: Mechanism and clinical study. Signal. Transduct Target. 2020, 5, 209. [Google Scholar] [CrossRef] [PubMed]
- Ouaaz, F.; Arron, J.; Zheng, Y.; Choi, Y.; Beg, A.A. Dendritic cell development and survival require distinct NF-kappaB subunits. Immunity 2002, 16, 257–270. [Google Scholar] [CrossRef] [Green Version]
- Hofer, S.; Rescigno, M.; Granucci, F.; Citterio, S.; Francolini, M.; Ricciardi-Castagnoli, P. Differential activation of NF-kappa B subunits in dendritic cells in response to Gram-negative bacteria and to lipopolysaccharide. Microbes Infect. 2001, 3, 259–265. [Google Scholar] [CrossRef]
- Rescigno, M.; Martino, M.; Sutherland, C.L.; Gold, M.R.; Ricciardi-Castagnoli, P. Dendritic cell survival and maturation are regulated by different signaling pathways. J. Exp. Med. 1998, 188, 2175–2180. [Google Scholar] [CrossRef] [PubMed]
- O’Keeffe, M.; Grumont, R.J.; Hochrein, H.; Fuchsberger, M.; Gugasyan, R.; Vremec, D.; Shortman, K.; Gerondakis, S. Distinct roles for the NF-kappaB1 and c-Rel transcription factors in the differentiation and survival of plasmacytoid and conventional dendritic cells activated by TLR-9 signals. Blood 2005, 106, 3457–3464. [Google Scholar] [CrossRef] [Green Version]
- Grumont, R.; Hochrein, H.; O’Keeffe, M.; Gugasyan, R.; White, C.; Caminschi, I.; Cook, W.; Gerondakis, S. c-Rel regulates interleukin 12 p70 expression in CD8(+) dendritic cells by specifically inducing p35 gene transcription. J. Exp. Med. 2001, 194, 1021–1032. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; D’Amico, A.; Winkel, K.D.; Suter, M.; Lo, D.; Shortman, K. RelB Is Essential for the Development of Myeloid-Related CD8α− Dendritic Cells but Not of Lymphoid-Related CD8α+ Dendritic Cells. Immunity 1998, 9, 839–847. [Google Scholar] [CrossRef] [Green Version]
- Castiglioni, P.; Janssen, E.M.; Prilliman, K.R.; Gerloni, M.; Schoenberger, S.; Zanetti, M. Cross-priming is under control of the relB gene. Scand J. Immunol. 2002, 56, 219–223. [Google Scholar] [CrossRef]
- Katakam, A.K.; Brightbill, H.; Franci, C.; Kung, C.; Nunez, V.; Jones, C.; Peng, I.; Jeet, S.; Wu, L.C.; Mellman, I.; et al. Dendritic cells require NIK for CD40-dependent cross-priming of CD8+ T cells. Proc. Natl. Acad. Sci. USA 2015, 112, 14664–14669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Speirs, K.; Lieberman, L.; Caamano, J.; Hunter, C.A.; Scott, P. Cutting edge: NF-kappa B2 is a negative regulator of dendritic cell function. J. Immunol. 2004, 172, 752–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, V.F.; Davis-Turak, J.; Macal, M.; Huang, J.Q.; Ponomarenko, J.; Kearns, J.D.; Yu, T.; Fagerlund, R.; Asagiri, M.; Zuniga, E.I.; et al. Control of RelB during dendritic cell activation integrates canonical and noncanonical NF-kappaB pathways. Nat. Immunol. 2012, 13, 1162–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayden, M.S. A less-canonical, canonical NF-kappaB pathway in DCs. Nat. Immunol. 2012, 13, 1139–1141. [Google Scholar] [CrossRef]
- Baratin, M.; Foray, C.; Demaria, O.; Habbeddine, M.; Pollet, E.; Maurizio, J.; Verthuy, C.; Davanture, S.; Azukizawa, H.; Flores-Langarica, A.; et al. Homeostatic NF-kappaB Signaling in Steady-State Migratory Dendritic Cells Regulates Immune Homeostasis and Tolerance. Immunity 2015, 42, 627–639. [Google Scholar] [CrossRef] [Green Version]
- Tas, S.W.; Vervoordeldonk, M.J.; Hajji, N.; Schuitemaker, J.H.; van der Sluijs, K.F.; May, M.J.; Ghosh, S.; Kapsenberg, M.L.; Tak, P.P.; de Jong, E.C. Noncanonical NF-kappaB signaling in dendritic cells is required for indoleamine 2,3-dioxygenase (IDO) induction and immune regulation. Blood 2007, 110, 1540–1549. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.D.; Baban, B.; Chandler, P.; Hou, D.Y.; Singh, N.; Yagita, H.; Azuma, M.; Blazar, B.R.; Mellor, A.L.; Munn, D.H. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. J. Clin. Investig. 2007, 117, 2570–2582. [Google Scholar] [CrossRef] [Green Version]
- Karyampudi, L.; Lamichhane, P.; Krempski, J.; Kalli, K.R.; Behrens, M.D.; Vargas, D.M.; Hartmann, L.C.; Janco, J.M.T.; Dong, H.; Hedin, K.E.; et al. PD-1 Blunts the Function of Ovarian Tumor-Infiltrating Dendritic Cells by Inactivating NF-κB. Cancer Res. 2016, 76, 239–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Y.; Liang, H.; Rao, E.; Zheng, W.; Huang, X.; Deng, L.; Zhang, Y.; Yu, X.; Xu, M.; Mauceri, H.; et al. Non-canonical NF-kappaB Antagonizes STING Sensor-Mediated DNA Sensing in Radiotherapy. Immunity 2018, 49, 490–503.e494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.M.; Mahtabifard, A.; Yamada, R.; Crystal, R.G.; Korst, R.J. Adenovirus vector-mediated overexpression of a truncated form of the p65 nuclear factor kappa B cDNA in dendritic cells enhances their function resulting in immune-mediated suppression of preexisting murine tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2002, 8, 3561–3569. [Google Scholar]
- Dougan, S.K.; Dougan, M. Regulation of innate and adaptive antitumor immunity by IAP antagonists. Immunotherapy 2018, 10, 787–796. [Google Scholar] [CrossRef] [Green Version]
- Garris, C.S.; Arlauckas, S.P.; Kohler, R.H.; Trefny, M.P.; Garren, S.; Piot, C.; Engblom, C.; Pfirschke, C.; Siwicki, M.; Gungabeesoon, J.; et al. Successful Anti-PD-1 Cancer Immunotherapy Requires T Cell-Dendritic Cell Crosstalk Involving the Cytokines IFN-γ and IL-12. Immunity 2018, 49, 1148–1161.e1147. [Google Scholar] [CrossRef] [Green Version]
- Javaid, N.; Choi, S. Toll-like Receptors from the Perspective of Cancer Treatment. Cancers 2020, 12, 297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urban-Wojciuk, Z.; Khan, M.M.; Oyler, B.L.; Fahraeus, R.; Marek-Trzonkowska, N.; Nita-Lazar, A.; Hupp, T.R.; Goodlett, D.R. The Role of TLRs in Anti-cancer Immunity and Tumor Rejection. Front. Immunol. 2019, 10, 2388. [Google Scholar] [CrossRef]
- Chiossone, L.; Dumas, P.-Y.; Vienne, M.; Vivier, E. Natural killer cells and other innate lymphoid cells in cancer. Nat. Rev. Immunol. 2018, 18, 671–688. [Google Scholar] [CrossRef] [PubMed]
- Pandey, R.; DeStephan, C.M.; Madge, L.A.; May, M.J.; Orange, J.S. NKp30 ligation induces rapid activation of the canonical NF-kappaB pathway in NK cells. J. Immunol. 2007, 179, 7385–7396. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.-J.; Choi, G.-E.; Ryu, S.; Kwon, S.J.; Kim, S.C.; Booth, C.; Nichols, K.E.; Kim, H.S. Stepwise phosphorylation of p65 promotes NF-κB activation and NK cell responses during target cell recognition. Nat. Commun. 2016, 7, 11686. [Google Scholar] [CrossRef] [PubMed]
- Gross, O.; Grupp, C.; Steinberg, C.; Zimmermann, S.; Strasser, D.; Hannesschläger, N.; Reindl, W.; Jonsson, H.; Huo, H.; Littman, D.R.; et al. Multiple ITAM-coupled NK-cell receptors engage the Bcl10/Malt1 complex via Carma1 for NF-κB and MAPK activation to selectively control cytokine production. Blood 2008, 112, 2421–2428. [Google Scholar] [CrossRef]
- Jyothi, M.D.; Khar, A. Regulation of CD40L expression on natural killer cells by interleukin-12 and interferon γ: Its role in the elicitation of an effective antitumor immune response. Cancer Immunol. Immunother. 2000, 49, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Chaix, J.; Tessmer, M.S.; Hoebe, K.; Fuseri, N.; Ryffel, B.; Dalod, M.; Alexopoulou, L.; Beutler, B.; Brossay, L.; Vivier, E.; et al. Cutting edge: Priming of NK cells by IL-18. J. Immunol. 2008, 181, 1627–1631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ardolino, M.; Azimi, C.S.; Iannello, A.; Trevino, T.N.; Horan, L.; Zhang, L.; Deng, W.; Ring, A.M.; Fischer, S.; Garcia, K.C.; et al. Cytokine therapy reverses NK cell anergy in MHC-deficient tumors. J. Clin. Investig. 2014, 124, 4781–4794. [Google Scholar] [CrossRef] [Green Version]
- Adachi, O.; Kawai, T.; Takeda, K.; Matsumoto, M.; Tsutsui, H.; Sakagami, M.; Nakanishi, K.; Akira, S. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity 1998, 9, 143–150. [Google Scholar] [CrossRef] [Green Version]
- Tato, C.M.; Villarino, A.; Caamaño, J.H.; Boothby, M.; Hunter, C.A. Inhibition of NF-κB Activity in T and NK Cells Results in Defective Effector Cell Expansion and Production of IFN-γ Required for Resistance to Toxoplasma gondii. J. Immunol. 2003, 170, 3139–3146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Evaristo, C.; Alegre, M.-L.; Gurbuxani, S.; Kee, B.L. Analysis of GzmbCre as a Model System for Gene Deletion in the Natural Killer Cell Lineage. PLoS ONE 2015, 10, e0125211. [Google Scholar] [CrossRef]
- Tato, C.M.; Mason, N.; Artis, D.; Shapira, S.; Caamano, J.C.; Bream, J.H.; Liou, H.C.; Hunter, C.A. Opposing roles of NF-kappaB family members in the regulation of NK cell proliferation and production of IFN-gamma. Int. Immunol. 2006, 18, 505–513. [Google Scholar] [CrossRef]
- Orange, J.S.; Brodeur, S.R.; Jain, A.; Bonilla, F.A.; Schneider, L.C.; Kretschmer, R.; Nurko, S.; Rasmussen, W.L.; Köhler, J.R.; Gellis, S.E.; et al. Deficient natural killer cell cytotoxicity in patients with IKK-gamma/NEMO mutations. J. Clin. Investig. 2002, 109, 1501–1509. [Google Scholar] [CrossRef] [PubMed]
- Pannicke, U.; Baumann, B.; Fuchs, S.; Henneke, P.; Rensing-Ehl, A.; Rizzi, M.; Janda, A.; Hese, K.; Schlesier, M.; Holzmann, K.; et al. Deficiency of innate and acquired immunity caused by an IKBKB mutation. N. Engl. J. Med. 2013, 369, 2504–2514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Zhang, J.; Lichtenheld, M.G.; Meadows, G.G. A role for NF-kappa B activation in perforin expression of NK cells upon IL-2 receptor signaling. J. Immunol. 2002, 169, 1319–1325. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Bi, E.; Hu, Y.; Deng, W.; Tian, Z.; Dong, C.; Hu, Y.; Sun, B. A novel NF-kappaB binding site controls human granzyme B gene transcription. J. Immunol. 2006, 176, 4173–4181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, J.; Wang, X.; Stojanovic, A.; Zhang, Q.; Wincher, M.; Buhler, L.; Arnold, A.; Correia, M.P.; Winkler, M.; Koch, P.S.; et al. Single-Cell RNA Sequencing of Tumor-Infiltrating NK Cells Reveals that Inhibition of Transcription Factor HIF-1alpha Unleashes NK Cell Activity. Immunity 2020, 52, 1075–1087.e1078. [Google Scholar] [CrossRef]
- Xie, X.; Ma, L.; Zhou, Y.; Shen, W.; Xu, D.; Dou, J.; Shen, B.; Zhou, C. Polysaccharide enhanced NK cell cytotoxicity against pancreatic cancer via TLR4/MAPKs/NF-kappaB pathway in vitro/vivo. Carbohydr. Polym. 2019, 225, 115223. [Google Scholar] [CrossRef]
- Kubo, M.; Morisaki, T.; Matsumoto, K.; Tasaki, A.; Yamanaka, N.; Nakashima, H.; Kuroki, H.; Nakamura, K.; Nakamura, M.; Katano, M. Paclitaxel probably enhances cytotoxicity of natural killer cells against breast carcinoma cells by increasing perforin production. Cancer Immunol. Immunother. 2005, 54, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Jasinski, D.L.; Medina, J.L.; Spencer, D.M.; Foster, A.E.; Bayle, J.H. Inducible MyD88/CD40 synergizes with IL-15 to enhance antitumor efficacy of CAR-NK cells. Blood Adv. 2020, 4, 1950–1964. [Google Scholar] [CrossRef]
- Zheng, Y.; Li, Y.; Lian, J.; Yang, H.; Li, F.; Zhao, S.; Qi, Y.; Zhang, Y.; Huang, L. TNF-alpha-induced Tim-3 expression marks the dysfunction of infiltrating natural killer cells in human esophageal cancer. J. Transl. Med. 2019, 17, 165. [Google Scholar] [CrossRef] [Green Version]
- Yuen, G.J.; Demissie, E.; Pillai, S. B lymphocytes and cancer: A love-hate relationship. Trends Cancer 2016, 2, 747–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brodt, P.; Gordon, J. Anti-tumor immunity in B lymphocyte-deprived mice. I. Immunity to a chemically induced tumor. J. Immunol. 1978, 121, 359–362. [Google Scholar]
- Ou, Z.; Wang, Y.; Liu, L.; Li, L.; Yeh, S.; Qi, L.; Chang, C. Tumor microenvironment B cells increase bladder cancer metastasis via modulation of the IL-8/androgen receptor (AR)/MMPs signals. Oncotarget 2015, 6, 26065–26078. [Google Scholar] [CrossRef] [Green Version]
- Ammirante, M.; Luo, J.L.; Grivennikov, S.; Nedospasov, S.; Karin, M. B-cell-derived lymphotoxin promotes castration-resistant prostate cancer. Nature 2010, 464, 302–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Visser, K.E.; Korets, L.V.; Coussens, L.M. De novo carcinogenesis promoted by chronic inflammation is B lymphocyte dependent. Cancer Cell 2005, 7, 411–423. [Google Scholar] [CrossRef] [Green Version]
- Inoue, S.; Leitner, W.W.; Golding, B.; Scott, D. Inhibitory effects of B cells on antitumor immunity. Cancer Res. 2006, 66, 7741–7747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tadmor, T.; Zhang, Y.; Cho, H.M.; Podack, E.R.; Rosenblatt, J.D. The absence of B lymphocytes reduces the number and function of T-regulatory cells and enhances the anti-tumor response in a murine tumor model. Cancer Immunol. Immunother. 2011, 60, 609–619. [Google Scholar] [CrossRef]
- Largeot, A.; Pagano, G.; Gonder, S.; Moussay, E.; Paggetti, J. The B-side of Cancer Immunity: The Underrated Tune. Cells 2019, 8, 449. [Google Scholar] [CrossRef] [Green Version]
- Griss, J.; Bauer, W.; Wagner, C.; Simon, M.; Chen, M.; Grabmeier-Pfistershammer, K.; Maurer-Granofszky, M.; Roka, F.; Penz, T.; Bock, C.; et al. B cells sustain inflammation and predict response to immune checkpoint blockade in human melanoma. Nat. Commun. 2019, 10, 4186. [Google Scholar] [CrossRef] [Green Version]
- Pinc, A.; Somasundaram, R.; Wagner, C.; Hormann, M.; Karanikas, G.; Jalili, A.; Bauer, W.; Brunner, P.; Grabmeier-Pfistershammer, K.; Gschaider, M.; et al. Targeting CD20 in melanoma patients at high risk of disease recurrence. Mol. Ther. J. Am. Soc. Gene Ther. 2012, 20, 1056–1062. [Google Scholar] [CrossRef] [Green Version]
- Hua, Z.; Hou, B. The role of B cell antigen presentation in the initiation of CD4+ T cell response. Immunol. Rev. 2020, 296, 24–35. [Google Scholar] [CrossRef]
- Schultz, K.R.; Klarnet, J.P.; Gieni, R.S.; HayGlass, K.T.; Greenberg, P.D. The role of B cells for in vivo T cell responses to a Friend virus-induced leukemia. Science 1990, 249, 921–923. [Google Scholar] [CrossRef]
- Bruno, T.C.; Ebner, P.J.; Moore, B.L.; Squalls, O.G.; Waugh, K.A.; Eruslanov, E.B.; Singhal, S.; Mitchell, J.D.; Franklin, W.A.; Merrick, D.T.; et al. Antigen-Presenting Intratumoral B Cells Affect CD4(+) TIL Phenotypes in Non-Small Cell Lung Cancer Patients. Cancer Immunol. Res. 2017, 5, 898–907. [Google Scholar] [CrossRef] [Green Version]
- Wouters, M.C.A.; Nelson, B.H. Prognostic Significance of Tumor-Infiltrating B Cells and Plasma Cells in Human Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 6125–6135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabrita, R.; Lauss, M.; Sanna, A.; Donia, M.; Skaarup Larsen, M.; Mitra, S.; Johansson, I.; Phung, B.; Harbst, K.; Vallon-Christersson, J.; et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 2020, 577, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Petitprez, F.; de Reynies, A.; Keung, E.Z.; Chen, T.W.; Sun, C.M.; Calderaro, J.; Jeng, Y.M.; Hsiao, L.P.; Lacroix, L.; Bougouin, A.; et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature 2020, 577, 556–560. [Google Scholar] [CrossRef] [PubMed]
- Helmink, B.A.; Reddy, S.M.; Gao, J.; Zhang, S.; Basar, R.; Thakur, R.; Yizhak, K.; Sade-Feldman, M.; Blando, J.; Han, G.; et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 2020, 577, 549–555. [Google Scholar] [CrossRef]
- Milanovic, M.; Heise, N.; De Silva, N.S.; Anderson, M.M.; Silva, K.; Carette, A.; Orelli, F.; Bhagat, G.; Klein, U. Differential requirements for the canonical NF-kappaB transcription factors c-REL and RELA during the generation and activation of mature B cells. Immunol. Cell Biol. 2017, 95, 261–271. [Google Scholar] [CrossRef] [Green Version]
- Pasparakis, M.; Schmidt-Supprian, M.; Rajewsky, K. IkappaB kinase signaling is essential for maintenance of mature B cells. J. Exp. Med. 2002, 196, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Heise, N.; De Silva, N.S.; Silva, K.; Carette, A.; Simonetti, G.; Pasparakis, M.; Klein, U. Germinal center B cell maintenance and differentiation are controlled by distinct NF-kappaB transcription factor subunits. J. Exp. Med. 2014, 211, 2103–2118. [Google Scholar] [CrossRef]
- De Silva, N.S.; Anderson, M.M.; Carette, A.; Silva, K.; Heise, N.; Bhagat, G.; Klein, U. Transcription factors of the alternative NF-kappaB pathway are required for germinal center B-cell development. Proc. Natl. Acad. Sci. USA 2016, 113, 9063–9068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almaden, J.V.; Liu, Y.C.; Yang, E.; Otero, D.C.; Birnbaum, H.; Davis-Turak, J.; Asagiri, M.; David, M.; Goldrath, A.W.; Hoffmann, A. B-cell survival and development controlled by the coordination of NF-kappaB family members RelB and cRel. Blood 2016, 127, 1276–1286. [Google Scholar] [CrossRef] [Green Version]
- Pages, F.; Berger, A.; Camus, M.; Sanchez-Cabo, F.; Costes, A.; Molidor, R.; Mlecnik, B.; Kirilovsky, A.; Nilsson, M.; Damotte, D.; et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. N. Engl. J. Med. 2005, 353, 2654–2666. [Google Scholar] [CrossRef] [PubMed]
- Barnes, T.A.; Amir, E. HYPE or HOPE: The prognostic value of infiltrating immune cells in cancer. Br. J. Cancer 2017, 117, 451–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Togashi, Y.; Shitara, K.; Nishikawa, H. Regulatory T cells in cancer immunosuppression - implications for anticancer therapy. Nat. Rev. Clin. Oncol. 2019, 16, 356–371. [Google Scholar] [CrossRef] [PubMed]
- deLeeuw, R.J.; Kost, S.E.; Kakal, J.A.; Nelson, B.H. The prognostic value of FoxP3+ tumor-infiltrating lymphocytes in cancer: A critical review of the literature. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 3022–3029. [Google Scholar] [CrossRef] [Green Version]
- Voisin, A.; Grinberg-Bleyer, Y. The many-sided contributions of NF-κB to T-cell biology in health and disease. Int. Rev. Cell Mol. Biol. 2020. [Google Scholar] [CrossRef]
- Kontgen, F.; Grumont, R.J.; Strasser, A.; Metcalf, D.; Li, R.; Tarlinton, D.; Gerondakis, S. Mice lacking the c-rel proto-oncogene exhibit defects in lymphocyte proliferation, humoral immunity, and interleukin-2 expression. Genes Dev. 1995, 9, 1965–1977. [Google Scholar] [CrossRef] [Green Version]
- Sriskantharajah, S.; Belich, M.P.; Papoutsopoulou, S.; Janzen, J.; Tybulewicz, V.; Seddon, B.; Ley, S.C. Proteolysis of NF-kappaB1 p105 is essential for T cell antigen receptor-induced proliferation. Nat. Immunol. 2009, 10, 38–47. [Google Scholar] [CrossRef]
- Shifrut, E.; Carnevale, J.; Tobin, V.; Roth, T.L.; Woo, J.M.; Bui, C.T.; Li, P.J.; Diolaiti, M.E.; Ashworth, A.; Marson, A. Genome-wide CRISPR Screens in Primary Human T Cells Reveal Key Regulators of Immune Function. Cell 2018, 175, 1958–1971.e1915. [Google Scholar] [CrossRef] [Green Version]
- Doi, T.S.; Takahashi, T.; Taguchi, O.; Azuma, T.; Obata, Y. NF-kappa B RelA-deficient lymphocytes: Normal development of T cells and B cells, impaired production of IgA and IgG1 and reduced proliferative responses. J. Exp. Med. 1997, 185, 953–961. [Google Scholar] [CrossRef] [PubMed]
- Grumont, R.; Lock, P.; Mollinari, M.; Shannon, F.M.; Moore, A.; Gerondakis, S. The mitogen-induced increase in T cell size involves PKC and NFAT activation of Rel/NF-kappaB-dependent c-myc expression. Immunity 2004, 21, 19–30. [Google Scholar] [CrossRef] [Green Version]
- Saibil, S.D.; Jones, R.G.; Deenick, E.K.; Liadis, N.; Elford, A.R.; Vainberg, M.G.; Baerg, H.; Woodgett, J.R.; Gerondakis, S.; Ohashi, P.S. CD4 + and CD8 + T Cell Survival Is Regulated Differentially by Protein Kinase Cθ, c-Rel, and Protein Kinase, B. J. Immunol. 2007, 178, 2932–2939. [Google Scholar] [CrossRef] [Green Version]
- Ruan, Q.; Kameswaran, V.; Zhang, Y.; Zheng, S.; Sun, J.; Wang, J.; DeVirgiliis, J.; Liou, H.C.; Beg, A.A.; Chen, Y.H. The Th17 immune response is controlled by the Rel-RORgamma-RORgamma T transcriptional axis. J. Exp. Med. 2011, 208, 2321–2333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Hardy, K.; Pagler, E.; Ma, L.; Lee, S.; Gerondakis, S.; Daley, S.; Shannon, M.F. The NF-κB Transcription Factor c-Rel Is Required for Th17 Effector Cell Development in Experimental Autoimmune Encephalomyelitis. J. Immunol. 2011, 187, 4483–4491. [Google Scholar] [CrossRef] [Green Version]
- Balasubramani, A.; Shibata, Y.; Crawford, G.E.; Baldwin, A.S.; Hatton, R.D.; Weaver, C.T. Modular Utilization of Distal cis-Regulatory Elements Controls Ifng Gene Expression in T Cells Activated by Distinct Stimuli. Immunity 2010, 33, 35–47. [Google Scholar] [CrossRef] [Green Version]
- Hilliard, B.A.; Mason, N.; Xu, L.; Sun, J.; Lamhamedi-Cherradi, S.-E.; Liou, H.-C.; Hunter, C.; Chen, Y.H. Critical roles of c-Rel in autoimmune inflammation and helper T cell differentiation. J. Clin. Investig. 2002, 110, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Henriksson, J.; Chen, X.; Gomes, T.; Ullah, U.; Meyer, K.B.; Miragaia, R.; Duddy, G.; Pramanik, J.; Yusa, K.; Lahesmaa, R.; et al. Genome-wide CRISPR Screens in T Helper Cells Reveal Pervasive Crosstalk between Activation and Differentiation. Cell 2019, 176, 882–896.e818. [Google Scholar] [CrossRef] [PubMed]
- Jash, A.; Sahoo, A.; Kim, G.-C.; Chae, C.-S.; Hwang, J.-S.; Kim, J.-E.; Im, S.-H. Nuclear factor of activated T cells 1 (NFAT1)-induced permissive chromatin modification facilitates nuclear factor-κB (NF-κB)-mediated interleukin-9 (IL-9) transactivation. J. Biol. Chem. 2012, 287, 15445–15457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, X.; Balasubramanian, S.; Liu, W.; Chu, X.; Wang, H.; Taparowsky, E.J.; Fu, Y.-X.; Choi, Y.; Walsh, M.C.; Li, X.C. OX40 signaling favors the induction of TH9 cells and airway inflammation. Nat. Immunol. 2012, 13, 981–990. [Google Scholar] [CrossRef] [Green Version]
- Knudson, K.M.; Pritzl, C.J.; Saxena, V.; Altman, A.; Daniels, M.A.; Teixeiro, E. NFκB-Pim-1-Eomesodermin axis is critical for maintaining CD8 T-cell memory quality. Proc. Natl. Acad. Sci. USA 2017, 114, E1659–E1667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaussant-Cohen, S.; Jaber, F.; Massaad, M.J.; Weeks, S.; Jones, J.; Alosaimi, M.F.; Wallace, J.; Al-Herz, W.; Geha, R.S.; Chou, J. Combined immunodeficiency in a patient with c-Rel deficiency. J. Allergy Clin. Immunol. 2019, 144, 606–608.e604. [Google Scholar] [CrossRef] [Green Version]
- Deenick, E.K.; Po, L.; Chapatte, L.; Murakami, K.; Lu, Y.-C.; Elford, A.R.; Saibil, S.D.; Ruland, J.; Gerondakis, S.; Mak, T.W.; et al. c-Rel phenocopies PKCθ but not Bcl-10 in regulating CD8+ T-cell activation versus tolerance. Eur. J. Immunol. 2010, 40, 867–877. [Google Scholar] [CrossRef]
- Ghosh, P.; Sica, A.; Young, H.A.; Ye, J.; Franco, J.L.; Wiltrout, R.H.; Longo, D.L.; Rice, N.R.; Komschlies, K.L. Alterations in NF kappa B/Rel family proteins in splenic T-cells from tumor-bearing mice and reversal following therapy. Cancer Res. 1994, 54, 2969–2972. [Google Scholar]
- Li, X.; Liu, J.; Park, J.K.; Hamilton, T.A.; Rayman, P.; Klein, E.; Edinger, M.; Tubbs, R.; Bukowski, R.; Finke, J. T cells from renal cell carcinoma patients exhibit an abnormal pattern of kappa B-specific DNA-binding activity: A preliminary report. Cancer Res. 1994, 54, 5424–5429. [Google Scholar] [PubMed]
- Clavijo, P.E.; Frauwirth, K.A. Anergic CD8+ T Lymphocytes Have Impaired NF-κB Activation with Defects in p65 Phosphorylation and Acetylation. J. Immunol. 2012, 188, 1213–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, S.E.; Wang, Y.; Chen, L.; Molinero, L.L.; Gajewski, T.F.; Evaristo, C.; Alegre, M.-L. T cell-NF-κB activation is required for tumor control in vivo. J. Immunother. Cancer 2015, 3, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evaristo, C.; Spranger, S.; Barnes, S.E.; Miller, M.L.; Molinero, L.L.; Locke, F.L.; Gajewski, T.F.; Alegre, M.-L. Cutting Edge: Engineering Active IKKβ in T Cells Drives Tumor Rejection. J. Immunol. 2016, 196, 2933–2938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giordano, M.; Roncagalli, R.; Bourdely, P.; Chasson, L.; Buferne, M.; Yamasaki, S.; Beyaert, R.; van Loo, G.; Auphan-Anezin, N.; Schmitt-Verhulst, A.-M.; et al. The tumor necrosis factor alpha-induced protein 3 (TNFAIP3, A20) imposes a brake on antitumor activity of CD8 T cells. Proc. Natl. Acad. Sci. USA 2014, 111, 11115–11120. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Klement, J.D.; Smith, A.D.; Yang, D.; Waller, J.L.; Browning, D.D.; Munn, D.H.; Liu, K. p50 suppresses cytotoxic T lymphocyte effector function to regulate tumor immune escape and response to immunotherapy. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Tay, R.E.; Richardson, E.K.; Toh, H.C. Revisiting the role of CD4(+) T cells in cancer immunotherapy-new insights into old paradigms. Cancer Gene 2020. [Google Scholar] [CrossRef] [PubMed]
- Deenick, E.K.; Elford, A.R.; Pellegrini, M.; Hall, H.; Mak, T.W.; Ohashi, P.S. c-Rel but not NF-kappaB1 is important for T regulatory cell development. Eur. J. Immunol. 2010, 40, 677–681. [Google Scholar] [CrossRef]
- Molinero, L.L.; Yang, J.; Gajewski, T.; Abraham, C.; Farrar, M.A.; Alegre, M.-L. CARMA1 Controls an Early Checkpoint in the Thymic Development of FoxP3 + Regulatory T Cells. J. Immunol. 2009, 182, 6736–6743. [Google Scholar] [CrossRef] [Green Version]
- Isomura, I.; Palmer, S.; Grumont, R.J.; Bunting, K.; Hoyne, G.; Wilkinson, N.; Banerjee, A.; Proietto, A.; Gugasyan, R.; Wu, L.; et al. c-Rel is required for the development of thymic Foxp3+ CD4 regulatory T cells. J. Exp. Med. 2009, 206, 3001–3014. [Google Scholar] [CrossRef]
- Oh, H.; Grinberg-Bleyer, Y.; Liao, W.; Maloney, D.; Wang, P.; Wu, Z.; Wang, J.; Bhatt, D.M.; Heise, N.; Schmid, R.M.; et al. An NF-κB Transcription-Factor-Dependent Lineage-Specific Transcriptional Program Promotes Regulatory T Cell Identity and Function. Immunity 2017, 47, 450–465.e455. [Google Scholar] [CrossRef]
- Di Pilato, M.; Kim, E.Y.; Cadilha, B.L.; Prüßmann, J.N.; Nasrallah, M.N.; Seruggia, D.; Usmani, S.M.; Misale, S.; Zappulli, V.; Carrizosa, E.; et al. Targeting the CBM complex causes Treg cells to prime tumours for immune checkpoint therapy. Nature 2019, 570, 112–116. [Google Scholar] [CrossRef]
- Rosenbaum, M.; Gewies, A.; Pechloff, K.; Heuser, C.; Engleitner, T.; Gehring, T.; Hartjes, L.; Krebs, S.; Krappmann, D.; Kriegsmann, M.; et al. Bcl10-controlled Malt1 paracaspase activity is key for the immune suppressive function of regulatory T cells. Nat. Commun. 2019, 10, 2352. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Deng, N.; Yang, N.; Zhao, X.; Lin, X. Malt1 Protease Is Critical in Maintaining Function of Regulatory T Cells and May Be a Therapeutic Target for Antitumor Immunity. J. Immunol. 2019, 202, 3008–3019. [Google Scholar] [CrossRef]
- Heuser, C.; Gotot, J.; Piotrowski, E.C.; Philipp, M.-S.; Courrèges, C.J.F.; Otte, M.S.; Guo, L.; Schmid-Burgk, J.L.; Hornung, V.; Heine, A.; et al. Prolonged IKKβ Inhibition Improves Ongoing CTL Antitumor Responses by Incapacitating Regulatory T Cells. Cell Rep. 2017, 21, 578–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polesso, F.; Sarker, M.; Anderson, A.; Parker, D.C.; Murray, S.E. Constitutive expression of NF-κB inducing kinase in regulatory T cells impairs suppressive function and promotes instability and pro-inflammatory cytokine production. Sci. Rep. 2017, 7, 14779. [Google Scholar] [CrossRef]
- Grinberg-Bleyer, Y.; Caron, R.; Seeley, J.J.; Silva, N.S.D.; Schindler, C.W.; Hayden, M.S.; Klein, U.; Ghosh, S. The Alternative NF-κB Pathway in Regulatory T Cell Homeostasis and Suppressive Function. J. Immunol. 2018, 200, 2362–2371. [Google Scholar] [CrossRef]
- Koliesnik, I.O.; Andreas, N.; Thuy, A.; Sreekantapuram, S.; Haenold, R.; Weih, F. Alternative NF-kappaB signaling controls peripheral homeostasis and function of regulatory T cells. Immunobiology 2019, 224, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, S.; Chen, W.; Ye, Q.; Dou, Y.; Xiao, Y.; Zhang, L.; Minze, L.J.; Li, X.C.; Xiao, X. Role of the NF-kappaB Family Member RelB in Regulation of Foxp3(+) Regulatory T Cells In Vivo. J. Immunol. 2018, 200, 1325–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grinberg-Bleyer, Y.; Oh, H.; Desrichard, A.; Bhatt, D.M.; Caron, R.; Chan, T.A.; Schmid, R.M.; Klein, U.; Hayden, M.S.; Ghosh, S. NF-κB c-Rel Is Crucial for the Regulatory T Cell Immune Checkpoint in Cancer. Cell 2017, 170, 1096–1108.e1013. [Google Scholar] [CrossRef] [Green Version]
- Gao, F.; Cheng, Q.; Liu, M.D.; Rong, L.; KLiu, C.J.; Zhang, X.Z. Local T regulatory cells depletion by an integrated nanodrug system for efficient chem-immunotherapy of tumor. Sci. China Chem. 2019, 62, 1230–1244. [Google Scholar] [CrossRef]
- Amato, C.M.; Hintzsche, J.D.; Wells, K.; Applegate, A.; Gorden, N.T.; Vorwald, V.M.; Tobin, R.P.; Nassar, K.; Shellman, Y.G.; Kim, J.; et al. Pre-Treatment Mutational and Transcriptomic Landscape of Responding Metastatic Melanoma Patients to Anti-PD1 Immunotherapy. Cancers 2020, 12, 1943. [Google Scholar] [CrossRef]
- Grasso, C.S.; Tsoi, J.; Onyshchenko, M.; Abril-Rodriguez, G.; Ross-Macdonald, P.; Wind-Rotolo, M.; Champhekar, A.; Medina, E.; Torrejon, D.Y.; Shin, D.S.; et al. Conserved Interferon-gamma Signaling Drives Clinical Response to Immune Checkpoint Blockade Therapy in Melanoma. Cancer Cell 2020, 38, 500–515.e503. [Google Scholar] [CrossRef]
- Roh, W.; Chen, P.L.; Reuben, A.; Spencer, C.N.; Prieto, P.A.; Miller, J.P.; Gopalakrishnan, V.; Wang, F.; Cooper, Z.A.; Reddy, S.M.; et al. Integrated molecular analysis of tumor biopsies on sequential CTLA-4 and PD-1 blockade reveals markers of response and resistance. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Harlin, H.; Hwang, K.W.; Palucki, D.A.; Kim, O.; Thompson, C.B.; Boothby, M.; Alegre, M.L. CTLA-4 engagement regulates NF-kappaB activation in vivo. Eur. J. Immunol. 2002, 32, 2095–2104. [Google Scholar] [CrossRef]
- Pioli, C.; Gatta, L.; Frasca, D.; Doria, G. Cytotoxic T lymphocyte antigen 4 (CTLA-4) inhibits CD28-induced IkappaBalpha degradation and RelA activation. Eur. J. Immunol. 1999, 29, 856–863. [Google Scholar] [CrossRef]
- Arasanz, H.; Gato-Canas, M.; Zuazo, M.; Ibanez-Vea, M.; Breckpot, K.; Kochan, G.; Escors, D. PD1 signal transduction pathways in T cells. Oncotarget 2017, 8, 51936–51945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jutz, S.; Hennig, A.; Paster, W.; Asrak, O.; Dijanovic, D.; Kellner, F.; Pickl, W.F.; Huppa, J.B.; Leitner, J.; Steinberger, P. A cellular platform for the evaluation of immune checkpoint molecules. Oncotarget 2017, 8, 64892–64906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamphorst, A.O.; Wieland, A.; Nasti, T.; Yang, S.; Zhang, R.; Barber, D.L.; Konieczny, B.T.; Daugherty, C.Z.; Koenig, L.; Yu, K.; et al. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science 2017, 355, 1423–1427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, L.Z.; Fu, T.; Guan, B.; Chen, J.; Blando, J.M.; Allison, J.P.; Xiong, L.; Subudhi, S.K.; Gao, J.; Sharma, P. Interdependent IL-7 and IFN-γ signalling in T-cell controls tumour eradication by combined α-CTLA-4+α-PD-1 therapy. Nat. Commun. 2016, 7, 12335. [Google Scholar] [CrossRef] [Green Version]
- Dong, M.B.; Wang, G.; Chow, R.D.; Ye, L.; Zhu, L.; Dai, X.; Park, J.J.; Kim, H.R.; Errami, Y.; Guzman, C.D.; et al. Systematic Immunotherapy Target Discovery Using Genome-Scale In Vivo CRISPR Screens in CD8 T Cells. Cell 2019, 178, 1189–1204.e1123. [Google Scholar] [CrossRef]
- Li, G.; Boucher, J.C.; Kotani, H.; Park, K.; Zhang, Y.; Shrestha, B.; Wang, X.; Guan, L.; Beatty, N.; Abate-Daga, D.; et al. 4-1BB enhancement of CAR T function requires NF-kappaB and TRAFs. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Philipson, B.I.; O’Connor, R.S.; May, M.J.; June, C.H.; Albelda, S.M.; Milone, M.C. 4-1BB costimulation promotes CAR T cell survival through noncanonical NF-kappaB signaling. Sci. Signal. 2020, 13. [Google Scholar] [CrossRef]
- Smith, M.; Garcia-Martinez, E.; Pitter, M.R.; Fucikova, J.; Spisek, R.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial Watch: Toll-like receptor agonists in cancer immunotherapy. Oncoimmunology 2018, 7, e1526250. [Google Scholar] [CrossRef] [PubMed]
- Fang, R.; Wang, C.; Jiang, Q.; Lv, M.; Gao, P.; Yu, X.; Mu, P.; Zhang, R.; Bi, S.; Feng, J.M.; et al. NEMO-IKKbeta Are Essential for IRF3 and NF-kappaB Activation in the cGAS-STING Pathway. J. Immunol. 2017, 199, 3222–3233. [Google Scholar] [CrossRef]
- Hostager, B.S.; Bishop, G.A. CD40-Mediated Activation of the NF-kappaB2 Pathway. Front. Immunol. 2013, 4, 376. [Google Scholar] [CrossRef] [Green Version]
- Ward-Kavanagh, L.K.; Lin, W.W.; Sedy, J.R.; Ware, C.F. The TNF Receptor Superfamily in Co-stimulating and Co-inhibitory Responses. Immunity 2016, 44, 1005–1019. [Google Scholar] [CrossRef] [PubMed]
- Schaer, D.A.; Hirschhorn-Cymerman, D.; Wolchok, J.D. Targeting tumor-necrosis factor receptor pathways for tumor immunotherapy. J. Immunother. Cancer 2014, 2, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharyya, S.; Md Sakib Hossain, D.; Mohanty, S.; Sankar Sen, G.; Chattopadhyay, S.; Banerjee, S.; Chakraborty, J.; Das, K.; Sarkar, D.; Das, T.; et al. Curcumin reverses T cell-mediated adaptive immune dysfunctions in tumor-bearing hosts. Cell Mol. Immunol. 2010, 7, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Churchill, M.; Chadburn, A.; Bilinski, R.T.; Bertagnolli, M.M. Inhibition of intestinal tumors by curcumin is associated with changes in the intestinal immune cell profile. J. Surg. Res. 2000, 89, 169–175. [Google Scholar] [CrossRef]
- Luo, F.; Song, X.; Zhang, Y.; Chu, Y. Low-dose curcumin leads to the inhibition of tumor growth via enhancing CTL-mediated antitumor immunity. Int. Immunopharmacol. 2011, 11, 1234–1240. [Google Scholar] [CrossRef]
- Shao, Y.; Zhu, W.; Da, J.; Xu, M.; Wang, Y.; Zhou, J.; Wang, Z. Bisdemethoxycurcumin in combination with alpha-PD-L1 antibody boosts immune response against bladder cancer. Onco Targets 2017, 10, 2675–2683. [Google Scholar] [CrossRef] [Green Version]
- Shiri, S.; Alizadeh, A.M.; Baradaran, B.; Farhanghi, B.; Shanehbandi, D.; Khodayari, S.; Khodayari, H.; Tavassoli, A. Dendrosomal curcumin suppresses metastatic breast cancer in mice by changing m1/m2 macrophage balance in the tumor microenvironment. Asian Pac. J. Cancer Prev. 2015, 16, 3917–3922. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; You, M.; Xu, Y.; Li, F.; Zhang, D.; Li, X.; Hou, Y. Inhibition of curcumin on myeloid-derived suppressor cells is requisite for controlling lung cancer. Int. Immunopharmacol. 2016, 39, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Fried, A.; Hussaini, R.; White, R.; Baidoo, J.; Yalamanchi, S.; Banerjee, P. Phytosomal curcumin causes natural killer cell-dependent repolarization of glioblastoma (GBM) tumor-associated microglia/macrophages and elimination of GBM and GBM stem cells. J. Exp. Clin. Cancer Res. 2018, 37, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, J.Y.; Su, C.H.; Luo, H.H.; Lei, Y.Y.; Zeng, B.; Zhu, H.S.; Chen, Z.G. Curcumin converts Foxp3+ regulatory T cells to T helper 1 cells in patients with lung cancer. J. Cell Biochem. 2018, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Chesi, M.; Mirza, N.N.; Garbitt, V.M.; Sharik, M.E.; Dueck, A.C.; Asmann, Y.W.; Akhmetzyanova, I.; Kosiorek, H.E.; Calcinotto, A.; Riggs, D.L.; et al. IAP antagonists induce anti-tumor immunity in multiple myeloma. Nat. Med. 2016, 22, 1411–1420. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.S.; Dastidar, H.; Zhang, C.; Zemp, F.J.; Lau, K.; Ernst, M.; Rakic, A.; Sikdar, S.; Rajwani, J.; Naumenko, V.; et al. Smac mimetics and oncolytic viruses synergize in driving anticancer T-cell responses through complementary mechanisms. Nat. Commun. 2017, 8, 344. [Google Scholar] [CrossRef] [Green Version]
- Shono, Y.; Tuckett, A.Z.; Ouk, S.; Liou, H.C.; Altan-Bonnet, G.; Tsai, J.J.; Oyler, J.E.; Smith, O.M.; West, M.L.; Singer, N.V.; et al. A small-molecule c-Rel inhibitor reduces alloactivation of T cells without compromising antitumor activity. Cancer Discov. 2014, 4, 578–591. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, L.Y.; Vo, D.D.; Garban, H.J.; Comin-Anduix, B.; Owens, S.K.; Dissette, V.B.; Glaspy, J.A.; McBride, W.H.; Bonavida, B.; Economou, J.S.; et al. Immunosensitization of tumor cells to dendritic cell-activated immune responses with the proteasome inhibitor bortezomib (PS-341, Velcade). J. Immunol. 2006, 176, 4757–4765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enzler, T.; Sano, Y.; Choo, M.K.; Cottam, H.B.; Karin, M.; Tsao, H.; Park, J.M. Cell-selective inhibition of NF-kappaB signaling improves therapeutic index in a melanoma chemotherapy model. Cancer Discov. 2011, 1, 496–507. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Z.; Su, Z.; Han, S.; Huang, J.; Lin, L.; Shuai, X. Dual pH-sensitive nanodrug blocks PD-1 immune checkpoint and uses T cells to deliver NF-kappaB inhibitor for antitumor immunotherapy. Sci. Adv. 2020, 6, eaay7785. [Google Scholar] [CrossRef] [Green Version]
- Stephan, P.; Lautraite, R.; Voisin, A.; Grinberg-Bleyer, Y. Transcriptional Control of Regulatory T Cells in Cancer: Toward Therapeutic Targeting? Cancers 2020, 12, 3194. [Google Scholar] [CrossRef]
- Wei, Y.; Zhao, Q.; Gao, Z.; Lao, X.M.; Lin, W.M.; Chen, D.P.; Mu, M.; Huang, C.X.; Liu, Z.Y.; Li, B.; et al. The local immune landscape determines tumor PD-L1 heterogeneity and sensitivity to therapy. J. Clin. Investig. 2019, 129, 3347–3360. [Google Scholar] [CrossRef] [Green Version]
- Hayakawa, T.; Yaguchi, T.; Kawakami, Y. Enhanced anti-tumor effects of the PD-1 blockade combined with a highly absorptive form of curcumin targeting STAT3. Cancer Sci. 2020, 111, 4326–4335. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Feng, X.; Wang, J.; Shao, N.; Ji, C.; Ma, D.; Henter, J.I.; Fadeel, B.; Zheng, C. Bortezomib and IL-12 produce synergetic anti-multiple myeloma effects with reduced toxicity to natural killer cells. Anticancer Drugs 2014, 25, 282–288. [Google Scholar] [CrossRef]
- Thirukkumaran, C.M.; Shi, Z.Q.; Nuovo, G.J.; Luider, J.; Kopciuk, K.A.; Dong, Y.; Mostafa, A.A.; Thakur, S.; Gratton, K.; Yang, A.; et al. Oncolytic immunotherapy and bortezomib synergy improves survival of refractory multiple myeloma in a preclinical model. Blood Adv. 2019, 3, 797–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Zheng, L.; Jiang, J.; Zhao, Y.; Wang, X.; Shen, M.; Zhu, F.; Tian, R.; Shi, C.; Xu, M.; et al. Blocking NF-kappaB Is Essential for the Immunotherapeutic Effect of Recombinant IL18 in Pancreatic Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 5939–5950. [Google Scholar] [CrossRef] [Green Version]
- Beug, S.T.; Tang, V.A.; LaCasse, E.C.; Cheung, H.H.; Beauregard, C.E.; Brun, J.; Nuyens, J.P.; Earl, N.; St-Jean, M.; Holbrook, J.; et al. Smac mimetics and innate immune stimuli synergize to promote tumor death. Nat. Biotechnol. 2014, 32, 182–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beug, S.T.; Beauregard, C.E.; Healy, C.; Sanda, T.; St-Jean, M.; Chabot, J.; Walker, D.E.; Mohan, A.; Earl, N.; Lun, X.; et al. Smac mimetics synergize with immune checkpoint inhibitors to promote tumour immunity against glioblastoma. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrucci, M.T.; Vozella, F. The Anti-CD38 Antibody Therapy in Multiple Myeloma. Cells 2019, 8, 1629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jelinek, T.; Paiva, B.; Hajek, R. Update on PD-1/PD-L1 Inhibitors in Multiple Myeloma. Front. Immunol. 2018, 9, 2431. [Google Scholar] [CrossRef] [Green Version]
- Jakubowiak, A.; Offidani, M.; Pegourie, B.; De La Rubia, J.; Garderet, L.; Laribi, K.; Bosi, A.; Marasca, R.; Laubach, J.; Mohrbacher, A.; et al. Randomized phase 2 study: Elotuzumab plus bortezomib/dexamethasone vs. bortezomib/dexamethasone for relapsed/refractory MM. Blood 2016, 127, 2833–2840. [Google Scholar] [CrossRef] [Green Version]
- Markowitz, J.; Luedke, E.A.; Grignol, V.P.; Hade, E.M.; Paul, B.K.; Mundy-Bosse, B.L.; Brooks, T.R.; Dao, T.V.; Kondalasula, S.V.; Lesinski, G.B.; et al. A phase I trial of bortezomib and interferon-alpha-2b in metastatic melanoma. J. Immunother. 2014, 37, 55–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrish, E.; Brumatti, G.; Silke, J. Future Therapeutic Directions for Smac-Mimetics. Cells 2020, 9, 406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaturvedi, M.M.; Sung, B.; Yadav, V.R.; Kannappan, R.; Aggarwal, B.B. NF-kappaB addiction and its role in cancer: ‘one size does not fit all’. Oncogene 2011, 30, 1615–1630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chau, C.H.; Steeg, P.S.; Figg, W.D. Antibody-drug conjugates for cancer. Lancet 2019, 394, 793–804. [Google Scholar] [CrossRef]
- Langone, P.; Debata, P.R.; Inigo Jdel, R.; Dolai, S.; Mukherjee, S.; Halat, P.; Mastroianni, K.; Curcio, G.M.; Castellanos, M.R.; Raja, K.; et al. Coupling to a glioblastoma-directed antibody potentiates antitumor activity of curcumin. Int. J. Cancer. J. Int. Du Cancer 2014, 135, 710–719. [Google Scholar] [CrossRef]
- Langone, P.; Debata, P.R.; Dolai, S.; Curcio, G.M.; Inigo Jdel, R.; Raja, K.; Banerjee, P. Coupling to a cancer cell-specific antibody potentiates tumoricidal properties of curcumin. Int. J. Cancer J. Int. Du Cancer 2012, 131, E569–E578. [Google Scholar] [CrossRef]



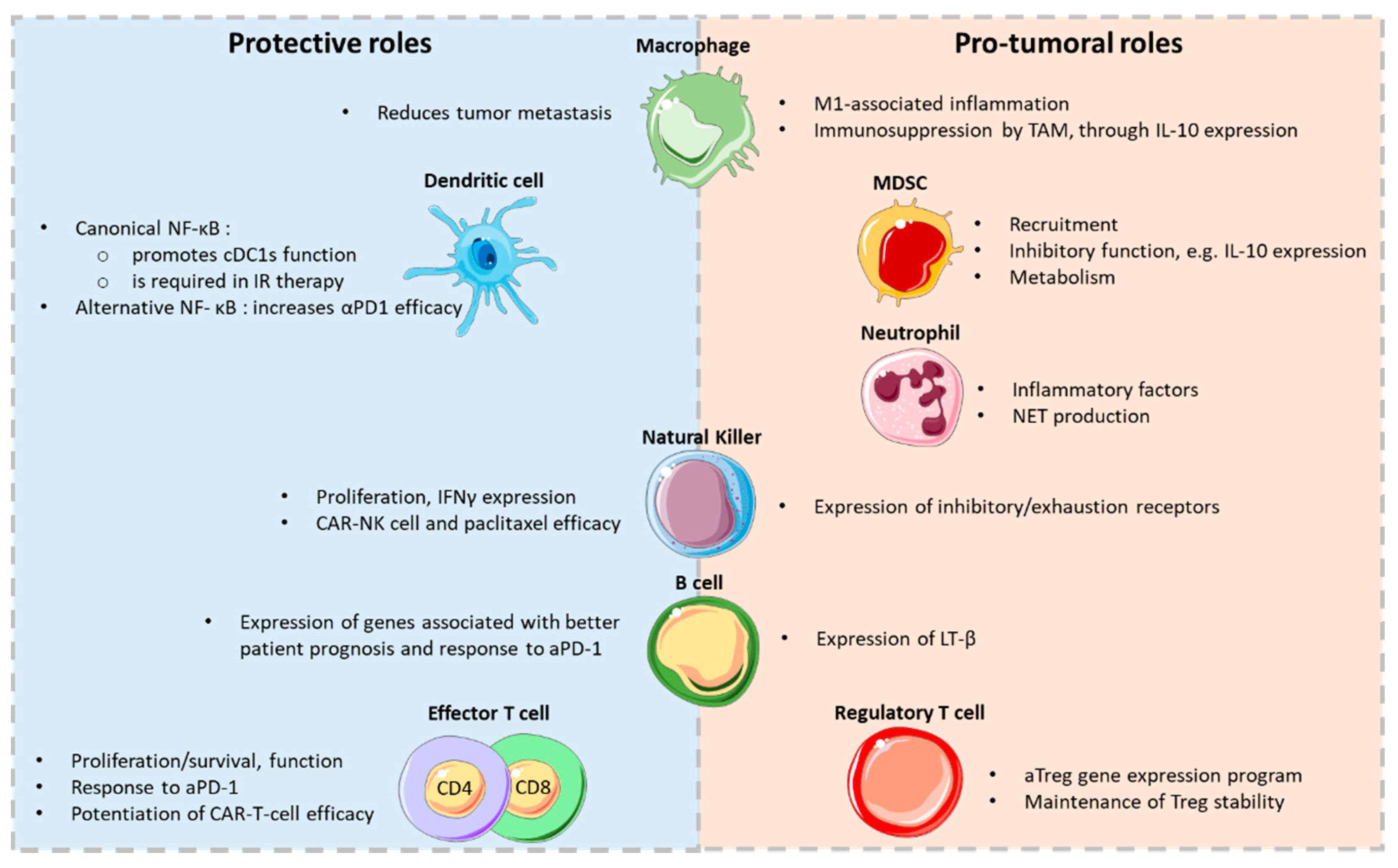{kind=link}
{kind=link}
| Agent/Mechanism | Tumor Model | Impact on Immune Cells | References |
|---|---|---|---|
| Mepazine/MI-2, MALT-1 inhibitor | Mouse melanoma | ↑ CD8+ T cells, ↑ IFNγ by Treg cells in TILs; efficacy lost in RAG−/− mice | [159] |
| Curcumin, prevents IκBα degradation, NF-κB translocation | Mouse breast, colon, lung cancer | ↑ T cells and IFNγ expression, ↓ Treg cell proportion in TILs | [186,187,188] |
| Mouse breast, bladder lung cancer | ↓ MDSCs in TILs, ↑M1 macrophages | [189,190,191] | |
| Mouse glioblastoma | ↑ NK cells; NK-cell-dependent therapeutic effect | [192] | |
| Patients with lung cancer | ↑ IFNγ expression by CD4+ T cells, ↓ Treg cell proportions in PBMCs | [193] | |
| KINK-1, IKKβ inhibitor | Mouse melanoma | ↑ CD8+ T-cell infiltration and function in tumors | [162] |
| SMAC mimetics (CIAP antagonists), inhibit canonical NF-κB, enhance alternative NF-κB | Mouse Multiple myeloma | ↑macrophage phagocytosis; macrophage-dependent therapeutic effect | [194] |
| Mouse glioblastoma, breast cancer | ↑ T-cell in TILs; CD8+ T-cell-dependent therapeutic effect | [195] | |
| IT-603, c-Rel inhibitor | Mouse thymoma, melanoma | ↓ circulating T cells; ↑ IFNγ expression by Teff/Treg cells in TILs | [196,167] |
| PTXF, c-Rel inhibitor | Mouse breast cancer, melanoma | ↑ CD8+ T cells, ↓ Treg cells in TILs; efficacy lost in RAG−/− mice | [168,167] |
| R96A, c-Rel inhibitor | Mouse melanoma | ↓ inhibitory function of MDSCs | [48] |
| Organism. | Combination Therapy | Cancer Type(s) | References |
|---|---|---|---|
| Mouse Studies | RELA shRNA + αPD-1 | Hepatocellular carcinoma | [201] |
| curcumin + αCTLA-4 | Breast cancer | [14] | |
| curcumin + αPD-1/PDL-1 | Diverse | [189,199,202] | |
| Bortezomib + IL12 | Multiple myeloma | [203] | |
| Bortezomib + reovirus | Multiple myeloma | [204] | |
| Mepazine + αPD-1/PD-L1 | Melanoma | [159,160] | |
| BAY11-7082 + rmIL-18 | PDAC | [205] | |
| SMAC mimetics + oncolytic VSV | Breast cancer | [206] | |
| SMAC mimetics + αPD-1 | Diverse | [81,207] | |
| PTXF/IT-603/R96A + αPD-1/PD-L1 | Diverse | [48,167] | |
| PTXF + αPD-1 + chemotherapy | Breast cancer | [168] | |
| Human Studies | Bortezomib + αCD38 + dexamethasone | Multiple myeloma | [208] |
| Bortezomib + αPD-1 | Multiple myeloma | [209] | |
| Bortezomib + αSLAMF7 | Multiple myeloma | [210] | |
| Bortezomib + Interferon α2B | Melanoma | [211] | |
| SMAC mimetics + αPD-1 | Diverse | [212] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lalle, G.; Twardowski, J.; Grinberg-Bleyer, Y. NF-κB in Cancer Immunity: Friend or Foe? Cells 2021, 10, 355. https://doi.org/10.3390/cells10020355
Lalle G, Twardowski J, Grinberg-Bleyer Y. NF-κB in Cancer Immunity: Friend or Foe? Cells. 2021; 10(2):355. https://doi.org/10.3390/cells10020355
Chicago/Turabian StyleLalle, Guilhem, Julie Twardowski, and Yenkel Grinberg-Bleyer. 2021. "NF-κB in Cancer Immunity: Friend or Foe?" Cells 10, no. 2: 355. https://doi.org/10.3390/cells10020355
APA StyleLalle, G., Twardowski, J., & Grinberg-Bleyer, Y. (2021). NF-κB in Cancer Immunity: Friend or Foe? Cells, 10(2), 355. https://doi.org/10.3390/cells10020355