
Ferroptosis and Its Modulation by Autophagy in Light of the Pathogenesis of Lysosomal Storage Diseases





Abstract
:1. Introduction
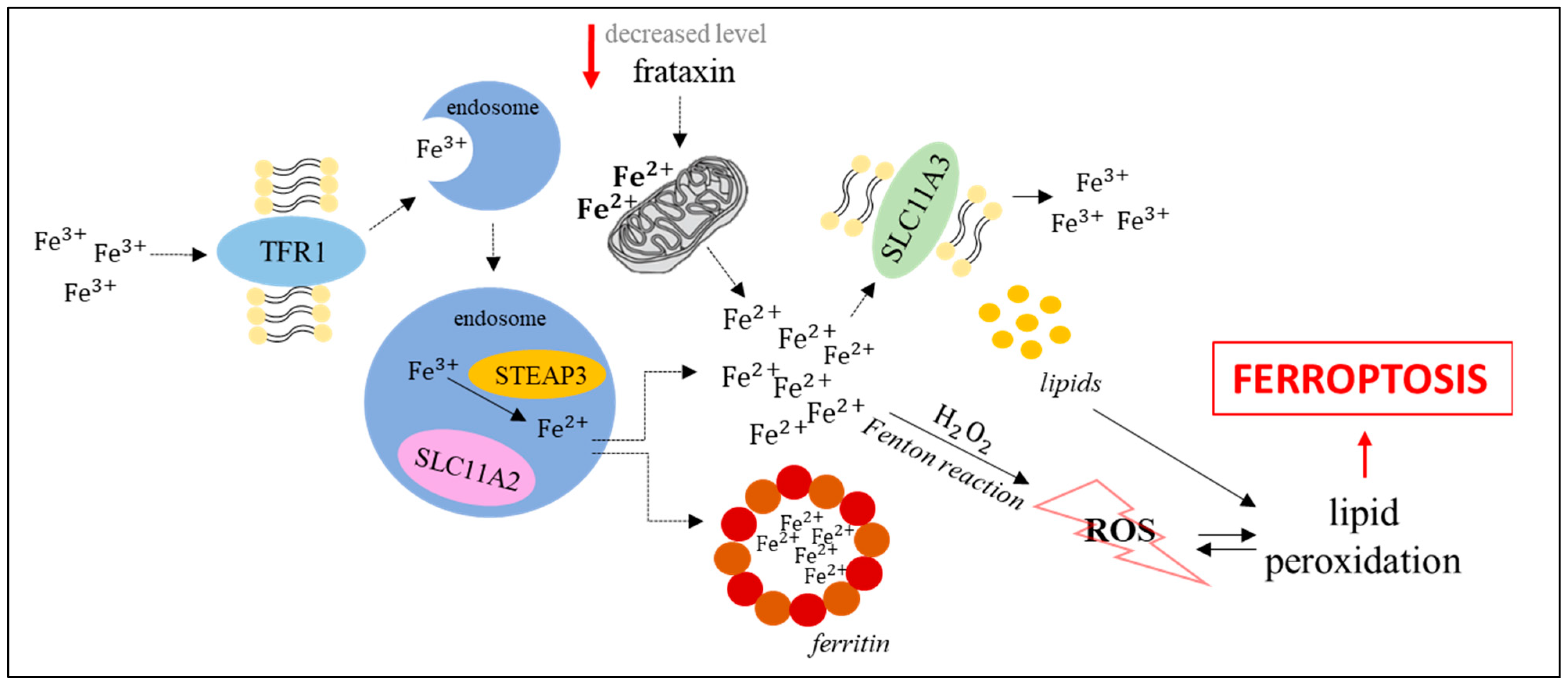
2. Molecular Mechanisms of Ferroptosis
3. Implication of Autophagy Process in Ferroptosis (Autophagy-Dependent Ferroptosis)
4. Links in the Ferroptosis Induction Network
5. Ferroptosis Disorders as a Mechanism for Pathogenesis of Lysosomal Storage Diseases
5.1. Modulation of Iron Levels in Lysosomal Storage Diseases
5.2. Lipid Peroxidation in Lysosomal Storage Diseases
5.3. Modulation of Activity of the GPX4-GSH-Xc− System in Lysosomal Storage Diseases
5.4. The Effectiveness of Ferroptosis Modulators in the Course of Lysosomal Storage Diseases: Already Published Reports and Perspectives
6. Autophagy Disorders in Lysosomal Storage Diseases
7. Possible Role of Autophagy in Ferroptosis Modulation in Lysosomal Storage Diseases
8. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, C.; Liu, Y.; Dai, R.; Ismail, N.; Su, W.; Li, B. Ferroptosis and Its Potential Role in Human Diseases. Front. Pharmacol. 2020, 11, 239. [Google Scholar]
- Liu, J.; Kuang, F.; Kroemer, G.; Klionsky, D.J.; Kang, R.; Tang, D. Autophagy-Dependent Ferroptosis: Machinery and Regulation. Cell Chem. Biol. 2020, 27, 420–435. [Google Scholar]
- Dixon, S.J.; Stockwell, B.R. The Role of Iron and Reactive Oxygen Species in Cell Death. Nat. Chem. Biol. 2014, 10, 9–17. [Google Scholar]
- Platt, F.M.; d’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal storage diseases. Nat. Rev. Dis. Primers 2018, 4, 27. [Google Scholar] [PubMed]
- Parenti, G.; Andria, G.; Ballabio, A. Lysosomal storage diseases: From pathophysiology to therapy. Annu. Rev. Med. 2015, 66, 471–486. [Google Scholar] [CrossRef] [PubMed]
- Winchester, B. Classification of lysosomal storage diseases. In Lysosomal Storage Disorders: A Practical Guide; Mehta, A., Winchester, B., Eds.; John Wiley & Sons, Ltd.: Hobokek, NJ, USA, 2012; pp. 37–46. [Google Scholar]
- Peters, H.; Ellaway, C.; Nicholls, K.; Reardon, K.; Szer, J. Treatable lysosomal storage diseases in the advent of disease-specific therapy. Intern. Med. J. 2020, 50, 5–27. [Google Scholar] [CrossRef]
- Leal, A.F.; Espejo-Mojica, A.J.; Sánchez, O.F.; Ramírez, C.M.; Reyes, L.H.; Cruz, J.C.; Alméciga-Díaz, C.J. Lysosomal storage diseases: Current therapies and future alternatives. J. Mol. Med. 2020, 98, 931–946. [Google Scholar] [PubMed]
- Sheth, J.; Nair, A. Treatment for lysosomal storage disorders. Curr. Pharm. Des. 2020, 26, 5110–5118. [Google Scholar] [CrossRef]
- Cotticelli, M.G.; Xia, S.; Lin, D.; Lee, T.; Terrab, L.; Wipf, P.; Huryn, D.M.; Wilson, R.B. Ferroptosis as a Novel Therapeutic Target for Friedreich’s Ataxia. J. Pharmacol. Exp. Ther. 2019, 369, 47–54. [Google Scholar] [CrossRef]
- La Rosa, P.; Petrillo, S.; Turchi, R.; Berardinelli, F.; Schirinzi, T.; Vasco, G.; Lettieri-Barbato, D.; Fiorenza, M.T.; Bertini, E.S.; Aquilano, K.; et al. The Nrf2 induction prevents ferroptosis in Friedreich’s Ataxia. Redox Biol. 2021, 38, 101791. [Google Scholar] [CrossRef] [PubMed]
- Turchi, R.; Tortolici, F.; Guidobaldi, G.; Iacovelli, F.; Falconi, M.; Rufini, S.; Faraonio, R.; Casagrande, V.; Federici, M.; De Angelis, L.; et al. Frataxin deficiency induces lipid accumulation and affects thermogenesis in brown adipose tissue. Cell Death Dis. 2020, 11, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, J.; Zhou, Y.; Li, Y.; Xia, J.; Chen, Y.; Chen, S.; Wang, X.; Sun, W.; Wang, T.; Ren, X.; et al. Identification of Frataxin as a regulator of ferroptosis. Redox Biol. 2020, 32, 101483. [Google Scholar] [CrossRef]
- Liu, J.; He, H.; Wang, J.; Guo, X.; Lin, H.; Chen, H.; Jiang, C.; Chen, L.; Yao, P.; Tang, Y. Oxidative stress-dependent frataxin inhibition mediated alcoholic hepatocytotoxicity through ferroptosis. Toxicology 2020, 445, 152584. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cel death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; Stockwell, B.R. Synthetic lethal screening identifies compounds activating irondependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef] [Green Version]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [Green Version]
- Ursini, F.; Maiorino, M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic. Biol. Med. 2020, 152, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Su, L.J.; Zhang, J.H.; Gomez, H.; Murugan, R.; Hong, X.; Xu, D.; Jiang, F.; Peng, Z.Y. Reactive Oxygen Species-Induced Lipid Peroxidation in Apoptosis, Autophagy, and Ferroptosis. Oxid. Med. Cell. Longev. 2019, 2019, 5080843. [Google Scholar] [CrossRef] [Green Version]
- Koppula, P.; Zhang, Y.; Zhuang, L.; Gan, B. Amino acid transporter SLC7A11/xCT at the crossroads of regulating redox homeostasis and nutrient dependency of cancer. Cancer Commun. 2018, 38, 12. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Zhu, S.; Chen, P.; Hou, W.; Wen, Q.; Liu, J.; Xie, Y.; Liu, J.; Klionsky, D.J.; Kroemer, G.; et al. AMPK-Mediated BECN1 Phosphorylation Promotes Ferroptosis by Directly Blocking System X c- Activity. Curr. Biol. 2018, 28, 2388–2399.e5. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef]
- Shin, D.; Kim, E.H.; Lee, J.; Roh, J.-L. Nrf2 inhibition reverses resistance to GPX4 inhibitor-induced ferroptosis in head and neck cancer. Free Radic. Biol. Med. 2018, 129, 454–462. [Google Scholar] [CrossRef]
- Chen, D.; Tavana, O.; Chu, B.; Erber, L.; Chen, Y.; Baer, R.; Gu, W. NRF2 Is a Major Target of ARF in p53-Independent Tumor Suppression. Mol. Cell. 2017, 68, 224–232.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarangelo, A.; Magtanong, L.; Bieging-Rolett, K.T.; Li, Y.; Ye, J.; Attardi, L.D.; Dixon, S.J. p53 suppresses metabolic stress-induced ferroptosis in cancer cells. Cell Rep. 2018, 22, 569–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarangelo, A.; Dixon, S. The p53-p21 pathway inhibits ferroptosis during metabolic stress. Oncotarget 2018, 9, 24572–24573. [Google Scholar] [CrossRef] [PubMed]
- Maddocks, O.D.K.; Berkers, C.R.; Mason, S.M.; Zheng, L.; Blyth, K.; Gottlieb, E.; Vousden, K.H. Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature 2013, 493, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Das, U.N. Saturated Fatty Acids, MUFAs and PUFAs Regulate Ferroptosis. Cell Chem. Biol. 2019, 26, 309–311. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Jiang, X.; Yang, C.; Zhang, J.; Chen, B.; Li, Y.; Yao, S.; Xie, Q.; Gomez, H.; Murugan, R.; et al. Pannexin 1 Mediates Ferroptosis That Contributes to Renal Ischemia/Reperfusion Injury. J. Biol. Chem. 2019, 294, 19395–19404. [Google Scholar] [CrossRef]
- Ye, F.; Chai, W.; Xie, M.; Yang, M.; Yu, Y.; Cao, Y.; Yang, L. HMGB1 regulates erastin-induced ferroptosis via RAS-JNK/p38 signaling in HL-60/NRASQ61L cells. Am. J. Cancer Res. 2019, 9, 730–739. [Google Scholar]
- Yagoda, N.; von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 864–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Guo, P.; Xie, X.; Wang, Y.; Chen, G. Ferroptosis, a new form of cell death, and its relationships with tumourous diseases. J. Cell Mol. Med. 2017, 21, 648–657. [Google Scholar] [CrossRef]
- Wen, Q.; Liu, J.; Kang, R.; Zhou, B.; Tang, D. The release and activity of HMGB1 in ferroptosis. Biochem. Biophys. Res. Commun. 2019, 510, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Hayano, M.; Yang, W.S.; Corn, C.K.; Pagano, N.C.; Stockwell, B.R. Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ. 2016, 23, 270–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Gai, C.; Ding, D.; Wang, F.; Li, W. Targeted p53 on Small-Molecules-Induced Ferroptosis in Cancers. Front. Oncol. 2018, 8, 507. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Tanaka, T.; Poyurovsky, M.V.; Nagano, H.; Mayama, T.; Ohkubo, S.; Lokshin, M.; Hosokawa, H.; Nakayama, T.; Suzuki, Y.; et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc. Natl. Acad. Sci. USA 2010, 107, 7461–7466. [Google Scholar] [CrossRef] [Green Version]
- Shintoku, R.; Takigawa, Y.; Yamada, K.; Kubota, C.; Yoshimoto, Y.; Takeuchi, T.; Koshiishi, I.; Torii, S. Lipoxygenase-mediated generation of lipid peroxides enhances ferroptosis induced by erastin and RSL3. Cancer Sci. 2017, 108, 2187–2194. [Google Scholar] [CrossRef]
- Xie, Y.; Zhu, S.; Song, X.; Sun, X.; Fan, Y.; Liu, J.; Zhong, M.; Yuan, H.; Zhang, L.; Billiar, T.R.; et al. The Tumor Suppressor p53 Limits Ferroptosis by Blocking DPP4 Activity. Cell Rep. 2017, 20, 1692–1704. [Google Scholar] [CrossRef] [Green Version]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Meng, L.; Han, L.; Jia, Y.; Zhao, Y.; Gao, H.; Kang, R.; Wang, X.; Tang, D.; Dai, E. Lipid storage and lipophagy regulates ferroptosis. Biochem. Biophys. Res. Commun. 2019, 508, 997–1003. [Google Scholar] [CrossRef]
- Yang, M.; Chen, P.; Liu, J.; Zhu, S.; Kroemer, G.; Klionsky, D.J.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. Clockophagy is a novel selective autophagy process favoring ferroptosis. Sci. Adv. 2019, 5, eaaw2238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, K.; Skouta, R.; Kaplan, A.; Yang, W.S.; Hayano, M.; Dixon, S.J.; Brown, L.M.; Valenzuela, C.A.; Wolpaw, A.J.; Stockwell, B.R. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol. 2016, 12, 497–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, S.; Zhang, Q.; Sun, X.; Zeh, H.J.; Lotze, M.T.; Kang, R.; Tang, D. HSPA5 Regulates Ferroptotic Cell Death in Cancer Cells. Cancer Res. 2017, 77, 2064–2077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Shen, S.; Chen, C.; Sui, X.; Yang, J.; Wang, L.; Zhou, J. The crosstalk between autophagy and ferroptosis: What can we learn to target drug resistance in cancer? Cancer Biol. Med. 2019, 16, 630–646. [Google Scholar]
- Lee, Y.-S.; Kalimuthu, K.; Park, Y.S.; Makala, H.; Watkins, S.C.; Choudry, M.H.A.; Bartlett, D.L.; Kwon, Y.T.; Lee, Y.J. Ferroptotic agent-induced endoplasmic reticulum stress response plays a pivotal role in the autophagic process outcome. J. Cell Physiol. 2020, 235, 6767–6778. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, X.; Lu, S.; He, C.; Wang, C.; Wang, L.; Wang, X.; Ge, P.; Song, D. Erastin triggers autophagic death of breast cancer cells by increasing intracellular iron levels. Oncol. Lett. 2020, 20, 57. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Masaldan, S.; Clatworthy, S.A.S.; Gamell, C.; Meggyesy, P.M.; Rigopoulos, A.T.; Haupt, S.; Haupt, Y.; Denoyer, D.; Adlard, P.A.; Bush, A.I.; et al. Iron accumulation in senescent cells is coupled with impaired ferritinophagy and inhibition of ferroptosis. Redox Biol. 2018, 14, 100–115. [Google Scholar] [CrossRef]
- Schreiber, R.; Buchholz, B.; Kraus, A.; Schley, G.; Scholz, J.; Ousingsawat, J.; Kunzelmann, K. Lipid peroxidation drives renal cyst growth in vitro through activation of TMEM16A. J. Am. Soc. Nephrol. 2019, 30, 228–242. [Google Scholar] [CrossRef] [Green Version]
- Gaffke, L.; Pierzynowska, K.; Podlacha, M.; Brokowska, J.; Węgrzyn, G. Changes in cellular processes occurring in mucopolysaccharidoses as underestimated pathomechanisms of these diseases. Cell Biol. Int. 2019. [Google Scholar] [CrossRef]
- Kant, S.; Atta, M.G. Therapeutic advances in Fabry disease: The future awaits. Biomed. Pharmacother. 2020, 131, 110779. [Google Scholar] [CrossRef]
- Meena, N.K.; Raben, N. Pompe Disease: New Developments in an Old Lysosomal Storage Disorder. Biomolecules 2020, 10, 1339. [Google Scholar] [CrossRef]
- Gutteridge, J.M.; Rowley, D.A.; Halliwell, B.; Westermarck, T. Increased non-protein-bound iron and decreased protection against superoxide-radical damage in cerebrospinal fluid from patients with neuronal ceroid lipofuscinoses. Lancet 1982, 2, 459–460. [Google Scholar] [CrossRef]
- Heiskala, H.; Gutteridge, J.M.; Westermarck, T.; Alanen, T.; Santavuori, P. Bleomycin-detectable iron and phenanthroline-detectable copper in the cerebrospinal fluid of patients with neuronal ceroid-lipofuscinoses. Am. J. Med. Genet. Suppl. 1988, 5, 193–202. [Google Scholar] [CrossRef]
- Christomanou, H.; Kellermann, J.; Linke, R.P.; Harzer, K. Deficient ferritin immunoreactivity in visceral organs from four patients with Niemann-Pick disease type C. Biochem. Mol. Med. 1995, 55, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Christomanou, H.; Harzer, K. Ouchterlony double immunodiffusion method demonstrates absence of ferritin immunoreactivity in visceral organs from nine patients with Niemann-Pick disease type C. Biochem. Mol. Med. 1996, 58, 176–183. [Google Scholar] [CrossRef]
- Christomanou, H.; Vanier, M.T.; Santambrogio, P.; Arosio, P.; Kleijer, W.J.; Harzer, K. Deficient ferritin immunoreactivity in tissues from niemann-pick type C patients: Extension of findings to fetal tissues, H and L ferritin isoforms, but also one case of the rare Niemann-Pick C2 complementation group. Mol. Genet. Metab. 2000, 70, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Dhami, R.; Passini, M.A.; Schuchman, E.H. Identification of novel biomarkers for Niemann-Pick disease using gene expression analysis of acid sphingomyelinase knockout mice. Mol. Ther. 2006, 13, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Argüello, G.; Martinez, P.; Peña, J.; Chen, O.; Platt, F.; Zanlungo, S.; González, M. Hepatic metabolic response to restricted copper intake in a Niemann-Pick C murine model. Metallomics 2014, 6, 1527–1539. [Google Scholar] [CrossRef] [PubMed]
- Hung, Y.H.; Lotan, A.; Yeshurun, S.; Schroeder, A.; Bush, A.I. Iron chelation by deferiprone does not rescue the Niemann-Pick Disease Type C1 mouse model. Biometals 2020, 33, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Iancu, T.C.; Perl, D.P.; Sternlieb, I.; Lerner, A.; Leshinsky, E.; Kolodny, E.H.; Hsu, A.; Good, P.F. The application of laser microprobe mass analysis to the study of biological material. Biometals 1996, 9, 57–65. [Google Scholar] [CrossRef]
- Stein, P.; Yu, H.; Jain, D.; Mistry, P.K. Hyperferritinemia and iron overload in type 1 Gaucher disease. Am. J. Hematol. 2010, 85, 472–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mekinian, A.; Stirnemann, J.; Belmatoug, N.; Heraoui, D.; Fantin, B.; Fain, O.; Charpentier, A.; Rose, C. Ferritinemia during type 1 Gaucher disease: Mechanisms and progression under treatment. Blood Cells Mol. Dis. 2012, 49, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Faucher, B.; Seguier, J.; Swiader, L.; Cuquemelle, C.; Cerutti, D.; Ebbo, M. Gaucher Disease type 1 mimicking immune thrombocytopenia: Role of hyperferritinemia and hypergammaglobulinemia in the initial evaluation of an isolated thrombopenia. Rev. Med. Interne 2019, 40, 680–683. [Google Scholar] [CrossRef]
- Regenboog, M.; Bohte, A.E.; Akkerman, E.M.; Stoker, J.; Hollak, C.E.M. Iron storage in liver, bone marrow and splenic Gaucheroma reflects residual disease in type 1 Gaucher disease patients on treatment. Br. J. Haematol. 2017, 179, 635–647. [Google Scholar] [CrossRef]
- Bohte, A.E.; van Dussen, L.; Akkerman, E.M.; Nederveen, A.J.; Sinkus, R.; Jansen, P.L.M.; Stoker, J.; Hollak, C.E.M. Liver fibrosis in type I Gaucher disease: Magnetic resonance imaging, transient elastography and parameters of iron storage. PLoS ONE 2013, 8, e57507. [Google Scholar] [CrossRef] [Green Version]
- Regenboog, M.; van Dussen, L.; Verheij, J.; Weinreb, N.J.; Santosa, D.; Dahl, S.V.; Häussinger, D.; Müller, M.N.; Canbay, A.; Rigoldi, M.; et al. Hepatocellular carcinoma in Gaucher disease: An international case series. J. Inherit. Metab. Dis. 2018, 41, 819–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefebvre, T.; Reihani, N.; Daher, R.; de Villemeur, T.B.; Belmatoug, N.; Rose, C.; Colin-Aronovicz, Y.; Puy, H.; Le Van Kim, C.; Franco, M.; et al. Involvement of hepcidin in iron metabolism dysregulation in Gaucher disease. Haematologica 2018, 103, 587–596. [Google Scholar] [CrossRef] [Green Version]
- Marchi, G.; Nascimbeni, F.; Motta, I.; Busti, F.; Carubbi, F.; Cappellini, M.D.; Pietrangelo, A.; Corradini, E.; Piperno, A.; Girelli, D. Hyperferritinemia and diagnosis of type 1 Gaucher disease. Am. J. Hematol. 2020, 95, 570–576. [Google Scholar] [CrossRef]
- Medrano-Engay, B.; Irun, P.; Gervas-Arruga, J.; Andrade-Campos, M.; Andreu, V.; Alfonso, P.; Pocovi, M.; Giraldo, P. Iron homeostasis and infIammatory biomarker analysis in patients with type 1 Gaucher disease. Blood Cells Mol. Dis. 2014, 53, 171–175. [Google Scholar] [CrossRef]
- Lorenz, F.; Pawłowicz, E.; Klimkowska, M.; Beshara, S.; Bulanda Brustad, A.; Skotnicki, A.B.; Wahlin, A.; Machaczka, M. Ferritinemia and serum inflammatory cytokines in Swedish adults with Gaucher disease type 1. Blood Cells Mol. Dis. 2018, 68, 35–42. [Google Scholar] [CrossRef]
- Brady, J.; Trehan, A.; Landis, D.; Toro, C. Mucopolysaccharidosis type IIIB (MPS IIIB) masquerading as a behavioural disorder. BMJ Case Rep. 2013, 2013, bcr2013009592. [Google Scholar] [CrossRef] [Green Version]
- Puy, V.; Darwiche, W.; Trudel, S.; Gomila, C.; Lony, C.; Puy, L.; Lefebvre, T.; Vitry, S.; Boullier, A.; Karim, Z.; et al. Predominant role of microglia in brain iron retention in Sanfilippo syndrome, a pediatric neurodegenerative disease. Glia 2018, 66, 1709–1723. [Google Scholar] [CrossRef]
- Dong, X.-P.; Cheng, X.; Mills, E.; Delling, M.; Wang, F.; Kurz, T.; Xu, H. The type IV mucolipidosis-associated protein TRPML1 is an endolysosomal iron release channel. Nature 2008, 455, 992–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiselyov, K.; Colletti, G.A.; Terwilliger, A.; Ketchum, K.; Lyons, C.W.P.; Quinn, J.; Muallem, S. TRPML: Transporters of metals in lysosomes essential for cell survival? Cell Calcium 2011, 50, 288–294. [Google Scholar] [CrossRef] [Green Version]
- Jeyakumar, M.; Williams, I.; Smith, D.; Cox, T.M.; Platt, F.M. Critical role of iron in the pathogenesis of the murine gangliosidoses. Neurobiol. Dis. 2009, 34, 406–416. [Google Scholar] [CrossRef]
- Gautschi, M.; Merlini, L.; Calza, A.-M.; Hayflick, S.; Nuoffer, J.-M.; Fluss, J. Late diagnosis of fucosidosis in a child with progressive fixed dystonia, bilateral pallidal lesions and red spots on the skin. Eur. J. Paediatr. Neurol. 2014, 18, 516–519. [Google Scholar] [CrossRef] [PubMed]
- Britton, R.S.; Bacon, B.R.; Recknagel, R.O. Lipid peroxidation and associated hepatic organelle dysfunction in iron overload. Chem. Phys. Lipids 1987, 45, 207–239. [Google Scholar] [CrossRef]
- Rider, J.A.; Dawson, G.; Siakotos, A.N. Perspective of biochemical research in the neuronal ceroid-lipofuscinosis. Am. J. Med. Genet. 1992, 42, 519–524. [Google Scholar] [CrossRef]
- Evans, J.; Katz, M.L.; Levesque, D.; Shelton, G.D.; de Lahunta, A.; O’Brien, D. A variant form of neuronal ceroid lipofuscinosis in American bulldogs. J. Vet. Intern. Med. 2005, 19, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Zampieri, S.; Mellon, S.H.; Butters, T.D.; Nevyjel, M.; Covey, D.F.; Bembi, B.; Dardis, A. Oxidative stress in NPC1 deficient cells: Protective effect of allopregnanolone. J. Cell Mol. Med. 2009, 13, 3786–3796. [Google Scholar] [CrossRef] [PubMed]
- Cielo Rech, V.; Feksa, L.R.; Arevalo do Amaral, M.F.; Waltereith Koch, G.; Wajner, M.; Dutra-Filho, C.S.; de Souza Wyse, A.T.; Duval Wannmacher, C.M. Promotion of oxidative stress in kidney of rats loaded with cystine dimethyl ester. Pediatr. Nephrol. 2007, 22, 1121–1128. [Google Scholar] [CrossRef]
- Wilmer, M.J.; Kluijtmans, L.A.J.; van der Velden, T.J.; Willems, P.H.; Scheffer, P.G.; Masereeuw, R.; Monnens, L.A.; van den Heuvel, L.P.; Levtchenko, E.N. Cysteamine restores glutathione redox status in cultured cystinotic proximal tubular epithelial cells. Biochim. Biophys. Acta 2011, 1812, 643–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonçalves Pereira, V.; Martins, A.M.; Micheletti, C.; D’Almeida, V. Mutational and oxidative stress analysis in patients with mucopolysaccharidosis type I undergoing enzyme replacement therapy. Clin. Chim. Acta 2008, 387, 75–79. [Google Scholar] [CrossRef]
- Reolon, G.K.; Reinke, A.; de Oliveira, M.R.; Braga, L.M.; Camassola, M.; Andrades, M.E.; Fonseca Moreira, J.C.; Nardi, N.B.; Roesler, R.; Dal-Pizzol, F. Alterations in oxidative markers in the cerebellum and peripheral organs in MPS I mice. Cell Mol. Neurobiol. 2009, 29, 443–448. [Google Scholar] [CrossRef]
- Hamano, K.; Hayashi, M.; Shioda, K.; Fukatsu, R.; Mizutani, S. Mechanisms of neurodegeneration in mucopolysaccharidoses II and IIIB: Analysis of human brain tissue. Acta Neuropathol. 2008, 115, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Filippon, L.; Vanzin, C.C.; Biancini, G.B.; Pereira, I.N.; Manfredini, V.; Sitta, A.; do Carmo R Peralba, M.; Schwartz, I.V.D.; Giugliani, R.; Vargas, C.R. Oxidative stress in patients with mucopolysaccharidosis type II before and during enzyme replacement therapy. Mol. Genet. Metab. 2011, 103, 121–127. [Google Scholar] [CrossRef]
- Diaz Jacques, C.E.; Donida, B.; Mescka, C.P.; Rodrigues, D.G.B.; Marchetti, D.P.; Bitencourt, F.H.; Burin, M.G.; de Souza, C.F.M.; Giugliani, R.G.; Vargas, C.R. Oxidative and nitrative stress and pro-inflammatory cytokines in Mucopolysaccharidosis type II patients: Effect of long-term enzyme replacement therapy and relation with glycosaminoglycan accumulation. Biochim. Biophys. Acta 2016, 1862, 1608–1616. [Google Scholar] [CrossRef] [PubMed]
- Villani, G.R.D.; Di Domenico, C.; Musella, A.; Cecere, F.; Di Napoli, D.; Di Natale, P. Mucopolysaccharidosis IIIB: Oxidative damage and cytotoxic cell involvement in the neuronal pathogenesis. Brain Res. 2009, 1279, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Biancini, G.B.; Jacques, C.E.; Hammerschmidt, T.; de Souza, H.M.; Donida, B.; Deon, M.; Vairo, F.P.; Lourenço, C.M.; Giugliani, R.; Vargas, C.R. Biomolecules damage and redox status abnormalities in Fabry patients before and during enzyme replacement therapy. Clin. Chim. Acta 2016, 461, 41–46. [Google Scholar] [CrossRef]
- Ravarotto, V.; Carraro, G.; Pagnin, E.; Bertoldi, G.; Simioni, F.; Maiolino, G.; Martinato, M.; Landini, L.; Davis, P.A.; Calò, L.A. Oxidative stress and the altered reaction to it in Fabry disease: A possible target for cardiovascular-renal remodeling? PLoS ONE 2018, 13, e0204618. [Google Scholar] [CrossRef] [PubMed]
- Coblentz, J.; St Croix, C.; Kiselyov, K. Loss of TRPML1 promotes production of reactive oxygen species: Is oxidative damage a factor in mucolipidosis type IV? Biochem. J. 2014, 457, 361–368. [Google Scholar] [CrossRef]
- Moraitou, M.; Dimitriou, E.; Dekker, N.; Monopolis, I.; Aerts, J.; Michelakakis, H. Gaucher disease: Plasmalogen levels in relation to primary lipid abnormalities and oxidative stress. Blood Cells Mol. Dis. 2014, 53, 30–33. [Google Scholar] [CrossRef]
- Paintlia, M.K.; Singh, I.; Singh, A.K. Effect of vitamin D3 intake on the onset of disease in a murine model of human Krabbe disease. J. Neurosci. Res. 2015, 93, 28–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardoso, B.R.; Hare, D.J.; Bush, A.I.; Roberts, B.R. Glutathione peroxidase 4: A new player in neurodegeneration? Mol. Psychiatry 2017, 22, 328–335. [Google Scholar] [CrossRef] [Green Version]
- Park, T.-J.; Park, J.H.; Lee, G.S.; Lee, J.-Y.; Shin, J.-H.; Kim, M.W.; Kim, Y.S.; Kim, J.Y.; Oh, K.-J.; Han, B.-S.; et al. Quantitative proteomic analyses reveal that GPX4 downregulation during myocardial infarction contributes to ferroptosis in cardiomyocytes. Cell Death Dis. 2019, 10, 835. [Google Scholar] [CrossRef] [Green Version]
- Magtanong, L.; Ko, P.J.; Dixon, S.J. Emerging roles for lipids in non-apoptotic cell death. Cell Death Differ. 2016, 23, 1099–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Cao, F.; Yin, H.-L.; Huang, Z.-J.; Lin, Z.-T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, P.; Druckrey-Fiskaaen, C.; Kouznetsova, E.; Heinitz, K.; Bigl, M.; Cotman, S.L.; Schliebs, R. Developmental impairments of select neurotransmitter systems in brains of Cln3(Deltaex7/8) knock-in mice, an animal model of juvenile neuronal ceroid lipofuscinosis. J. Neurosci. Res. 2008, 86, 1857–1870. [Google Scholar] [CrossRef]
- Biancini, G.B.; Vanzin, C.S.; Rodrigues, D.B.; Deon, M.; Ribas, G.S.; Barschak, A.G.; Manfredini, V.; Netto, C.B.O.; Jardim, L.B.; Giugliani, R.; et al. Globotriaosylceramide is correlated with oxidative stress and inflammation in Fabry patients treated with enzyme replacement therapy. Biochim. Biophys. Acta 2012, 1822, 226–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chol, M.; Nevo, N.; Cherqui, S.; Antignac, C.; Rustin, P. Glutathione precursors replenish decreased glutathione pool in cystinotic cell lines. Biochem. Biophys. Res. Commun. 2004, 324, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Mannucci, L.; Pastore, A.; Rizzo, C.; Piemonte, F.; Rizzoni, G.; Emma, F. Impaired activity of the gamma-glutamyl cycle in nephropathic cystinosis fibroblasts. Pediatr. Res. 2006, 59, 332–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donida, B.; Marchetti, D.P.; Biancini, G.B.; Deon, M.; Manini, P.R.; da Rosa, H.T.; Moura, D.J.; Saffi, J.; Bender, F.; Burin, M.G.; et al. Oxidative stress and inflammation in mucopolysaccharidosis type IVA patients treated with enzyme replacement therapy. Biochim. Biophys. Acta 2015, 1852, 1012–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J.-S.; Arning, E.; West, M.L.; Day, T.S.; Chen, S.; Meng, X.-L.; Forni, S.; McNeill, N.; Goker-Alpan, O.; Wang, X.; et al. Tetrahydrobiopterin deficiency in the pathogenesis of Fabry disease. Hum. Mol. Genet. 2017, 26, 1182–1192. [Google Scholar] [CrossRef] [PubMed]
- Gaffke, L.; Pierzynowska, K.; Podlacha, M.; Hoinkis, D.; Rintz, E.; Brokowska, J.; Cyske, Z.; Wegrzyn, G. Underestimated Aspect of Mucopolysaccharidosis Pathogenesis: Global Changes in Cellular Processes Revealed by Transcriptomic Studies. Int. J. Mol. Sci. 2020, 21, 1204. [Google Scholar] [CrossRef] [Green Version]
- Gaffke, L.; Pierzynowska, K.; Krzelowska, K.; Piotrowska, E.; Węgrzyn, G. Changes in expressions of genes involved in the regulation of cellular processes in mucopolysaccharidoses as assessed by fibroblast culture-based transcriptomic analyses. Metab Brain Dis. 2020, 35, 1353–1360. [Google Scholar] [CrossRef]
- Pierzynowska, K.; Gaffke, L.; Jankowska, E.; Rintz, E.; Witkowska, J.; Wieczerzak, E.; Podlacha, M.; Węgrzyn, G. Proteasome Composition and Activity Changes in Cultured Fibroblasts Derived From Mucopolysaccharidoses Patients and Their Modulation by Genistein. Front. Cell Dev. Biol. 2020, 8, 540726. [Google Scholar] [CrossRef]
- Pierzynowska, K.; Gaffke, L.; Podlacha, M.; Węgrzyn, G. Genetic Base of Behavioral Disorders in Mucopolysaccharidoses: Transcriptomic Studies. Int. J. Mol. Sci. 2020, 21, 1156. [Google Scholar] [CrossRef] [Green Version]
- Brokowska, J.; Pierzynowska, K.; Gaffke, L.; Rintz, E.; Węgrzyn, G. Expression of genes involved in apoptosis is dysregulated in mucopolysaccharidoses as revealed by pilot transcriptomic analyses. Cell Biol. Int. 2020. [Google Scholar] [CrossRef]
- Rintz, E.; Gaffke, L.; Podlacha, M.; Brokowska, J.; Cyske, Z.; Węgrzyn, G.; Pierzynowska, K. Transcriptomic Changes Related to Cellular Processes with Particular Emphasis on Cell Activation in Lysosomal Storage Diseases from the Group of Mucopolysaccharidoses. Int. J. Mol. Sci. 2020, 21, 3194. [Google Scholar] [CrossRef]
- Skouta, R.; Dixon, S.J.; Wang, J.; Dunn, D.E.; Orman, M.; Shimada, K.; Rosenberg, P.A.; Lo, D.C.; Weinberg, J.M.; Linkermann, A.; et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J. Am. Chem. Soc. 2014, 136, 4551–4556. [Google Scholar] [CrossRef]
- Guiney, S.J.; Adlard, P.A.; Lei, P.; Mawal, C.H.; Bush, A.I.; Finkelstein, D.I.; Ayton, S. Fibrillar α-synuclein toxicity depends on functional lysosomes. J. Biol. Chem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Seranova, E.; Connolly, K.J.; Zatyka, M.; Rosenstock, T.R.; Barrett, T.; Tuxworth, R.I.; Sarkar, S. Dysregulation of autophagy as a common mechanism in lysosomal storage diseases. Essays Biochem. 2017, 61, 733–749. [Google Scholar]
- Chévrier, M.; Brakch, N.; Céline, L.; Genty, D.; Ramdani, Y.; Moll, S.; Djavaheri-Mergny, M.; Brasse-Lagnel, C.; Laquerrière, L.; Barbey, F.; et al. Autophagosome maturation is impaired in Fabry disease. Autophagy 2010, 6, 589–599. [Google Scholar] [CrossRef] [Green Version]
- Micsenyi, M.C.; Dobrenis, K.; Stephney, G.; Pickel, J.; Vanier, M.T.; Slaugenhaupt, S.A.; Walkley, S.U. Neuropathology of the Mcoln1(-/-) knockout mouse model of mucolipidosis type IV. J Neuropathol. Exp. Neurol. 2009, 68, 125–135. [Google Scholar] [CrossRef] [Green Version]
- Curcio-Morelli, C.; Charles, F.A.; Micsenyi, M.A.; Cao, Y.; Venugopal, B.; Browning, M.F.; Dobrenis, K.; Cotman, S.L.; Walkley, S.U.; Slaugenhaupt, S.A. Macroautophagy is defective in mucolipin-1-deficient mouse neurons. Neurobiol. Dis. 2010, 40, 370–377. [Google Scholar] [CrossRef] [Green Version]
- Otomo, T.; Higaki, K.; Nanba, E.; Ozono, K.; Sakai, N. Inhibition of autophagosome formation restores mitochondrial function in mucolipidosis II and III skin fibroblasts. Mol. Genet. Metab. 2009, 98, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Nascimbeni, A.C.; Fanin, M.; Masiero, E.; Angelini, C.; Sandri, M. The role of autophagy in the pathogenesis of glycogen storage disease type II (GSDII). Cell Death Differ. 2012, 19, 1698–1708. [Google Scholar] [CrossRef] [Green Version]
- Sansanwal, P.; Sarwal, M.M. p62/SQSTM1 prominently accumulates in renal proximal tubules in nephropathic cystinosis. Pediatr. Nephrol. 2012, 27, 2137–2144. [Google Scholar] [CrossRef] [PubMed]
- Osellame, L.D.; Rahim, A.A.; Hargreaves, I.P.; Gegg, M.E.; Richard-Londt, A.; Brandner, S.; Waddington, S.N.; Schapira, A.H.V.; Duchen, M.R. Mitochondria and quality control defects in a mouse model of Gaucher disease--links to Parkinson’s disease. Cell Metab. 2013, 17, 941–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pergande, M.R.; Zarate, E.; Haney-Ball, C.; Davidson, C.D.; Scesa, G.; Givogri, M.I.; Bongarzone, E.R.; Cologna, S.M. Standard-flow LC and thermal focusing ESI elucidates altered liver proteins in late stage Niemann-Pick, type C1 disease. Bioanalysis 2019, 11, 1067–1083. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Features | Apoptosis | Autophagy | Ferroptosis | |
|---|---|---|---|---|
| morphology | cell membrane | plasma membrane blebs forming | no changes | cell rounding up (lack of plasma membrane blebs) |
| cytoplasm | pseudopods retraction and cellular volume reduction | accumulation of autophagosomes and autophagolysosomes | reduction in size of mitochondria, condensed densities of mitochondrial membrane, rupture of the mitochondrial membrane, reduction in mitochondrial cristae | |
| nucleus | nuclear volume reduction, fragmentation of nucleus and chromatin condensation | lack of chromatin condensation | lack of nuclear volume reduction and chromatin condensation | |
| biochemistry | caspases activation, DNA fragmentation, exposure of phosphatidylserine, mitochondrial membrane potential (ΔΨm) dissipation | conversion of LC3-I to LC3-II form, increased levels of the LAMP2 protein, increased levels of Atg proteins and the Beclin-1 protein | reactive oxygen species accumulation, lipid peroxidation, iron accumulation, activation of MAP kinases, inhibition of cystine-glutamate antiporter, increased NADPH oxidation, glutathione depletion, arachidonic acid mediators, mitochondrial membrane potential (ΔΨm) dissipation | |
| Ferroptosis Marker | Disease | Model | Material/Organ | Reference(s) | |
|---|---|---|---|---|---|
| increased iron concentration | NCL | Patients | cerebrospinal fluid | [54,55] | |
| Niemann–Pick Disease | ASMKO mice | lung and brain | [59] | ||
| BALB/cJ Npc1nih (Npc1−/−) mice | brain | [61] | |||
| Gaucher disease | Cells | patient-derived skin fibroblasts | [62] | ||
| Patients | liver or spleen biopsy serum MRI imaging of liver, bone, marrow, spleen | [62,63,67] [64,65,69] [66,71] | |||
| MPS type III | Patients | MRI imaging of brain | [73] | ||
| C57Bl/6 Naglu−/− mice | brain | [74] | |||
| ML type IV | Cells | patient-derived skin fibroblasts | [75] | ||
| Fucosidosis | Patients | MRI imaging of brain | [78] | ||
| increased lipid peroxidation | NCL | Patients | serum, brain | [80] | |
| English setters American bulldogs | brain brain, eyes | [80] [81] | |||
| Niemann–Pick disease | Cells | patient-derived skin fibroblasts | [82] | ||
| Cystinosis | rats loaded with cystine dimethyl ester | kidney | [83] | ||
| Cells | proximal tubule epithelial cells taken from patients | [84] | |||
| MPS type I | Patients | serum | [85] | ||
| C57BL/6 Idua−/− mice | brain, heart, lung, diaphragm, liver, kidney, spleen | [86] | |||
| MPS type II | Patients | plasma urine | [88,89] [89] | ||
| MPS type IIIB | C57Bl/6 Naglu−/− mice | brain | [90] | ||
| Fabry disease | Patients | blood, urine plasma | [91] [92] | ||
| ML type IV | cells | TRPML1−/− cells | [93] | ||
| Gaucher disease | patients | erythrocytes and plasma | [94] | ||
| Krabbe disease | GALC twi−/− mice | brain | [95] | ||
| inhibition of activity or decreased level of GPX4-GSH-Xc− system components | GPX4 | NCL | Cln3Dex7/8 knock-in mice | brain | [100] |
| Fabry disease | patients | urine and blood | [101] | ||
| glutathione (GSH) | infantile cystinosis | cells | patient-derived skin fibroblasts | [102] | |
| nephropathic cystinosis | cells | patient-derived skin fibroblasts | [103] | ||
| Fabry disease | patients | urine and blood | [101] | ||
| B6/129-B57BL/6 Fabry mice | heart, kidney, liver, plasma | [105] | |||
| MPS type IVA | patients | urine and blood | [104] | ||
| Xc− | no data | - | - | - | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pierzynowska, K.; Rintz, E.; Gaffke, L.; Węgrzyn, G. Ferroptosis and Its Modulation by Autophagy in Light of the Pathogenesis of Lysosomal Storage Diseases. Cells 2021, 10, 365. https://doi.org/10.3390/cells10020365
Pierzynowska K, Rintz E, Gaffke L, Węgrzyn G. Ferroptosis and Its Modulation by Autophagy in Light of the Pathogenesis of Lysosomal Storage Diseases. Cells. 2021; 10(2):365. https://doi.org/10.3390/cells10020365
Chicago/Turabian StylePierzynowska, Karolina, Estera Rintz, Lidia Gaffke, and Grzegorz Węgrzyn. 2021. "Ferroptosis and Its Modulation by Autophagy in Light of the Pathogenesis of Lysosomal Storage Diseases" Cells 10, no. 2: 365. https://doi.org/10.3390/cells10020365
APA StylePierzynowska, K., Rintz, E., Gaffke, L., & Węgrzyn, G. (2021). Ferroptosis and Its Modulation by Autophagy in Light of the Pathogenesis of Lysosomal Storage Diseases. Cells, 10(2), 365. https://doi.org/10.3390/cells10020365