Functional and Structural Characterization of ClC-1 and Nav1.4 Channels Resulting from CLCN1 and SCN4A Mutations Identified Alone and Coexisting in Myotonic Patients

 , , , , ,
, , , , ,  ,
,  , and
, and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Clinical Diagnosis
2.3. Genetic Analysis
2.4. Molecular Biology
2.5. Heterologous Expression and Electrophysiological Recordings
2.6. Structural Analysis of CLCN1 and SCN4A Gene Mutations
2.7. Statistical Analysis
3. Results
3.1. Clinical Results
3.2. Molecular Results
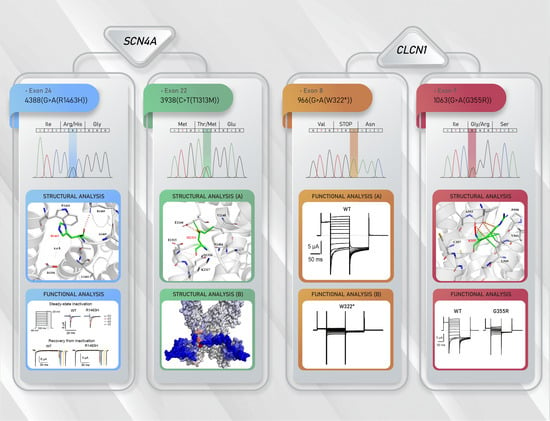
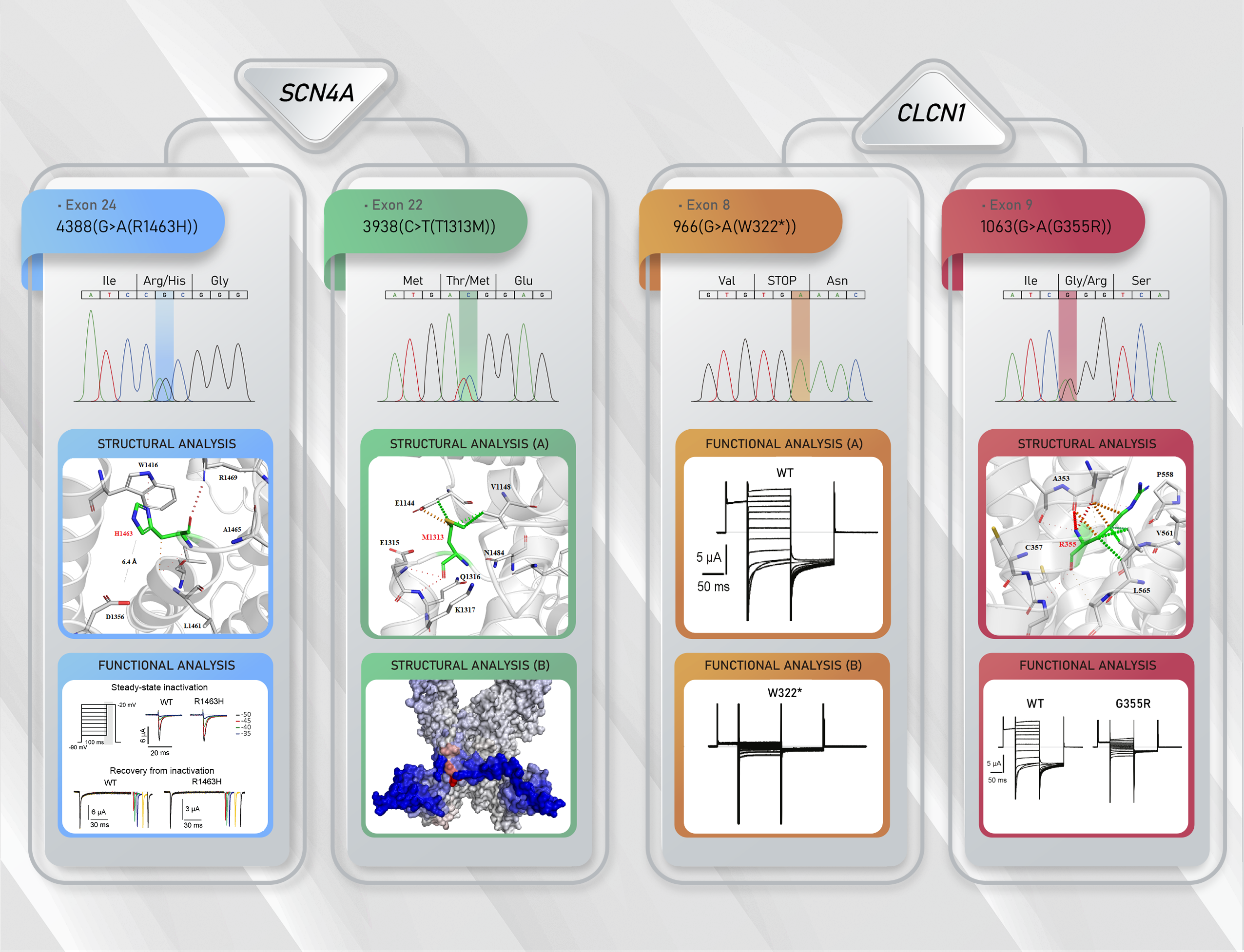
3.2.1. Family 1
3.2.2. Family 2
3.2.3. Family 3
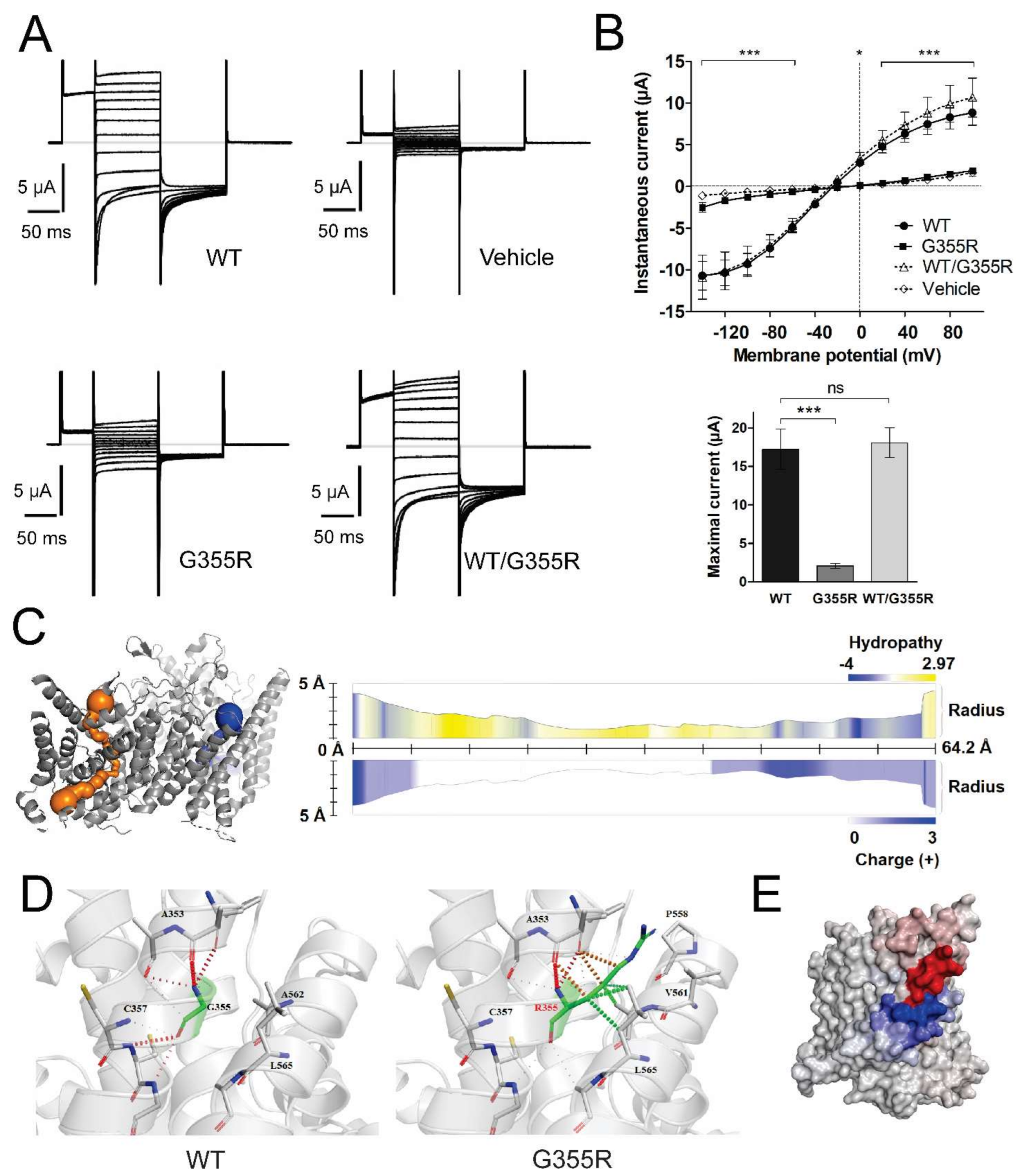
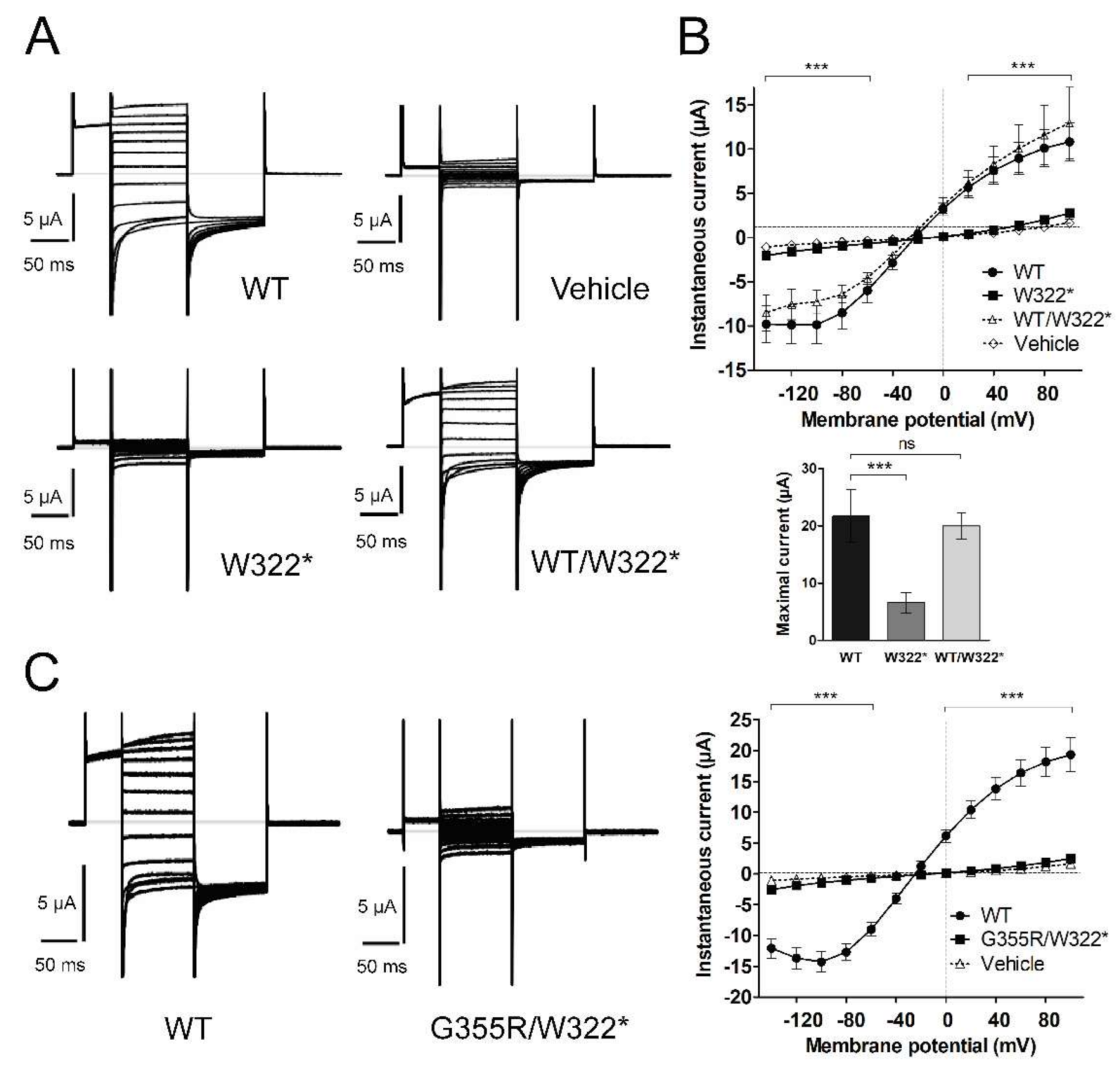
3.3. Functional Characterization of Mutant Chloride Channels
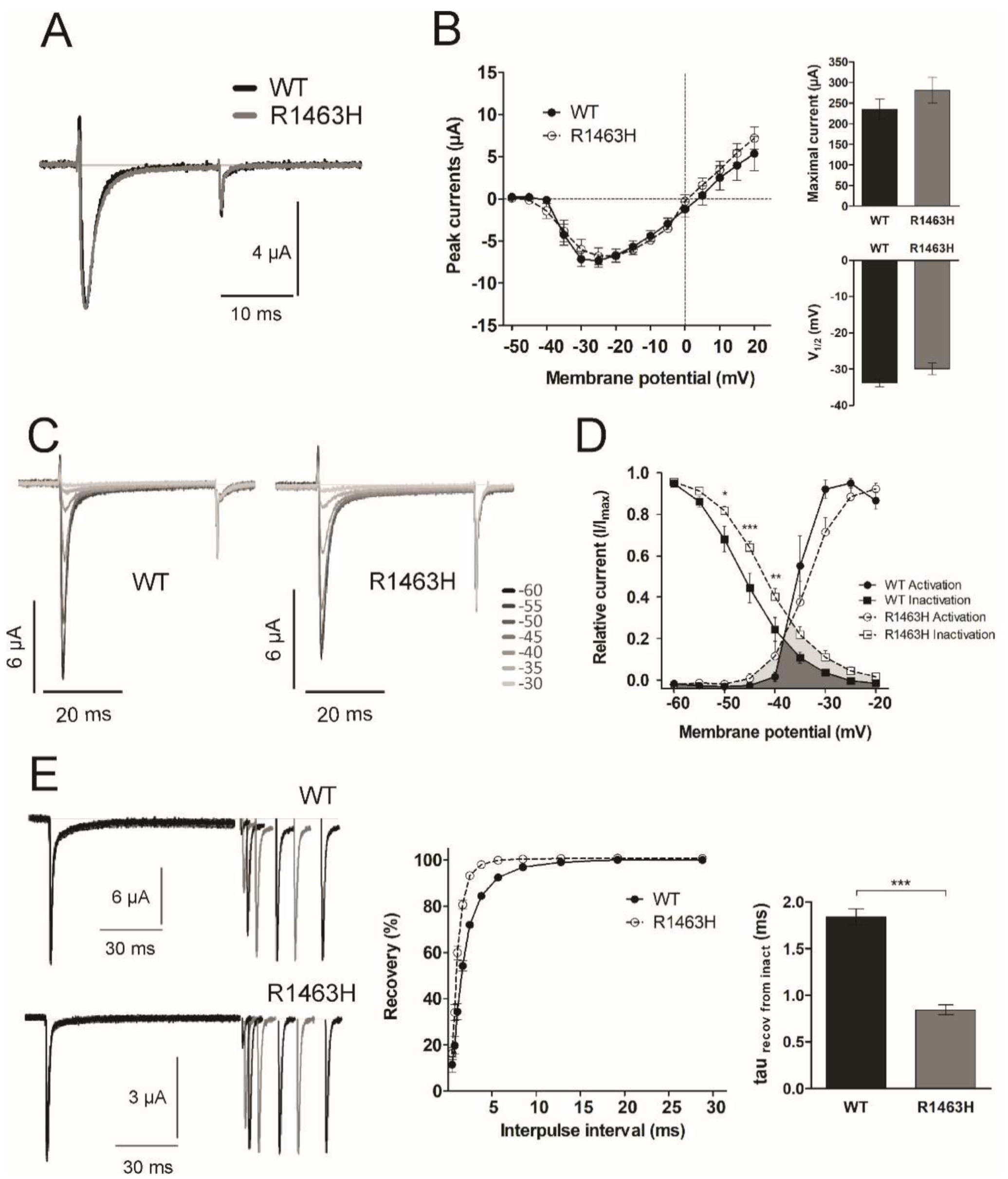
3.4. Functional Characterization of Mutant Sodium Channel
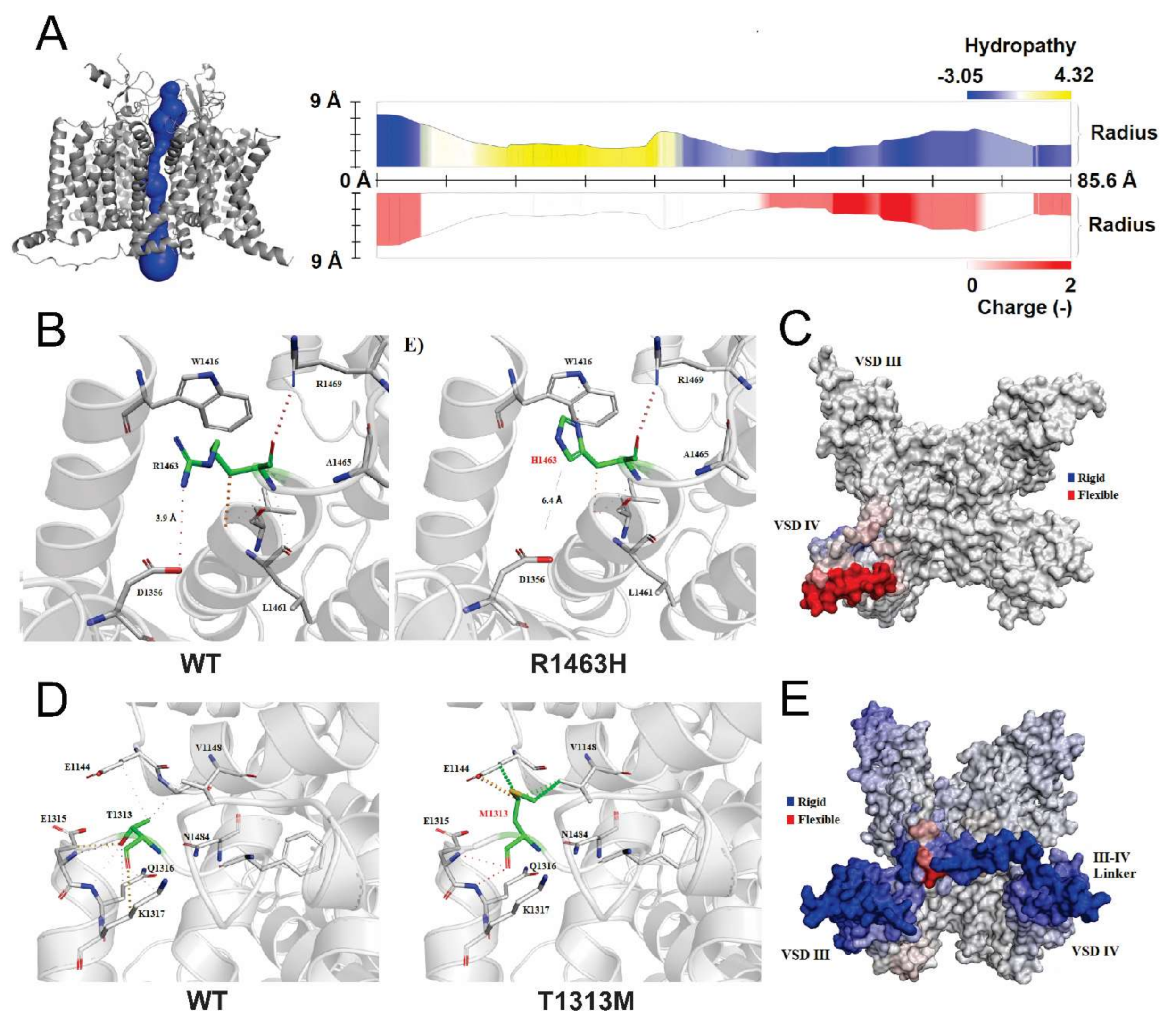
3.5. Structural Analysis of CLCN1 and SCN4A Gene Mutations
3.5.1. Analysis of G355R Mutation
3.5.2. Analysis of R1463H and T1313M Mutations
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Heatwole, C.R.; Statland, J.M.; Logigian, E.L. The diagnosis and treatment of myotonic disorders. Muscle Nerve 2013, 47, 632–648. [Google Scholar] [CrossRef]
- Cannon, S.C. Channelopathies of skeletal muscle excitability. Compr. Physiol. 2015, 5, 761–790. [Google Scholar] [CrossRef] [Green Version]
- Morales, F.; Pusch, M. An Up-to-Date Overview of the Complexity of Genotype-Phenotype Relationships in Myotonic Channelopathies. Front. Neurol 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Jurkat-Rott, K.; Holzherr, B.; Fauler, M.; Lehmann-Horn, F. Sodium channelopathies of skeletal muscle result from gain or loss of function. Pflugers Arch. 2010, 460, 239–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthews, E.; Fialho, D.; Tan, S.V.; Venance, S.L.; Cannon, S.C.; Sternberg, D.; Fontaine, B.; Amato, A.A.; Barohn, R.J.; Griggs, R.C.; et al. The non-dystrophic myotonias: Molecular pathogenesis, diagnosis and treatment. Brain 2010, 133, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Koch, M.C.; Ricker, K.; Otto, M.; Grimm, T.; Bender, K.; Zoll, B.; Harper, P.S.; Lehmann-Horn, F.; Rudel, R.; Hoffman, E.P. Linkage data suggesting allelic heterogeneity for paramyotonia congenita and hyperkalemic periodic paralysis on chromosome 17. Hum. Genet. 1991, 88, 71–74. [Google Scholar] [CrossRef]
- Koch, M.C.; Steinmeyer, K.; Lorenz, C.; Ricker, K.; Wolf, F.; Otto, M.; Zoll, B.; Lehmann-Horn, F.; Grzeschnik, K.-H.; Jentsch, T.J. The skeletal muscle chloride channel in dominant and recessive human myotonia. Science 1992, 257, 797–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imbrici, P.; Maggi, L.; Mangiatordi, G.F.; Dinardo, M.M.; Altamura, C.; Brugnoni, R.; Alberga, D.; Pinter, G.L.; Ricci, G.; Siciliano, G.; et al. ClC-1 mutations in myotonia congenita patients: Insights into molecular gating mechanisms and genotype-phenotype correlation. J. Physiol. 2015, 593, 4181–4199. [Google Scholar] [CrossRef] [Green Version]
- Jentsch, T.J.; Pusch, M. CLC Chloride Channels and Transporters: Structure, Function, Physiology, and Disease. Physiol. Rev. 2018, 98, 1493–1590. [Google Scholar] [CrossRef]
- Pusch, M. Analysis of Electrophysiological Data. In Expression and Analysis of Recombinant Ion Channels; Clare, J., Trezise, D., Eds.; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2006; pp. 111–144. [Google Scholar]
- Stuhmer, W. Electrophysiologic recordings from Xenopus oocytes. Methods Enzymol. 1998, 293, 280–300. [Google Scholar]
- Pusch, M.; Steinmeyer, K.; Koch, M.C.; Jentsch, T.J. Mutations in dominant human myotonia congenita drastically alter the voltage dependence of the CIC-1 chloride channel. Neuron 1995, 15, 1455–1463. [Google Scholar] [CrossRef] [Green Version]
- Vindas-Smith, R.; Fiore, M.; Vasquez, M.; Cuenca, P.; Del Valle, G.; Lagostena, L.; Gaitan-Penas, H.; Estevez, R.; Pusch, M.; Morales, F. Identification and Functional Characterization of CLCN1 Mutations Found in Nondystrophic Myotonia Patients. Hum. Mutat. 2016, 37, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Kwong, K.; Carr, M.J. Voltage-gated sodium channels. Curr. Opin. Pharmacol. 2015, 22, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Adrian, R.H.; Bryant, S.H. On the repetitive discharge in myotonic muscle fibres. J. Physiol. 1974, 240, 505–515. [Google Scholar] [CrossRef]
- Bryant, S.H.; Morales-Aguilera, A. Chloride conductance in normal and myotonic muscle fibres and the action of monocarboxylic aromatic acids. J. Physiol. 1971, 219, 367–383. [Google Scholar] [CrossRef] [Green Version]
- Morales, F.; Cuenca, P.; Del Valle, G.; Brian, R.; Sittenfeld, M.; Montoya, O.; Ashizawa, T.; Rosa, A.; Johnson, K. Miotonía congénita: Caracterización clínica de una familia costarricense afectada por la Enfermedad de Thomsen. Neuroeje 2003, 17, 82–86. [Google Scholar]
- Morales, F.; Cuenca, P.; del Valle, G.; Vasquez, M.; Brian, R.; Sittenfeld, M.; Johnson, K.; Lin, X.; Ashizawa, T. Clinical and molecular diagnosis of a Costa Rican family with autosomal recessive myotonia congenita (Becker disease) carrying a new mutation in the CLCN1 gene. Rev. Biol. Trop. 2008, 56, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Barbieri, R.; Bertelli, S.; Pusch, M.; Gavazzo, P. Late sodium current blocker GS967 inhibits persistent currents induced by familial hemiplegic migraine type 3 mutations of the SCN1A gene. J. Headache Pain 2019, 20, 107. [Google Scholar] [CrossRef]
- Estevez, R.; Schroeder, B.C.; Accardi, A.; Jentsch, T.J.; Pusch, M. Conservation of chloride channel structure revealed by an inhibitor binding site in ClC-1. Neuron 2003, 38, 47–59. [Google Scholar] [CrossRef] [Green Version]
- Pan, X.; Li, Z.; Zhou, Q.; Shen, H.; Wu, K.; Huang, X.; Chen, J.; Zhang, J.; Zhu, X.; Lei, J.; et al. Structure of the human voltage-gated sodium channel Nav1.4 in complex with beta1. Science 2018, 362. [Google Scholar] [CrossRef]
- Wang, K.; Preisler, S.S.; Zhang, L.; Cui, Y.; Missel, J.W.; Gronberg, C.; Gotfryd, K.; Lindahl, E.; Andersson, M.; Calloe, K.; et al. Structure of the human ClC-1 chloride channel. PLoS Biol. 2019, 17, e3000218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, C.H.; Pires, D.E.; Ascher, D.B. DynaMut: Predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res. 2018, 46, W350–W355. [Google Scholar] [CrossRef]
- Chen, V.B.; Arendall, W.B., 3rd; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pravda, L.; Sehnal, D.; Tousek, D.; Navratilova, V.; Bazgier, V.; Berka, K.; Svobodova Varekova, R.; Koca, J.; Otyepka, M. MOLEonline: A web-based tool for analyzing channels, tunnels and pores (2018 update). Nucleic Acids Res. 2018, 46, W368–W373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morales, F.; Cuenca, P.; Brian, R.; Sittenfeld, M.; Del Valle, G. Diagnóstico molecular de la Distrofia Miotónica (DM) en Costa Rica. Appl. Math. Comput. 2001, 43, 159–167. [Google Scholar]
- Hecht, B.K.; Donnelly, A.; Gedeon, A.K.; Byard, R.W.; Haan, E.A.; Mulley, J.C. Direct Molecular Diagnosis Of Myotonic Dystrophy. Clin. Genet. 1993, 43, 276–285. [Google Scholar] [CrossRef]
- Shelbourne, P.; Davies, J.; Buxton, J.; Anvret, M.; Blennow, E.; Bonduelle, M.; Schmedding, E.; Glass, I.; Lindenbaum, R.; Lane, R.; et al. Direct Diagnosis Of Myotonic Dystrophy with a Disease-Specific DNA Marker. N. Engl. J. Med. 1993, 328, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Shelbourne, P.; Winqvist, R.; Kunert, E.; Davies, J.; Leisti, J.; Thiele, H.; Bachmann, H.; Buxton, J.; Williamson, B.; Johnson, K. Unstable DNA may be responsible for the incomplete penetrance of the myotonic dystrophy phenotype. Hum. Mol. Genet. 1992, 1, 467–473. [Google Scholar] [CrossRef]
- Desaphy, J.F.; Gramegna, G.; Altamura, C.; Dinardo, M.M.; Imbrici, P.; George, A.L., Jr.; Modoni, A.; Lomonaco, M.; Conte Camerino, D. Functional characterization of ClC-1 mutations from patients affected by recessive myotonia congenita presenting with different clinical phenotypes. Exp. Neurol. 2013, 248, 530–540. [Google Scholar] [CrossRef] [Green Version]
- Ittisoponpisan, S.; Islam, S.A.; Khanna, T.; Alhuzimi, E.; David, A.; Sternberg, M.J.E. Can Predicted Protein 3D Structures Provide Reliable Insights into whether Missense Variants Are Disease Associated? J. Mol. Biol. 2019, 431, 2197–2212. [Google Scholar] [CrossRef]
- Petukh, M.; Kucukkal, T.G.; Alexov, E. On human disease-causing amino acid variants: Statistical study of sequence and structural patterns. Hum. Mutat. 2015, 36, 524–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, M.; Zhou, H.; Skolnick, J. Insights into Disease-Associated Mutations in the Human Proteome through Protein Structural Analysis. Structure 2015, 23, 1362–1369. [Google Scholar] [CrossRef] [Green Version]
- Brogna, S.; Wen, J. Nonsense-mediated mRNA decay (NMD) mechanisms. Nat. Struct. Mol. Biol. 2009, 16, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhou, L.; Hu, L.; Zhu, Y.; Xu, H.; Liu, Y.; Chen, X.; Yi, X.; Kong, X.; Hurst, L.D. Nonsense-mediated decay targets have multiple sequence-related features that can inhibit translation. Mol. Syst. Biol. 2010, 6, 442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deymeer, F.; Cakirkaya, S.; Serdaroglu, P.; Schleithoff, L.; Lehmann-Horn, F.; Rudel, R.; Ozdemir, C. Transient weakness and compound muscle action potential decrement in myotonia congenita. Muscle Nerve 1998, 21, 1334–1337. [Google Scholar]
- Matthews, E.; Manzur, A.Y.; Sud, R.; Muntoni, F.; Hanna, M.G. Stridor as a neonatal presentation of skeletal muscle sodium channelopathy. Arch. Neurol. 2011, 68, 127–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahmoush, A.J.; Schaller, K.L.; Zhang, P.; Hyslop, T.; Heiman-Patterson, T.; Caldwell, J.H. Muscle sodium channel inactivation defect in paramyotonia congenita with the thr1313met mutation. Neuromuscul. Disord. 1994, 4, 447–454. [Google Scholar] [CrossRef]
- Yang, N.; Ji, S.; Zhou, M.; Ptacek, L.J.; Barchi, R.L.; Horn, R.; George, A.L., Jr. Sodium channel mutations in paramyotonia congenita exhibit similar biophysical phenotypes in vitro. Proc. Natl. Acad. Sci. USA 1994, 91, 12785–12789. [Google Scholar] [CrossRef] [Green Version]
- Dice, M.S.; Abbruzzese, J.L.; Wheeler, J.T.; Groome, J.R.; Fujimoto, E.; Ruben, P.C. Temperature-sensitive defects in paramyotonia congenita mutants R1448C and T1313M. Muscle Nerve 2004, 30, 277–288. [Google Scholar] [CrossRef]
- Patton, D.E.; West, J.W.; Catterall, W.A.; Goldin, A.L. Amino acid residues required for fast Na(+)-channel inactivation: Charge neutralizations and deletions in the III-IV linker. Proc. Natl. Acad. Sci. USA 1992, 89, 10905–10909. [Google Scholar] [CrossRef] [Green Version]
- Stunnenberg, B.C.; Raaphorst, J.; Deenen, J.C.W.; Links, T.P.; Wilde, A.A.; Verbove, D.J.; Kamsteeg, E.J.; van den Wijngaard, A.; Faber, C.G.; van der Wilt, G.J.; et al. Prevalence and mutation spectrum of skeletal muscle channelopathies in the Netherlands. Neuromuscul. Disord. 2018, 28, 402–407. [Google Scholar] [CrossRef] [PubMed]
- Mannikko, R.; Wong, L.; Tester, D.J.; Thor, M.G.; Sud, R.; Kullmann, D.M.; Sweeney, M.G.; Leu, C.; Sisodiya, S.M.; FitzPatrick, D.R.; et al. Dysfunction of NaV1.4, a skeletal muscle voltage-gated sodium channel, in sudden infant death syndrome: A case-control study. Lancet 2018, 391, 1483–1492. [Google Scholar] [CrossRef] [Green Version]
- Mitrovic, N.; George, A.L., Jr.; Lerche, H.; Wagner, S.; Fahlke, C.; Lehmann-Horn, F. Different effects on gating of three myotonia-causing mutations in the inactivation gate of the human muscle sodium channel. J. Physiol. 1995, 487, 107–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitrovic, N.; George, A.L., Jr.; Heine, R.; Wagner, S.; Pika, U.; Hartlaub, U.; Zhou, M.; Lerche, H.; Fahlke, C.; Lehmann-Horn, F. K(+)-aggravated myotonia: Destabilization of the inactivated state of the human muscle Na+ channel by the V1589M mutation. J. Physiol. 1994, 478 Pt 3, 395–402. [Google Scholar] [CrossRef]
- Nakajima, T.; Kaneko, Y.; Dharmawan, T.; Kurabayashi, M. Role of the voltage sensor module in Nav domain IV on fast inactivation in sodium channelopathies: The implication of closed-state inactivation. Channels 2019, 13, 331–343. [Google Scholar] [CrossRef] [Green Version]
- Erisir, A.; Lau, D.; Rudy, B.; Leonard, C.S. Function of specific K(+) channels in sustained high-frequency firing of fast-spiking neocortical interneurons. J. Neurophysiol. 1999, 82, 2476–2489. [Google Scholar] [CrossRef] [Green Version]
- Jaffe, D.B.; Wang, B.; Brenner, R. Shaping of action potentials by type I and type II large-conductance Ca(2)+-activated K+ channels. Neuroscience 2011, 192, 205–218. [Google Scholar] [CrossRef] [Green Version]
- Shruti, S.; Clem, R.L.; Barth, A.L. A seizure-induced gain-of-function in BK channels is associated with elevated firing activity in neocortical pyramidal neurons. Neurobiol. Dis. 2008, 30, 323–330. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Xiao, Z.; Xu, Y.; Zhang, Y.; Tang, D.; Wu, X.; Tang, C.; Chen, M.; Shi, X.; Chen, P.; et al. Electrophysiological and Pharmacological Analyses of Nav1.9 Voltage-Gated Sodium Channel by Establishing a Heterologous Expression System. Front. Pharmacol. 2017, 8, 852. [Google Scholar] [CrossRef] [Green Version]
- Furby, A.; Vicart, S.; Camdessanche, J.P.; Fournier, E.; Chabrier, S.; Lagrue, E.; Paricio, C.; Blondy, P.; Touraine, R.; Sternberg, D.; et al. Heterozygous CLCN1 mutations can modulate phenotype in sodium channel myotonia. Neuromuscul. Disord. 2014, 24, 953–959. [Google Scholar] [CrossRef]
- Kato, H.; Kokunai, Y.; Dalle, C.; Kubota, T.; Madokoro, Y.; Yuasa, H.; Uchida, Y.; Ikeda, T.; Mochizuki, H.; Nicole, S.; et al. A case of non-dystrophic myotonia with concomitant mutations in the SCN4A and CLCN1 genes. J. Neurol. Sci. 2016, 369, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Maggi, L.; Ravaglia, S.; Farinato, A.; Brugnoni, R.; Altamura, C.; Imbrici, P.; Conte Camerino, D.; Padovani, A.; Mantegazza, R.; Bernasconi, P.; et al. Coexistence of CLCN1 and SCN4A mutations in one family suffering from myotonia. Neurogenetics 2017, 18, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Tang, D.; Huang, H.; Tang, H.; Yang, Y.; Yang, M.; Luo, Y.; Tao, H.; Tang, J.; Zhou, X.; et al. Myotonia congenita and periodic hypokalemia paralysis in a consanguineous marriage pedigree: Coexistence of a novel CLCN1 mutation and an SCN4A mutation. PLoS ONE 2020, 15, e0233017. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient/Code | Sex | Onset | MuscularStiffness | MuscularPhenotype | EMG Test 1 | Other Features | Most likely Clinical Diagnosis |
|---|---|---|---|---|---|---|---|
| F1-NDM1 a | F | Youthhood | Yes, with warm-upphenomenon |
|
|
| Thomsen’s disease |
| F1-NDM6 a | F | Youthhood | Yes, with warm-upphenomenon |
|
|
| Thomsen’s disease |
| F1-NDM15 a | M | Birth |
|
|
|
| Severe Thomsen’s disease |
| F2-NDM16 | F | Childhood | Unknown | Muscle weakness |
|
| Becker’s type myotonia |
| F3-NDM17 | M | Youthhood |
|
|
|
| Paramyotonia congenita |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brenes, O.; Barbieri, R.; Vásquez, M.; Vindas-Smith, R.; Roig, J.; Romero, A.; Valle, G.d.; Bermúdez-Guzmán, L.; Bertelli, S.; Pusch, M.; et al. Functional and Structural Characterization of ClC-1 and Nav1.4 Channels Resulting from CLCN1 and SCN4A Mutations Identified Alone and Coexisting in Myotonic Patients. Cells 2021, 10, 374. https://doi.org/10.3390/cells10020374
Brenes O, Barbieri R, Vásquez M, Vindas-Smith R, Roig J, Romero A, Valle Gd, Bermúdez-Guzmán L, Bertelli S, Pusch M, et al. Functional and Structural Characterization of ClC-1 and Nav1.4 Channels Resulting from CLCN1 and SCN4A Mutations Identified Alone and Coexisting in Myotonic Patients. Cells. 2021; 10(2):374. https://doi.org/10.3390/cells10020374
Chicago/Turabian StyleBrenes, Oscar, Raffaella Barbieri, Melissa Vásquez, Rebeca Vindas-Smith, Jeffrey Roig, Adarli Romero, Gerardo del Valle, Luis Bermúdez-Guzmán, Sara Bertelli, Michael Pusch, and et al. 2021. "Functional and Structural Characterization of ClC-1 and Nav1.4 Channels Resulting from CLCN1 and SCN4A Mutations Identified Alone and Coexisting in Myotonic Patients" Cells 10, no. 2: 374. https://doi.org/10.3390/cells10020374
APA StyleBrenes, O., Barbieri, R., Vásquez, M., Vindas-Smith, R., Roig, J., Romero, A., Valle, G. d., Bermúdez-Guzmán, L., Bertelli, S., Pusch, M., & Morales, F. (2021). Functional and Structural Characterization of ClC-1 and Nav1.4 Channels Resulting from CLCN1 and SCN4A Mutations Identified Alone and Coexisting in Myotonic Patients. Cells, 10(2), 374. https://doi.org/10.3390/cells10020374