1. Introduction
Uptake of biomolecules from the extracellular environment is the most fundamental cellular process. Clathrin-mediated endocytosis (CME) is the most important and best characterized endocytic pathway and regulates nutrient uptake, protein and lipid turnover at the plasma membrane, cell signaling, cell motility and cellular polarity [
1,
2,
3]. CME is a very complex process that relies on the coordinated recruitment of multiple proteins to initiate the formation of a clathrin-coated pit (CCP) at the plasma membrane, which will drive membrane invagination and ultimately lead to the release of clathrin-coated vesicles (CCVs) into the cytosol [
4,
5]. The building blocks for CCP and CCV formation are clathrin triskelia. A clathrin triskelion consists of three clathrin heavy chains (CHCs) and three clathrin light chains (CLCs) [
3]. CHCs are fundamental for CME as they form the structural backbone of the triskelia while CLCs appear to usually be dispensable for CME [
6,
7].
Vertebrates possess two clathrin light chain isoforms, clathrin light chain A (CLCA) and clathrin light chain B (CLCB), which are the result of the duplication of an ancient gene [
8]. CLCA and CLCB are ubiquitously expressed in mammalian tissue, with the exception of lymphoid cells that seem to have only CLCA [
9]. CLCA and CLCB are similarly organized and have about 60% sequence identity. CLCB has a unique serine phosphorylation site close to the N terminus of the protein; whereas, CLCA has an additional Hsc70 stimulating region [
2,
10]. The functions of CLCA and CLCB remain poorly understood. Silencing experiments of both CLCA and CLCB reported little to no impact on CME of transferrin or epidermal growth factor (EGF) [
6,
7]. However, recent work exploiting CRISPR/Cas9 editing technology shows that cells expressing only CLCB exhibit an increased rate of CME compared to cells expressing only CLCA or cells expressing both CLCs [
11]. CLCs were also reported to be important for the uptake of a subset of G protein-coupled receptors (GPCRs) [
9,
12] and, more recently, it has shown that site specific phosphorylation of CLCB regulates GPCR endocytosis but not endocytosis of transferrin, by promoting the transition from flat to curve clathrin lattices [
13]. Transition from flat to curved lattices represents a key step during CME [
14] and in vitro studies have proposed that CLCs contribute to the tensile strength of the clathrin coat, which will help bending artificial membranes [
15]. CLCs were reported to interact with the actin cytoskeleton via binding to huntingtin interacting protein 1 (Hip1R) [
2,
16]. Coupling of CLCs to the actin-cytoskeleton has been shown to be important for endocytosis of unconventionally large cargo such as bacteria or certain viruses [
17,
18] or at sites of increased membrane tension [
19]. Furthermore, CLCA acts together with Myosin VI to generate the force required for endocytosis at polarized tissue [
20]. As such, there is growing evidence for a light chain specific function in regulating CME.
Beside their role in endocytosis, CLCs have been described to have an important function in intracellular trafficking and cargo recycling to/from the plasma membrane. Gene silencing of both CLCA and CLCB expression results in a perturbed trafficking of the cation-independent mannose-6-phosphate receptor which was found to be trapped in the trans-Golgi compartment [
7]. Interestingly, knock-out of both CLCA and CLCB was also found to induce the accumulation of actin patches but their identity and function remains unclear [
7,
21,
22]. Similarly, CLCs were shown to be important for the recycling of β1 integrin back to the plasma membrane [
21] and, thereby, influencing cell migration [
21] and cell spreading [
11,
23]. Finally, deletion of CLCA in B cells impairs both their maturation in the germinal center and antibody isotype switching. Selective recycling and internalization of signaling receptors like TGFβR2, CXCR4, and DOR mediated by CLCA were proposed to be the underlying mechanisms [
9].
In this study, we use CRISPR/Cas9 mediated genome editing to sequentially deplete both CLCs. We confirm the appearance of aberrant actin accumulations following both CLC isoforms deletion. Using qualitative and functional assays, we describe these actin structures as functional invadopodia based on the presence of common invadopodia markers as well as their association with discrete proteolytic activity of the extracellular matrix. We could show that deletion of both CLCs induces the accumulation of the matrix metalloproteinase MMP14 at the plasma membrane, which, in turn, promotes invadopodia formation. Accumulation of MMP14 was due to its increased trafficking from intracellular compartments to the plasma membrane and not to a defect in internalization. As such, our work further highlights the importance of CLCs in regulating intracellular trafficking of cargo molecules and describes a novel function of CLCs in regulating invadopodia formation.
2. Materials and Methods
2.1. Cell lines and Cell Culture
U373 and HEK293T cells were maintained in DMEM (Thermo Fisher, Waltham, MA, USA, #41965-039) and in IMDM (Thermo Fisher, #21980-032) media, respectively. Both cell culture media were supplemented with 10% FBS (Biochrom, S0615) and penicillin/streptomycin (Thermo Fisher, 15140). Cells were grown at 37 °C in the presence of 5% CO2. U373 cells stably expressing AP2-eGFP were created by transfection of a plasmid expressing the sigma2 subunit of AP2 fused to eGFP (Ehrlich et al., 2004). Selection of stable KO cell lines was performed using a final concentration of 500 µg/mL Geneticin/G418 (Thermo Fisher, 10131-027), 2 µg/mL Puromycin (Merck-Sigma Aldrich, St. Louis, MO, USA, P9620) and/or 10 µg/mL Blasticidin (Thermo Fisher, R21001).
2.2. Antibodies
Mouse monoclonal antibody against clathrin light chains (Con.1) (Merck-Sigma Aldrich, C1985) was used at 1:1000 for immunofluorescence (IF) and (1:200) Western blotting (WB). Mouse monoclonal antibody against clathrin heavy chains (TD.1) (Abcam, Cambridge, UK, ab24578) was used 1:1000 for WB. Mouse monoclonal antibody against Arp3 (Merck-Sigma Aldrich, A-5979) was used at 1:1000 for IF. Rabbit polyclonal anti-MMP14 antibody (Abcam, ab51074) was used at 1:1000 for IF and WB and monoclonal antibody against β actin (A5441); used 1:5000 for WB). Rabbit polyclonal anti-HIP1R (Merck-Millipore, Burlington, MA, USA, AB9882) was used 1:100 for IF. Mouse monoclonal antibody against Cortactin (p80/85) (Merck-Millipore, 05-180) was used at 1:1000 for IF. Phalloidin coupled with Alexa fluorophore 647 (Thermo Fisher, A-22287) was used at 1:100 for IF. Secondary anti-mouse antibodies coupled to Horseradish Peroxidase (HRP) (GE Healthcare, Chicago, IL, USA, NA931) or anti-rabbit IgG HRP (GE Healthcare, NA934) was used 1:50000 for WB. Secondary anti-mouse antibodies coupled to Alexa Fluorophore 568 (Thermo Fisher, A-11004) and anti-rabbit antibodies coupled to Alexa Fluorophore 568 (Thermo Fisher, A-11011) were used (1:1000) for IF.
2.3. Plasmids, Viral Vectors and gRNA
The lentiviral backbones lentiCRISPR v2 (#52961), lentiCRISPR v2-Blast (#83480) and plko.1 neo (13425) as well as helper plasmids pMDG.2 (#12259) and psPAX (#12260) were purchased from Addgene. MMP14 pHluorin plasmid was kindly provided by Kay Oliver Schink, Harald Stenmark and Philippe Chavrier. The BacMam mCherry-zyxin viral vector was used for transient zyxin expression [
24]. CLCB QQN RFP vector was provided by P. McPherson. The Gateway Destination vector pDest RFP-C was used to clone CLCA and CLCB at the C-terminal end of RFP as well as the cDNA of human CLCA and CLCB (pENTR CLCA and pENTR CLCB) were obtained from the Vector and Clone Repository and repository genome wide cDNA library Gateway Full ORF Clones distributed by the GPCF available at the DKFZ. The following target gRNA sequences were used for genome editing: For single KO clones: CLCA−− (5’GCCTGGACGGCGGCGCCCCC3`) and CLCB−/− (5’AGAGAACGACGAGGGCTTCG3´). For double KO clones: CLCA−/− (5´GACGCGCCCGCACTCTCACC 3)´ and CLCB−/− (5´AGAGAACGACGAGGGCTTCG 3´). Following target sequence was used for shRNA mediated protein knock-down: MMP14 (5’CATTGCATCTTCCCTAGATAG 3´).
2.4. Western Blot (WB)
Protein samples for Western blot analysis (WB) were produced from cell lysates, cells were detached and washed twice with PBS and lysed for 30 min on ice by resuspending them in appropriate volume of RIPA buffer (150 mM sodium chloride, 1.0% (v/v) NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulphate (SDS) and 50 mM Tris, pH 8.0) containing the complete protease inhibitor cocktail (Roche, #118735800001)). Protein concentration was measured using the Protein assay Kit II (BioRad, Hercules, CA, USA, #500112). Sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) gels were prepared with appropriate concentrations of the separating gel. Protein samples were mixed with 4x Laemmli buffer (0.2 M Tris-HCl pH 6.8, 0.05 M EDTA,40% 8v/v) glycerol, 8% 8w/v) SDS, 4% 8% (w/v) SDS, 4% (v/v) β-mercaptoethanol, 0.03% (w/v) bromophenol blue), heated for 10 min at 95 °C. 5 µL Precision Plus Protein Dual Color Standards marker (BioRad, 1610374) was used together with the protein samples in SDS-Tris-Glycine buffer (25 mM Tris-base, 200 mM Glycine, 1% (w/v) SDS). SDS-PAGE was performed at 80 V until samples reached the running gel and the voltage was changed to 120 V until desired separation was achieved. Proteins were transferred to a nitrocellulose blotting membrane (0.45 µm; GE Healthcare, #10600003) by wet blot transfer at 100 V for 80 min, nitrocellulose membrane were blocked with 5% milk in TBST (50 mM Tris-HCL pH 7.5, 150 mM NaCl, 0.1% Tween) for 1 h at room temperature. Incubation with primary antibody diluted in TBST with 5% milk was performed overnight at 4 °C. After four washes for 5 min with TBST, membranes were incubated with secondary antibody diluted in TBST with 5% milk for 1 h at room temperature. The membranes were washed four times for 5 min with TBST and developed using the ECL Western Blotting detection reagents (GE Healthcare, RNP2106) or Western Bright Chemiluminescence Substrate Sirius (Biozym, 541021) using high performance chemiluminescence films (GE Healthcare, #28906837).
2.5. Cloning of Guide RNA (gRNA) or shRNA Encoding Vector for Lentivirus Production
Forward and reverse oligonucleotides containing guide RNA (gRNA) sequence were designed based on the open source gRNA design tools (
https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design (accessed on 15 July 2015) and
http://chopchop.cbu.uib.no/) (accessed on 15 July 2015). Forward and reverse oligonucleotides contaning shRNA sequence were designed according to RNA interference (RNAi) consortium (TRC) (
https://www.broadinstitute.org/scientific-community/science/projects/rnai-consortium/rnai-consortium) (accessed on 17 August 2020). All oligonucleotides were ordered from Eurofins. Complementary oligonucleotides were designed to generate 5′ and 3′ overhangs after annealing complementary to BSMBI restriction sites for gRNA or AgeI and EcoRI restriction sites for shRNA, respectively, for direct ligation into linearized vectors. Oligonucleotides were diluted in H
2O at a concentration of 1 µg/µL. 2.5 µL of forward and reverse oligonucleotides were mixed with 5 µL NEB Buffer 2 (New England Biolabs, Ipswich, MA, USA, B7002S) and 40 µL H
2O. The oligonucleotide mix was heated at 95 °C for 5 min and slowly cooled down to room temperature for annealing of both strands. For the ligation reaction, 150 ng of lentiCRISPR v2 or lentiCRISPR v2-Blast backbone for gRNA cloning or 150 ng of plko.1 neo backbone for shRNA, which has been digested with either BSMBI (R0580) or AgeI-HF (R3552S) and EcoRI-HF (R3101S) purchased from NEB, and 2 µL annealed oligonucleotides were used. A 20 µL ligation reaction was set up using T4 Ligase (New England Biolabs, M0202S) and incubated for 1 h at room temperature. The whole ligation mix was used for transformation into DH5α (Thermo Fisher, 18265017). Positive colonies were confirmed by sequencing.
2.6. Gateway Cloning
To generate expression vectors, Gateway LR Clonase II Enzyme Mix (Thermo Fisher, 11791) was used. In brief, LR-recombination reactions between Gateway entry vectors containing attL-sites and Gateway destination plasmids containing attR-sites were set up by mixing 150 ng of the entry vector with 150 ng of the destination vector. TE buffer was added to reach a final volume of 8 µL. Then, 2 µL of LR Clonase II Enzyme Mix were added and incubated for 1–3 h at 25 °C. 1 µL of Proteinase K was added to the reaction mix and incubated for 10 min at 37 °C. The reaction mix was transformed into DH5α.
2.7. Lentivirus Production
For lentivirus production, Hek293T cells were cultured in 10 cm dishes until 80% confluence. The cells were transfected with pMDG.2, psPAX, and the shRNA encoding pLKO.1 vector or the pWPI vector containing the gene of interest using PEI. 50 µL of PEI (1 mg/mL) was diluted in 200 µL Opti-MEM (Thermo Fisher, 31985062) and mixed well. An amount of 4 µg pMDG.2, 4 µg psPAX, and 8 µg pLKO.1 or pWPI were mixed with Opti-MEM to a final volume of 250 µL and mixed well. After 5 min, both solutions were mixed, vortexed and incubated for 20 min at room temperature. Then the transfection mix was added dropwise to cells. Culture medium was exchanged the next day. After another two days, the supernatant was harvested, centrifuged (10 min, 4000× g) and filtered through a syringe filter (Millex-HA, 0.45 µm, Merck-Millipore, SLHA033SS). For short time storage, the lentivirus containing supernatant was kept at 4 °C; for long time storage, the supernatant was aliquoted and stored at −80 °C.
2.8. Transfection and Viral Transduction
Transfection of cells was done using Lipofectamine 3000 (Thermo Fisher, L300008) if not stated otherwise. Cells were plated in 6-well plates one day before transfection. The next day, cells were transfected at 70–80% confluence. 2.5 µg DNA together with 10µL of P3000 Reagent and 7.5 µL Lipofectamine 3000 were separately mixed with 125 µL Opti-MEM. The two solutions were mixed together. After incubation for 15 min at room temperature, the transfection mix was added dropwise onto the cells. For live-cell imaging of cells transiently expressing fluorescently tagged focal adhesion (FA) proteins, the growth medium was exchanged for fresh growth medium after 6 h. The transfected cells were seeded 24 h after transfection and imaged 6–8 h after seeding.
For transient expression of mCherry-zyxin, cells were transduced with BacMam mCherry-zyxin. Cells were seeded with BacMam mCherry-zyxin in appropriate dishes and were used for experiments 1 day after seeding. 3.5 µL of the P3 stock was used per 10,000 cells. For lentiviral transduction for generation of stable cell lines, 10,000 cells per well were seeded into 6-well plates together with 500 µL of lentivirus containing supernatant. After 2 to 3 days, growth medium was exchanged to selection medium with appropriate antibiotics. Cells were sub-cultured in selection medium for 2 weeks and then used for experiments.
2.9. Wide Field Epifluorescence Microscopy
Widefield epifluorescence microscopy was performed with an inverted Nikon Ti microscope with a 20× (0.75 numerical aperture, Plan Apo λ, Nikon Instruments, Amstelveen, Netherlands) “air” objective or a 40× (1.3 numerical aperture, Plan Fluor, Nikon Instruments) oil immersion objective. The microscope is equipped with a digital camera (DS-Qi1MC, Nikon Instruments) and a LED fluorescence source LED (Lumencor-Sola).
2.10. Total Internal Reflection Fluorescence (TIRF) Microscopy
TIRF microscopy was performed with an inverted Ti microscope (Nikon Instruments) with objective TIRF illumination, with a 60× (1.49 numerical aperture, Apo TIRF, Nikon Instruments) oil immersion objective and an EMCCD camera (Andor iXon Ultra DU-897U) and an inverted Axiovert200M (Zeiss) Orbital-TIRF system, using an alpha Plan-Apochromat 100×/1,46 Oil DIC oil immersion objective and an Hamamatsu EMCCD 9100-50 camera.
2.11. Spinning Disc Confocal Microscopy
Confocal live fluorescence cell imaging was performed with an inverted spinning disc confocal microscope (Nikon Instruments, PerkinElmer) equipped with 60× (1.42 numerical aperture, Apo TIRF, Nikon) oil immersion objective and a CMOS camera (Hamamatsu Ocra Flash 4) or an EMCCD camera (Hamamatsu C9100-23B). The biological samples were kept in an environment control chamber at 37 °C with 5% CO2.
2.12. Fluorescence Recovery after Photobleaching (FRAP)
MMP14-pHluorin was imaged and photobleached using a 488-nm laser light. A defined region of interest (ROI) was photobleached at high laser power. Live-cell confocal imaging was performed for a total of 25 min with at least 3 min and 30 s pre-bleach phase and 20 min recovery phase and at a frame rate of 5 s per frame.The fluorescence recovery in ROI was normalized and analyzed using the easyfrab-web tool:
https://easyfrap.vmnet.upatras.gr/ (accessed on 6 April 2020).
2.13. Immunofluorescence
Cells growing on glass coverslips (#1.5, diameter 12 mm, purchased from Thermo Scientific or 24 mm purchased from Marienfeld) were washed twice with PBS before being fixed with 2% PFA in PBS for 20 min at room temperature or overnight at 4 °C. All following steps were performed at room temperature. After three washes with PBS, cells were permeabilized with 0.5% TritonX in PBS for 15 min followed by blocking with 1% BSA in PBS for 30 min. Samples were incubated with appropriate primary antibody diluted in 1% BSA in PBS for 1 h. After three washes with PBS, samples were stained with appropriate secondary antibody or Phalloidin labeled with Alexa Fluor 647 for 45 min. After four washes with PBS, samples were quickly rinsed H2O and mounted with ProLong Gold antifade containing DAPI (Thermo Fisher, P36931).
2.14. ECM Degradation Assay
ECM degradation assay was performed as published before [
25]. In short, a solution of 0.2% gelatin (from porcine skin; Sigma, G-2500) was prepared in PBS. To dissolve the gelatin, the solution was heated up to 37 °C for 30 min. For sterilization, the solution was filtered using a syringe filter membrane (Millex-GS, 0.22 µm, Merck-Millipore, SLGS033SB). The gelatin was labelled with Alexa Fluor 647 NHS Ester solution (10 mg/mL, Thermo Fisher, A37573) for one hour at room temperature. Labelled gelatin was dialyzed against PBS first for 2 h at room temperature then overnight at 4 °C using Slide-A-Lyzer MINI Dialysis Device (20kMWCO, 0.5 mL; Thermo Scientific, #88402) to remove the free dye.
Coverslips for cell seeding were first coated using 50 µg/mL of poly-L-lysine (PLL Sigma, P8920) for 20 min at room temperature. Coverslips were washed three times with PBS and then fixed with 0.5% glutaraldehyde (Merck-Sigma Aldrich, G5882) in PBS for 15 min at room temperature. Following extensive washes with PBS, solution of Alexa Fluor 647 labelled gelatin and unlabeled gelatin (1:8 ratio) was preheated to 37 °C for 30 min and used to coat the PLL-coated coverslips. Coverslips were inverted on a drop of gelatin mix (80 µL for 12 mm diameter coverslips; 120 µL for 25 mm diameter coverslips) and incubated for 10 min at room temperature. Coverslips were washed three times with PBS. The coated coverslips were directly used for experiments.
2.15. Microscopy-Based Internalization Assay of MMP14
Antibody based internalization assay was performed using a similar protocol as previously described (Majeed et al., 2014). In short, cells were plated on glass coverslips overnight. Following serum starvation for 1 h, cells were labelled with MMP14 antibody (1:100) on ice for 15 min. Cells were then washed 3 times with ice cold PBS to remove unbound antibodies. To determine “total surface MMP14” levels, cells were directly fixed using 2% PFA in PBS and immunostained using the anti-rabbit secondary antibody (without permeabilization). To determine “internal MMP14” levels, following incubation of the cells with MMP14 antibody (1:100) on ice for 15 min, cells were incubated at 37 °C for 30 min in order to start MMP14 internalization. For the measurement of “Internalized MMP14” levels, following incubation at 37 °C for 30 min, cell surface was stripped using ice-cold PBS at pH 2.5, to removed non-internalized primary antibodies, followed by fixation with 2% PFA in PBS and immunostaining using the anti-rabbit secondary antibody (with permeabilization). Imaging was performed using confocal microscopy. Z-stack series were acquired every 0.5 µm for a total of 7.5 µm. Fiji was used to quantify MMP14 levels. Following background subtraction, mean fluorescent intensity was measured for individual cells.
2.16. Transferrin Uptake
Cells were incubated with 50 µg/mL Alexa Fluor 647 labelled Transferrin (Invitrogen, T-23366) for 5 min at 37 °C. Unbound transferrin was removed by washing 3 times with ice-cold PBS and cell surface bound transferrin was washed off using ice-cold PBS at pH 2.5 (acid wash). Following fixation of cells with 2% PFA, internalized transferrin was visualized using confocal microscopy and quantified using Fiji.
2.17. Flow Cytometry-Based Internalization Assay
Internalization assay was, in principle, performed as described before (Pujol et al., 2016 and Figure 5A). Cells were dissociated from cell culture flasks using StemPro AccutaseTM (Thermo Fisher, A1110501) and resuspended in serum free culture medium and kept on ice. Cells were incubated with saturating concentration of MMP14 antibody (1:100) on ice for 40 min, and then washed with cold serum free culture medium before being shifted to 37 °C for the respective time intervals between 0 and 40 min. Following MMP14 internalization, cells were incubated with anti-rabbit Alexa Fluor 647 (1:10000) for 30 min, washed and analyzed by flow cytometry (FACS Canto II, BD Biosciences). Data was analyzed using FlowJoTM version 10.
2.18. Flow Cytometry-Based MMP14 Surface Detection
Similar to the Internalization assay, cells were dissociated from cell culture flasks using StemPro AccutaseTM (Life Technologies, A1110501) and resuspended in serum free culture medium, kept on ice. Cells were incubated with rabbit anti-MMP14 antibodies for 30 min followed by fixation and permeabilization with Cytofix/CytopermTM fixation/Permeabilization Kit (BD Biosciences, 554714). Cells were then incubated with anti-rabbit Alexa Fluor 647 for 30 min and analyzed by flow cytometry (FACS Canto II, BD Biosciences). Data was analyzed using FlowJoTM version 10.
2.19. Surface Biotinylation Assay
A total of 5 × 105 U373 cells (WT and CLC dKO) were seeded in T25 flasks and left to adhere overnight. Afterwards, cells were put on ice and washed three times with cold PBS supplemented with 1mM CaCl2 and 0.5 mM MgCl2 (PBS-Ca/Mg). Cells were then treated with 1mg/mL Ez-Link© Sulfo-NHS-SS-biotin (Thermo Fisher) in PBS-Ca/Mg on ice for 15 min, followed by two 5 min incubations with cold 100mM glycine in PBS-Ca/Mg to quench free biotin. A total of three washes with PBS-Ca/Mg were performed and cells were incubated for 30 min at 37 °C to allow internalization of MMP14. Following the incubation, cells were put back on ice and surface biotin was stripped by three 10 min incubations with 50 mM L-glutathione (Merck-Sigma Aldrich), in solution with 75 mM NaCl, and 10 mM EDTA (pH 7.5). Free –SH groups were then alkylated by three successive incubations with 5mg/mL Iodoacetamide (IAA) in PBS-Ca/Mg. To allow for the recycling of internalized MMP14, cells were put back at 37 °C for an additional 30 min. After this short incubation the recycled surface biotin was again stripped as described above. Cells were lysed using a buffer composed of 1% Triton X-100, 0.1% SDS, 150 mM NaCl, 5 mM EDTA and 50 mM HEPES at pH 7.5, and incubated on ice for 30 min, followed by short sonication and centrifugation to remove cell debris. Protein concentration was assessed and normalized using a DC protein assay kit (Biorad) and then the same amount of protein per sample was added to high capacity NeutrAvidin agarose beads (Thermo Fisher), and incubated overnight at 4 °C under gentle agitation. The following day, the supernatant was removed and beads were washed two times with 1M NaCl, 50mM HEPES, 0.1% Triton X-100 (pH 7.4) and two times with 50mM HEPES (pH 7.4), followed by a 20 min incubation at RT with Laemmli buffer containing 5% SDS, 100 mM NaCl, 100 mM DTT, and 5% B-Mercaptoethanol. The eluted biotinylated proteins were then boiled and loaded onto acrylamide gels for Western blotting.
2.20. Inhibition of Protein Synthesis by Cycloheximide
Cells were seeded on coverslips (IFA) or appropriate cell culture dishes 16h prior experiment. Cells were incubated for 1, 6 and 12 h with 10µg/mL cycloheximide (Sigma). For surface MMP14 samples, cells were incubated on ice with MMP14 antibody for 30 min prior fixation with 2% PFA/PBS. Total MMP14 samples were either lysed with Laemmli buffer or fixed with 2% PFA/PBS and processed for WB analysis or IFA, respectively.
2.21. Quantification of Actin Patches
Confocal microscopy images of cells stained with actin marker Alexa Fluor 647-labelled Phalloidin were analyzed by eye whether cells show aberrant actin patches in addition to normal actin filaments.
2.22. Tracking of Clathrin Structures
The software ilastik (
http://ilastik.org (accessed on 17 August 2020)) was used for tracking CME events as described in [
24], in brief: Images were segmented using pixel classification and object classification workflows. The automatic tracking workflow was used for tracking single clathrin events. The maximum distance of two neighboring objects was set to 5 to avoid merging. Tracking results were analyzed using an automated KNIME workflow (lifetime, maximal fluorescence intensity and average position).
4. Discussion
Deletion of both CLCs in cells causes the assembly of abnormal actin patches. In this work, we identify the nature of these actin structures and could demonstrate that they show all characteristics of invadopodia rather than disorganized actin-cytoskeleton patches. Identification of these actin patches as functional invadopodia was based on the presence of common invadopodia markers as well as spatially increased proteolytic activity (gelatine digestion). Formation of these invadopodia structures requires the loss of both CLCA and CLCB but could be rescued by overexpression of either CLCs, suggesting that both CLCA and CLCB have a redundant function in regulating their formation. Finally, we found that formation of these structures was due to the accumulation of MMP14 at the plasma membrane in CLCA and CLCB depleted cells. This accumulation was not due to a defect in CME and MMP14 turn over from the plasma membrane but rather due to an increased intracellular trafficking of MMP14 towards the plasma membrane. Furthermore, we also showed that knock-down of MMP14 reduced invadopodia formation in CLC depleted cells, demonstrating that MMP14 is key for invadopodia formation upon CLC deletion. Thus, our work further highlights the fact that CLCs have limited functions in regulating endocytosis but play a critical role in regulating intracellular trafficking and sorting of cargo to the plasma membrane.
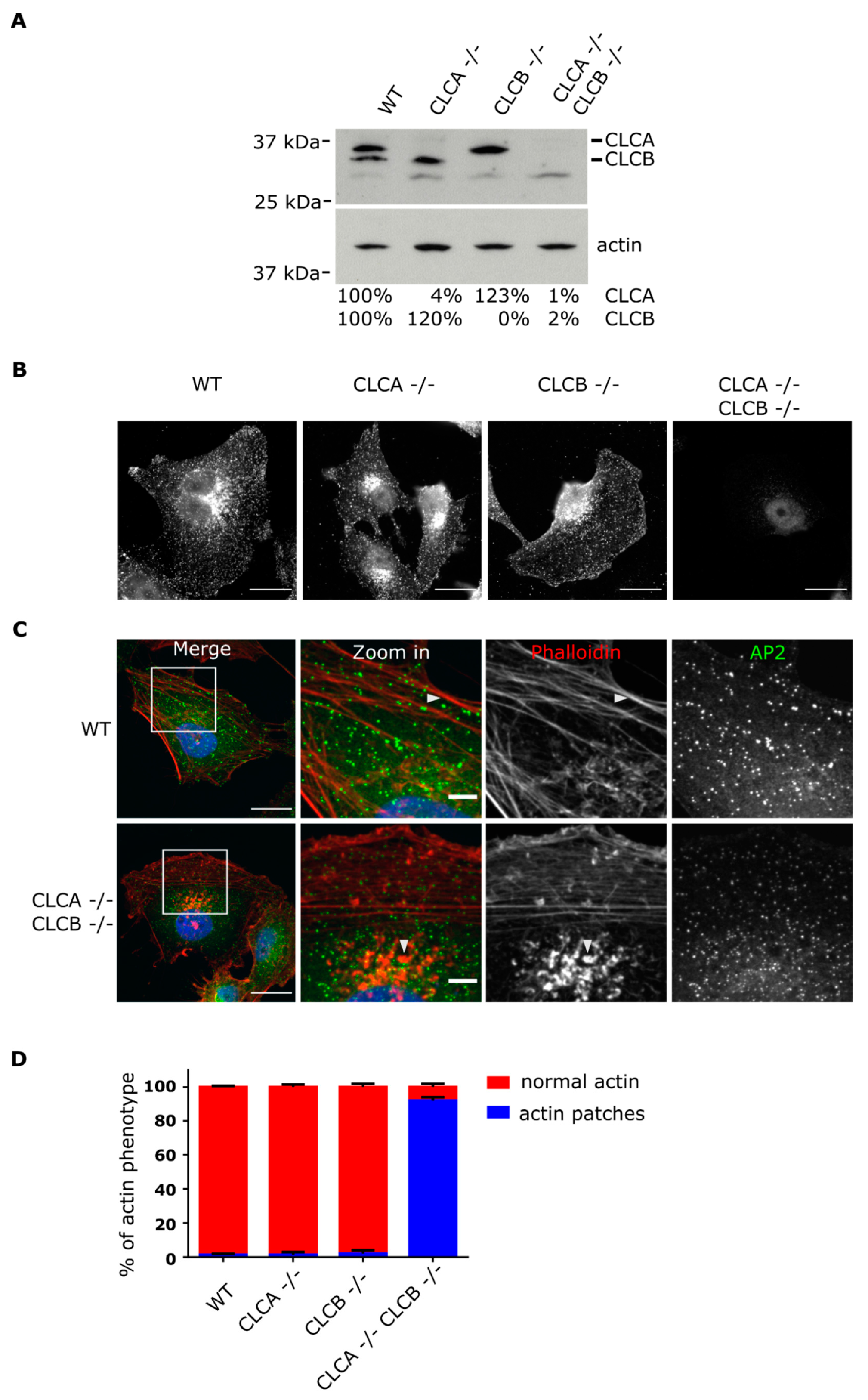
Deletion of both CLCs induced actin patch formation at the ventral plasma membrane (
Figure 1). These observations confirm the long known interplay between CLCs and the actin cytoskeleton as similar actin accumulations at the plasma membrane were previously reported upon siRNA mediated knockdown of CLCs [
7,
21,
22]. Accumulation of these actin structures at the plasma membrane required the deletion of both CLCA and CLCB. Individual knock-out of CLCA or CLCB was found to be not sufficient for inducing the formation of these invadopodia structures and this actin patch phenotype could be rescued by expression of either one of the CLC isoforms (
Figure 2), highlighting the redundant function of both CLCs in regulating actin patch/invadopodia formation. These findings are in agreement with previous works [
7,
21]; however, it was shown that in the HeLa cell line, transient knock down of CLCA alone was sufficient for inducing the formation of actin structures at the plasma membrane [
23]. While we cannot exclude that these discrepancies might reflect cell type specific different functions of CLCA and CLCB, it is possible that these discrepancies arise from the different strategies used to deplete CLCs: knock-out (in this work) vs. siRNA mediated knock-down CLCs [
23]. In cells transiently depleted of one CLC, the remaining CLC does not have the time to take over the impaired function of the depleted one. On the contrary, in cells knock-out for one CLC, the remaining CLC will be able to functionally replace the depleted one due to the time necessary to perform the single cell clonal expansion of the knock-out cell line. We actually observe a higher expression level of CLCB in cells knocked-out for CLCA, and vice versa (
Figure 1A). Within this possibility, this would suggest that although both CLCs can compensate for each other loss, in physiological condition, CLCA would be responsible for regulating actin patch formation. However, a recent study reported decreasing levels of MMP14 upon CLCB silencing [
39] and we observe the same finding in decrease in MMP14 expression in CLCB−/− cells (
Supplementary Figure S5B), suggesting that CLCB is also involved in regulating invadopodia formation.
Overexpression in cells of a CLCB mutant lacking the binding site of the actin organizer HIP1R [
16] (
Supplementary Figure S4) and siRNA-mediated knock-down of HIP1R also induced the formation of similar actin patches [
7,
21,
40,
41]. As HIP1R was shown to interact with cortactin at endocytic sites and regulate actin filament formation [
42] it was proposed that these aberrant actin structures upon HIP1R deletion/depletion are due to impaired actin filament formation through loss of HIP1R binding to cortactin [
7,
40]. Indeed, we observe an accumulation of cortactin at actin structures in cells (
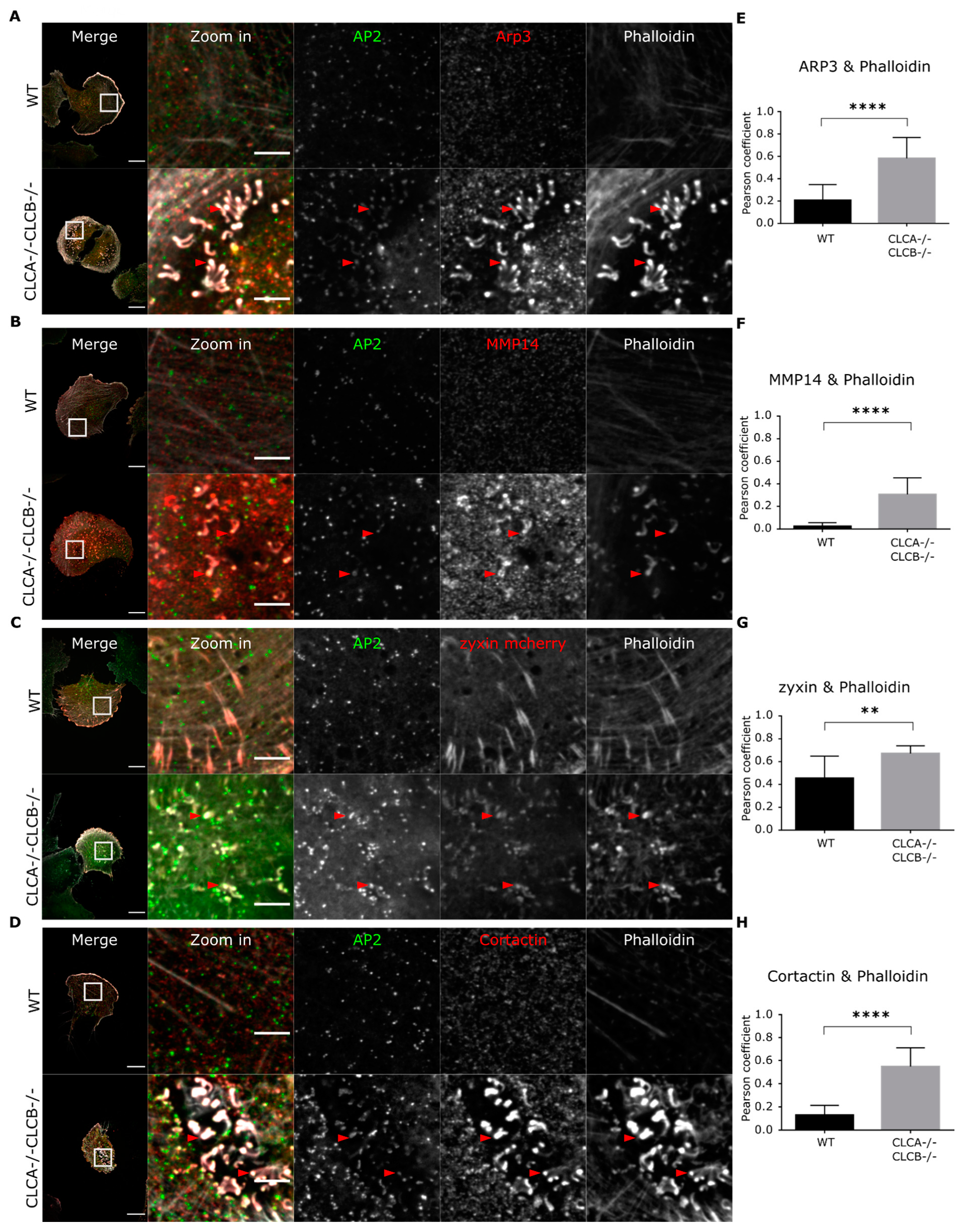
Figure 3D) as well as an attenuation of HIP1R in clathrin-coated structures in CLC depleted cells (
Supplementary Figure S8). Here we described that the actin structures that accumulate upon deletion of CLCA and CLCB are functional invadopodia [
7,
43,
44]. These invadopodia structures contain integrins, focal adhesion-related proteins (i.e., talin, zyxin), actin and actin polymerization and branching mediating proteins (Cortactin, WASP, Arp2/3). Interestingly, we also observed the presence of AP2 in these invadopodia structures (
Figure 3 and
Supplementary Figure S5A), and recruitment of AP2 seems to take place after digestion of the extracellular environment by MMP14 (
Figure 4E,F). In some rare instances, AP2 forms unusual filamentous “tail-like” structures which appear to follow actin (
Figure 3A). The origin and function of these structures remain to be determined, but we hypothesize that clathrin structures are involved in the activity of invadopodia, probably by mediating local recycling of membranes and internalization of extracellular compounds. This is consistent with the observation that clathrin structures are found at invadopodia [
31,
32].
We could show that upon CLC deletion, the trans-membrane metalloproteinase MMP14, a key component of invadopodia and essential for ECM degradation [
41], was upregulated at the protein level (
Supplementary Figure S4) and was enriched at the ventral plasma membrane (
Figure 3B,
Figure 5B and
Figure 6B). The reasons for MMP14 accumulation into cells are not clear but are very likely associated with its accumulation at the plasma membrane in invadopodia structures. MMP14 surface expression is tightly regulated and upregulation of MMP14 coincides with malignant cancer progression [
45,
46,
47]. Interestingly, a recent publication demonstrated that the MMP14 interaction with the ECM alone, independently of the catalytic activity is important for assembly of mature invadopodia [
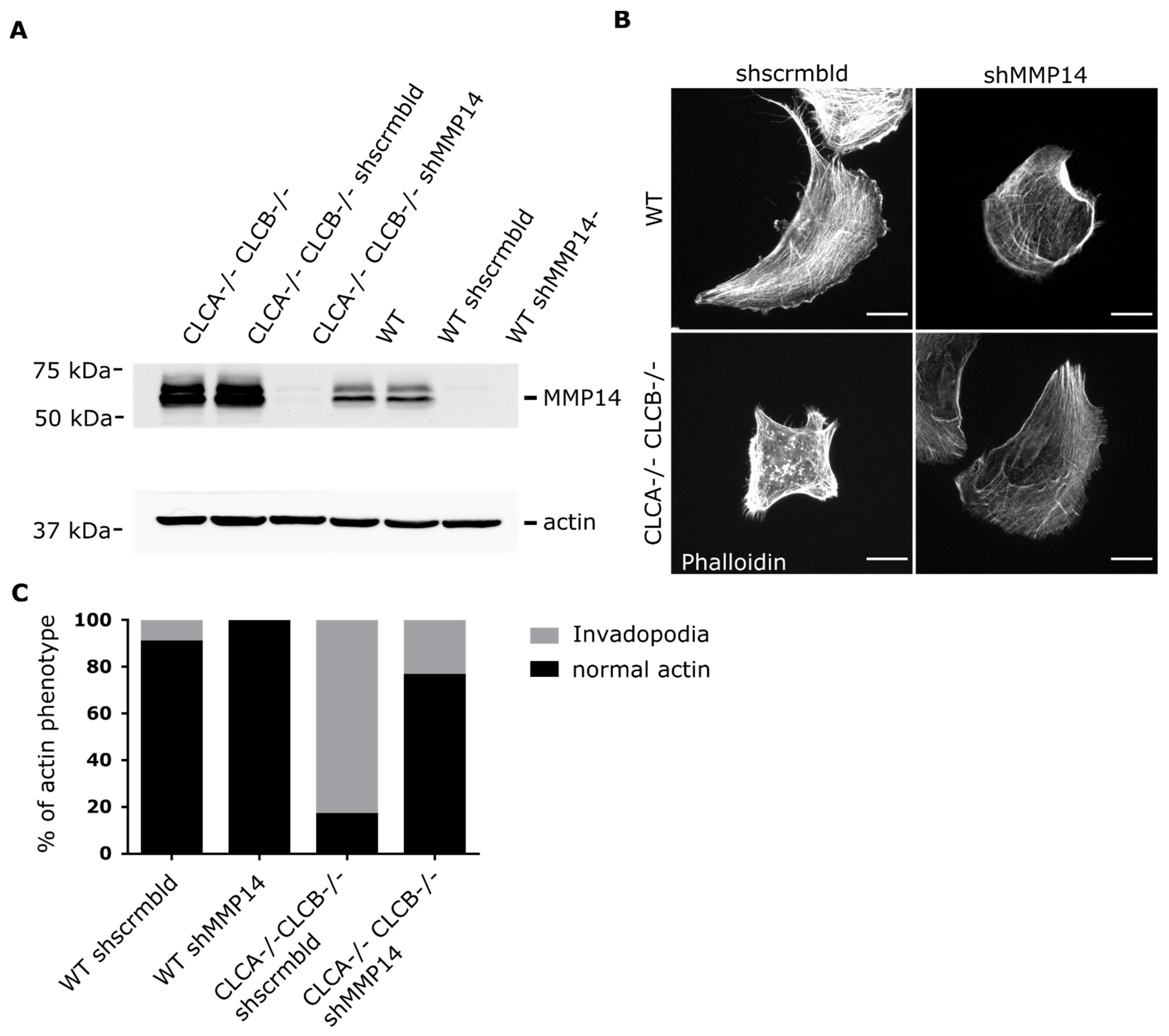
36]. Thus, the described elevated MMP14 surface levels of CLC knock-out cells (
Figure 3B,
Figure 5B and
Figure 6B) alone might be sufficient to prime invadopodia formation, since silencing of MMP14 in CLCA−/− CLCB−/− cells abolished invadopodia formation (
Figure 8). Given the importance of CME to regulate the surface expression of MMP14 [
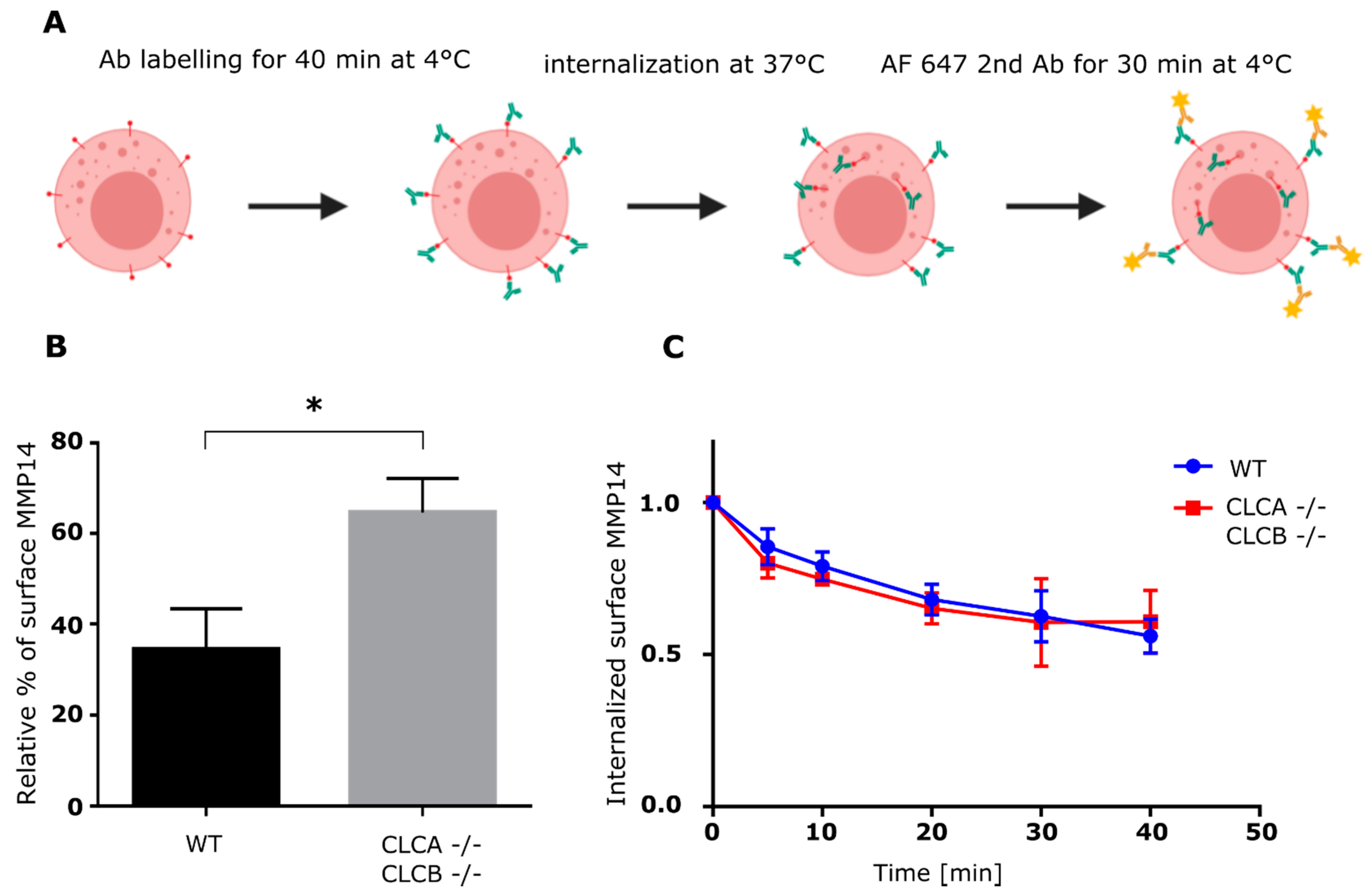
34], we monitored the kinetic of MMP14 uptake from the plasma membrane using either flow-cytometry or confocal microscopy (
Figure 5 and
Figure 6). We could show that the endocytic rate of MMP14 was not significantly different between WT and cells depleted of CLCA and CLCB (
Figure 5C and
Figure 6C). Thus, altered endocytosis of MMP14 in CLCs depleted cells is likely not the reason for MMP14 surface accumulation.
Using a FRAP microscopy approach, we were able to show that the surface accumulation of MMP14 was the result of an increased trafficking of MMP14 from intracellular compartments to the plasma membrane (
Figure 7). The half-time of fluorescence was the same between WT and CLCA−/− CLCB−/− cells, indicating similar intracellular trafficking rates (
Figure 7D). Interestingly, we could show that while WT cells only recovered to about 80% of their initial fluorescence, cells depleted of both CLCs fully recovered, indicating that deletion of CLCA and CLCB increases the mobile fraction of MMP14 (
Figure 7B,C). Recently, it was shown that knockdown of the Phosphatidylinositol 4-kinase IIβ (PI4KIIβ) similarly induces the formation of invadopodia [
35]. This was caused by increased trafficking of MMP14 via exocytic endosomal compartments to the plasma membrane leading to invadosome formation. As such, it is possible that PI4P levels and CLCs co-operate to regulate MMP14 trafficking to the plasma membrane. Upon CLC deletion, trafficking of several cargo was reported to be perturbed: CIMPR was trapped in the TGN and recycling of β1 integrin as well as signaling receptors like TGFβR2, CXCR4, and DOR were impaired [
7,
9,
21]. As such, we propose that CLCs negatively regulate MMP14 to prevent MMP14 accumulation and invadopodia formation.
Interestingly, the actin binding protein cortactin was proposed to not only be involved in forming the actin scaffold for invadopodia but rather promote the secretion of secretory vesicles containing MMPs including MMP14 [
46,
48]. Together with the previously described mechanisms that HIP1R binding to cortactin inhibits actin assemblies at endocytic sites, and with the fact that CLC deletion disrupts the localization of HIP1R to endocytic sites [
7,
42], we propose the following model: Deletion of CLCA and CLCB causes upregulation of MMP14 recycling to the plasma membrane (
Figure 9). The consequence of CLC deletion is the disruption of HIP1R recruitment to endocytic sites, more specifically to cortactin. In turn, the unbound cortactin drives actin polymerization, which causes actin patch formation. Cortactin simultaneously promotes secretory vesicles containing MMP14, thereby causing accumulation of both actin and MMP14. The actin structures as well as MMP14 clustering form the scaffold for further invadopodia maturation.

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








