
Upregulation of COX4-2 via HIF-1α in Mitochondrial COX4-1 Deficiency

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Tissue Cultures
2.2. RNA Interference
2.3. Quantitative Reverse Transcription Polymerase Chain Reaction (RT-qPCR)
2.4. COX Enzymatic Activity
2.5. Oxygen Consumption Rate
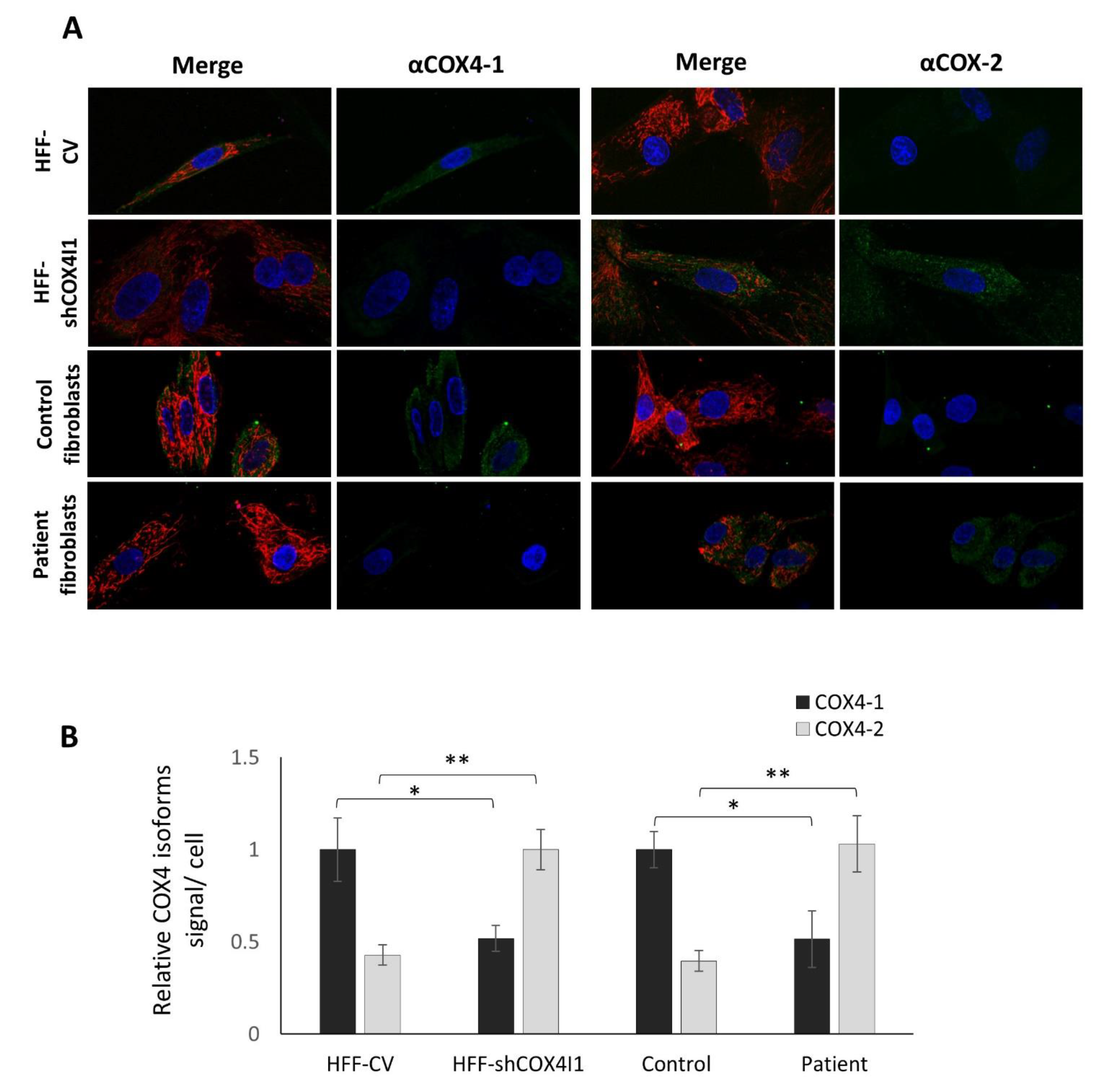
2.6. Immunofluorescence Staining
2.7. Western Blotting
2.8. Expression by Linear Amplification and Sequencing: CEL-Seq2
2.9. Statistical Analysis
3. Results
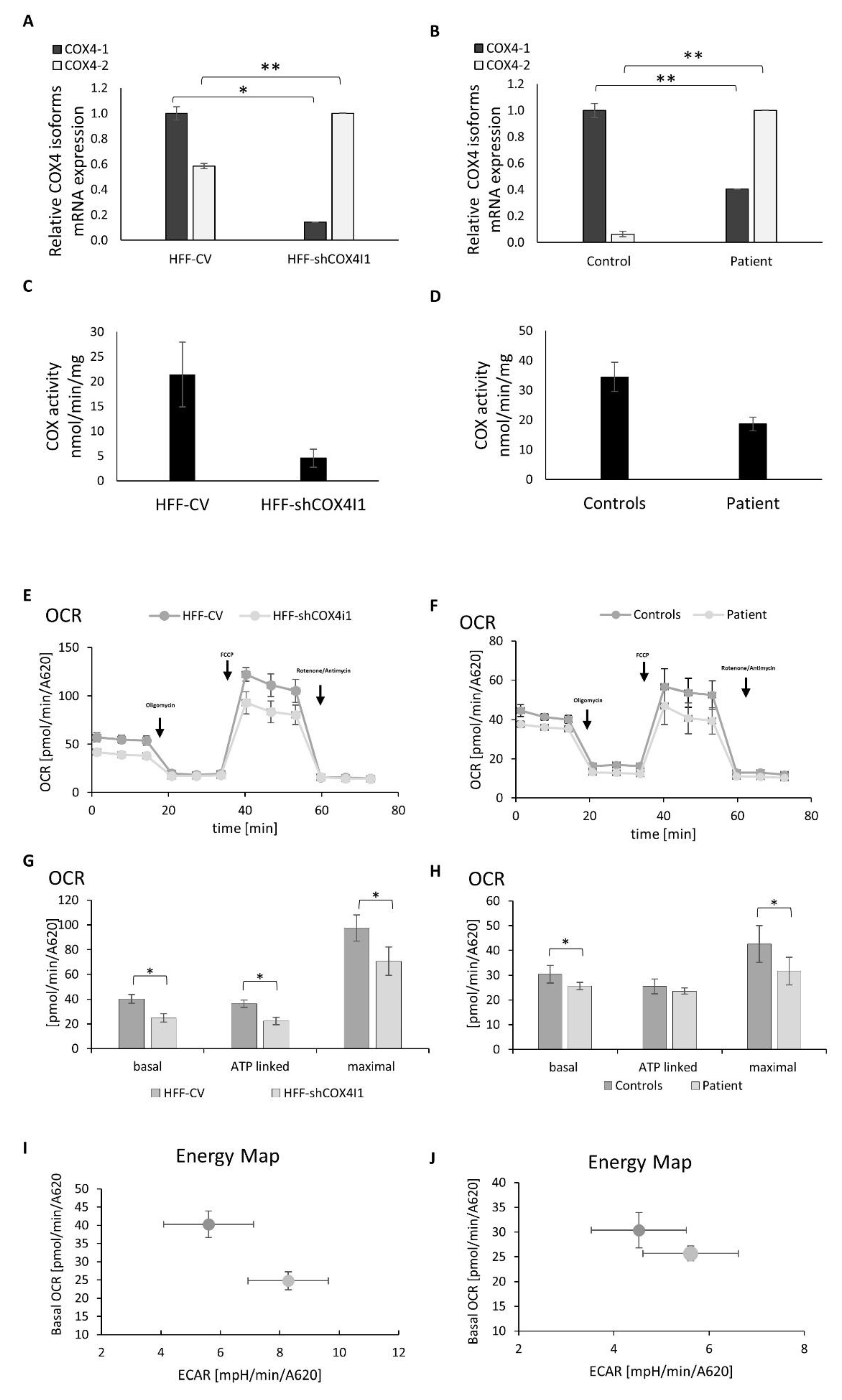
3.1. COX4 Isoform Switch and Altered Energetic Profile in COX4-1-Deficient Cells
3.2. Upregulation of Glycolytic and Hypoxia Pathways in COX4-1-Deficient Cells
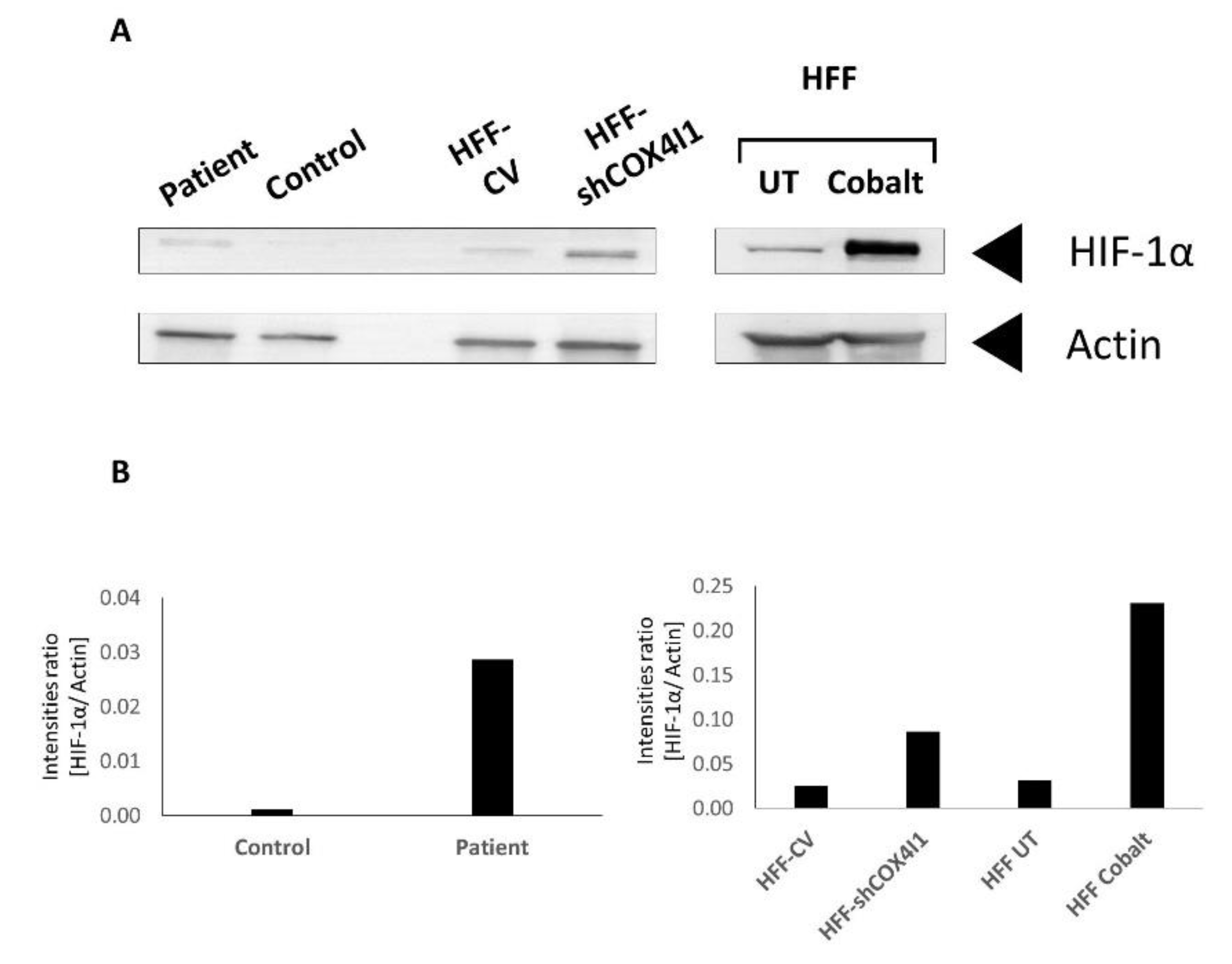
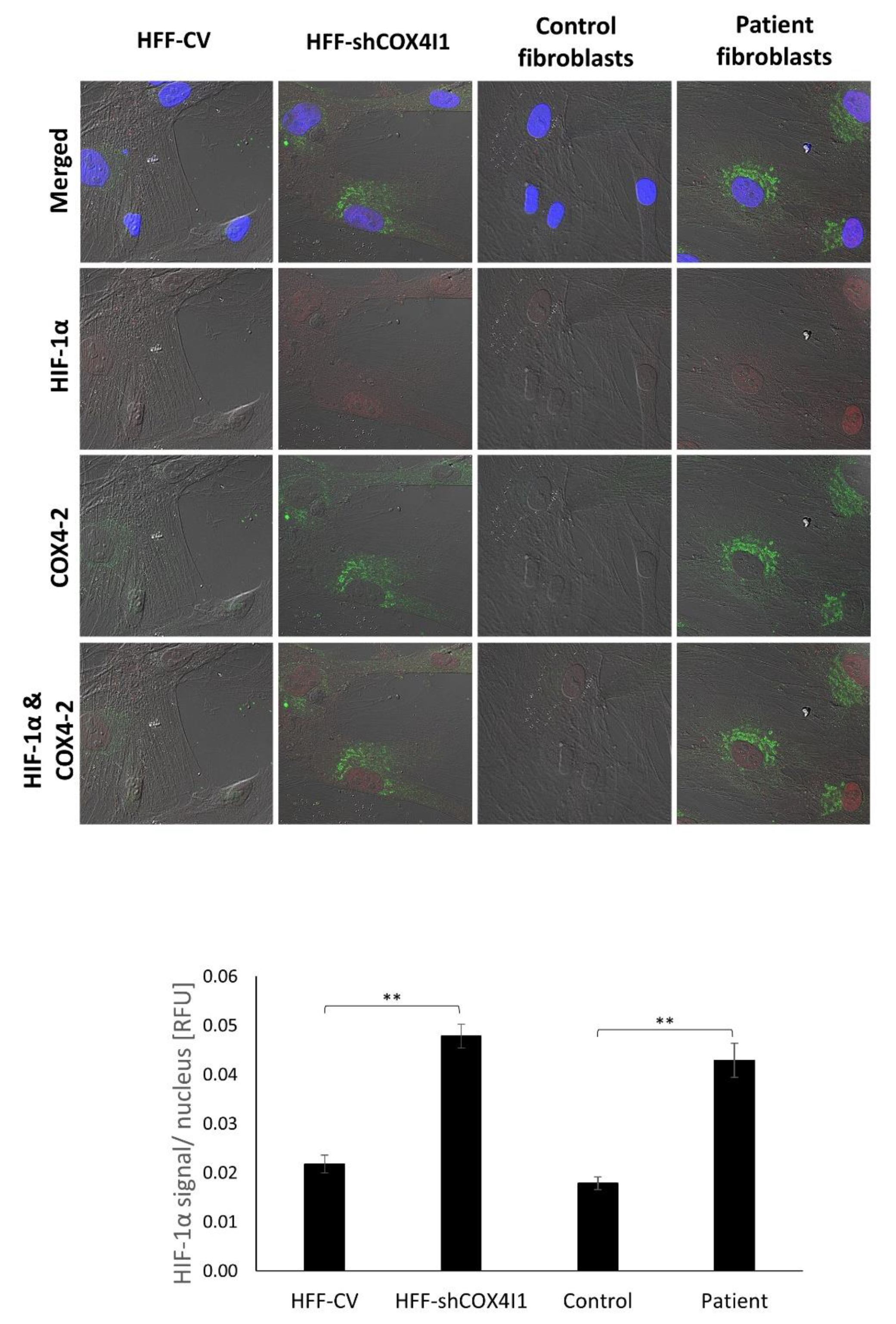
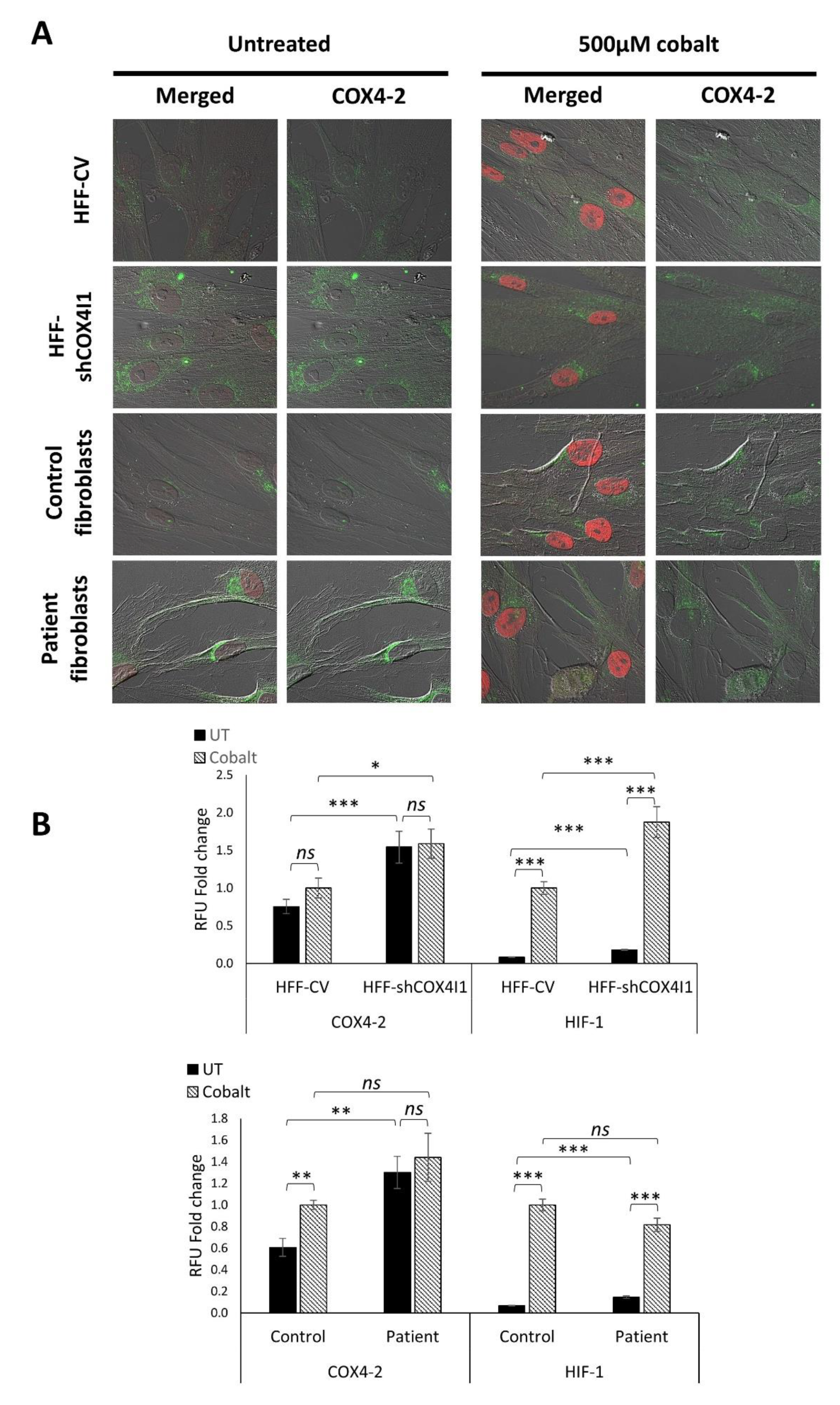
3.3. HIF-1α Is Elevated and Translocated to the Nucleus in COX4-1-Deficient Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arnold, S. The power of life--cytochrome c oxidase takes center stage in metabolic control, cell signalling and survival. Mitochondrion 2012, 12, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Sinkler, C.A.; Kalpage, H.; Shay, J.; Lee, I.; Malek, M.H.; Grossman, L.I.; Hüttemann, M. Tissue- and Condition-Specific Isoforms of Mammalian Cytochrome c Oxidase Subunits: From Function to Human Disease. Oxid. Med. Cell. Longev. 2017, 2017, 1534056. [Google Scholar] [CrossRef] [Green Version]
- Timón-Gómez, A.; Bartley-Dier, E.L.; Fontanesi, F.; Barrientos, A. HIGD-Driven Regulation of Cytochrome c Oxidase Biogenesis and Function. Cells 2020, 9, 2620. [Google Scholar] [CrossRef] [PubMed]
- Abu-Libdeh, B.; Douiev, L.; Amro, S.; Shahrour, M.; Ta-Shma, A.; Miller, C.; Elpeleg, O.; Saada, A. Mutation in the COX4I1 gene is associated with short stature, poor weight gain and increased chromosomal breaks, simulating Fanconi anemia. Eur. J. Hum. Genet. 2017, 25, 1142–1146. [Google Scholar] [CrossRef]
- Douiev, L.; Abu-Libdeh, B.; Saada, A. Cytochrome c oxidase deficiency, oxidative stress, possible antioxidant therapy and link to nuclear DNA damage. Eur. J. Hum. Genet. 2018, 26, 579–581. [Google Scholar] [CrossRef]
- Pillai, N.R.; AlDhaheri, N.S.; Ghosh, R.; Lim, J.; Streff, H.; Nayak, A.; Graham, B.H.; Hanchard, N.A.; Elsea, S.H.; Scaglia, F. Biallelic variants in COX4I1 associated with a novel phenotype resembling Leigh syndrome with developmental regression, intellectual disability, and seizures. Am. J. Med. Genet. A 2019, 179, 2138–2143. [Google Scholar] [CrossRef]
- Rak, M.; Bénit, P.; Chrétien, D.; Bouchereau, J.; Schiff, M.; El-Khoury, R.; Tzagoloff, A.; Rustin, P. Mitochondrial cytochrome c oxidase deficiency. Clin. Sci. 2016, 130, 393–407. [Google Scholar] [CrossRef] [Green Version]
- Shoubridge, E.A. Cytochrome c oxidase deficiency. Am. J. Med. Genet. 2001, 106, 46–52. [Google Scholar] [CrossRef]
- Hock, D.H.; Robinson, D.R.L.; Stroud, D.A. Blackout in the powerhouse: Clinical phenotypes associated with defects in the assembly of OXPHOS complexes and the mitoribosome. Biochem. J. 2020, 477, 4085–4132. [Google Scholar] [CrossRef]
- Shteyer, E.; Saada, A.; Shaag, A.; Al-Hijawi, F.A.; Kidess, R.; Revel-Vilk, S.; Elpeleg, O. Exocrine pancreatic insufficiency, dyserythropoeitic anemia, and calvarial hyperostosis are caused by a mutation in the COX4I2 gene. Am. J. Hum. Genet. 2009, 84, 412–417. [Google Scholar] [CrossRef] [Green Version]
- Golubitzky, A.; Dan, P.; Weissman, S.; Link, G.; Wikstrom, J.D.; Saada, A. Screening for active small molecules in mitochondrial complex I deficient patient’s fibroblasts, reveals AICAR as the most beneficial compound. PLoS ONE 2011, 6, e26883. [Google Scholar] [CrossRef] [Green Version]
- Hashimshony, T.; Senderovich, N.; Avital, G.; Klochendler, A.; de Leeuw, Y.; Anavy, L.; Gennert, D.; Li, S.; Livak, K.J.; Rozenblatt-Rosen, O.; et al. CEL-Seq2: Sensitive highly-multiplexed single-cell RNA-Seq. Genome Biol. 2016, 17, 77. [Google Scholar] [CrossRef] [Green Version]
- Marcel, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Arnold, S.; Kadenbach, B. Cell respiration is controlled by ATP, an allosteric inhibitor of cytochrome-c oxidase. Eur. J. Biochem. 1997, 249, 350–354. [Google Scholar] [CrossRef]
- Aras, S.; Pak, O.; Sommer, N.; Finley RJr Hüttemann, M.; Weissmann, N.; Grossman, L.I. Oxygen-dependent expression of cytochrome c oxidase subunit 4-2 gene expression is mediated by transcription factors RBPJ, CXXC5 and CHCHD2. Nucleic Acids Res. 2013, 41, 2255–2266. [Google Scholar] [CrossRef] [Green Version]
- Tam, S.Y.; Wu, V.W.C.; Law, H.K.W. Hypoxia-Induced Epithelial-Mesenchymal Transition in Cancers: HIF-1α and Beyond. Front. Oncol. 2020, 10, 486. [Google Scholar] [CrossRef]
- Fukuda, R.; Zhang, H.; Kim, J.W.; Shimoda, L.; Dang, C.V.; Semenza, G.L. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 2007, 129, 111–122. [Google Scholar] [CrossRef] [Green Version]
- Hervouet, E.; Pecina, P.; Demont, J.; Vojtísková, A.; Simonnet, H.; Houstek, J.; Godinot, C. Inhibition of cytochrome c oxidase subunit 4 precursor processing by the hypoxia mimic cobalt chloride. Biochem. Biophys. Res. Commun. 2006, 344, 1086–1093. [Google Scholar] [CrossRef]
- Douiev, L.; Saada, A. The pathomechanism of cytochrome c oxidase deficiency includes nuclear DNA damage. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 893–900. [Google Scholar] [CrossRef]
- Pajuelo Reguera, D.; Čunátová, K.; Vrbacký, M.; Pecinová, A.; Houštěk, J.; Mráček, T.; Pecina, P. Cytochrome c Oxidase Subunit 4 Isoform Exchange Results in Modulation of Oxygen Affinity. Cells 2020, 9, 443. [Google Scholar] [CrossRef] [Green Version]
- Acin-Perez, R.; Gatti, D.L.; Bai, Y.; Manfredi, G. Protein phosphorylation and prevention of cytochrome oxidase inhibition by ATP: Coupled mechanisms of energy metabolism regulation. Cell Metab. 2011, 13, 712–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mreisat, A.; Kanaani, H.; Saada, A.; Horowitz, M. Heat acclimation mediated cardioprotection is controlled by mitochondrial metabolic remodeling involving HIF-1α. J. Therm. Biol. 2020, 93, 102691. [Google Scholar] [CrossRef] [PubMed]
- Keller, G.; Binyamin, O.; Frid, K.; Saada, A.; Gabizon, R. Mitochondrial dysfunction in preclinical genetic prion disease: A target for preventive treatment? Neurobiol. Dis. 2019, 124, 57–66. [Google Scholar] [CrossRef]
- Čunátová, K.; Reguera, D.P.; Houštěk, J.; Mráček, T.; Pecina, P. Role of cytochrome c oxidase nuclear-encoded subunits in health and disease. Physiol. Res. 2020, 69, 947–965. [Google Scholar] [CrossRef]
- McElroy, G.S.; Chandel, N.S. Mitochondria control acute and chronic responses to hypoxia. Exp. Cell Res. 2017, 356, 217–222. [Google Scholar] [CrossRef]
- Iommarini, L.; Porcelli, A.M.; Gasparre, G.; Kurelac, I. Non-Canonical Mechanisms Regulating Hypoxia-Inducible Factor 1 Alpha in Cancer. Front. Oncol. 2017, 7, 286. [Google Scholar] [CrossRef] [Green Version]
- Koshiji, M.; To, K.K.; Hammer, S.; Kumamoto, K.; Harris, A.L.; Modrich, P.; Huang, L.E. HIF-1alpha induces genetic instability by transcriptionally downregulating MutSalpha expression. Mol. Cell 2005, 17, 793–803. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| COX4I1 (NM_001861.6) | 5′-TTTCACCGCGCTCGTTAT-3′ | 5′-CTTCATGTCCAGCATCCTCTT-3′ |
| COX4I2 (NM_032609) | 5′-GAAGACGAGGGATGCACAG-3′ | 5′-GGCTCTTCTGGCATGGG-3′ |
| PDK1 (NM_001278549.2) | 5′- CAGGACACCATCCGTTCAAT-3′ | 5′- AGCTTTAGCATCCTCAGCAC-3′ |
| GLUT1 (NM_006516.4) | 5′-GCTACAACACTGGAGTCATCAA-3′ | 5′-ACTGAGAGGGACCAGAGC-3′ |
| HK1 (NM_000188.3) | 5′- CCCTAAATGCTGGGAAACAAAG-3′ | 5′- CCCTTCTTGGTGAAGTCGATTA-3′ |
| HK2 (NM_000189.5) | 5′- CATCCTCCTCAAGTGGACAAA-3′ | 5′- ACCACATCCAGGTCAAACTC-3′ |
| GUSB (NM_000181.4) | 5′-GAAAATATGTGGTTGGAGAGCTCATT-3′ | 5′-CCGAGTGAAGATCCCCTTTTTA-3′ |
| GAPDH (NM_001357943.2) | 5′-CAAGAGCACAAGAGGAAGAGAG-3′ | 5′-CTACATGGCAACTGTGAGGAG-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Douiev, L.; Miller, C.; Ruppo, S.; Benyamini, H.; Abu-Libdeh, B.; Saada, A. Upregulation of COX4-2 via HIF-1α in Mitochondrial COX4-1 Deficiency. Cells 2021, 10, 452. https://doi.org/10.3390/cells10020452
Douiev L, Miller C, Ruppo S, Benyamini H, Abu-Libdeh B, Saada A. Upregulation of COX4-2 via HIF-1α in Mitochondrial COX4-1 Deficiency. Cells. 2021; 10(2):452. https://doi.org/10.3390/cells10020452
Chicago/Turabian StyleDouiev, Liza, Chaya Miller, Shmuel Ruppo, Hadar Benyamini, Bassam Abu-Libdeh, and Ann Saada. 2021. "Upregulation of COX4-2 via HIF-1α in Mitochondrial COX4-1 Deficiency" Cells 10, no. 2: 452. https://doi.org/10.3390/cells10020452
APA StyleDouiev, L., Miller, C., Ruppo, S., Benyamini, H., Abu-Libdeh, B., & Saada, A. (2021). Upregulation of COX4-2 via HIF-1α in Mitochondrial COX4-1 Deficiency. Cells, 10(2), 452. https://doi.org/10.3390/cells10020452