
HIV-1 Latency and Viral Reservoirs: Existing Reversal Approaches and Potential Technologies, Targets, and Pathways Involved in HIV Latency Studies

 , ,
, , {kind=link}
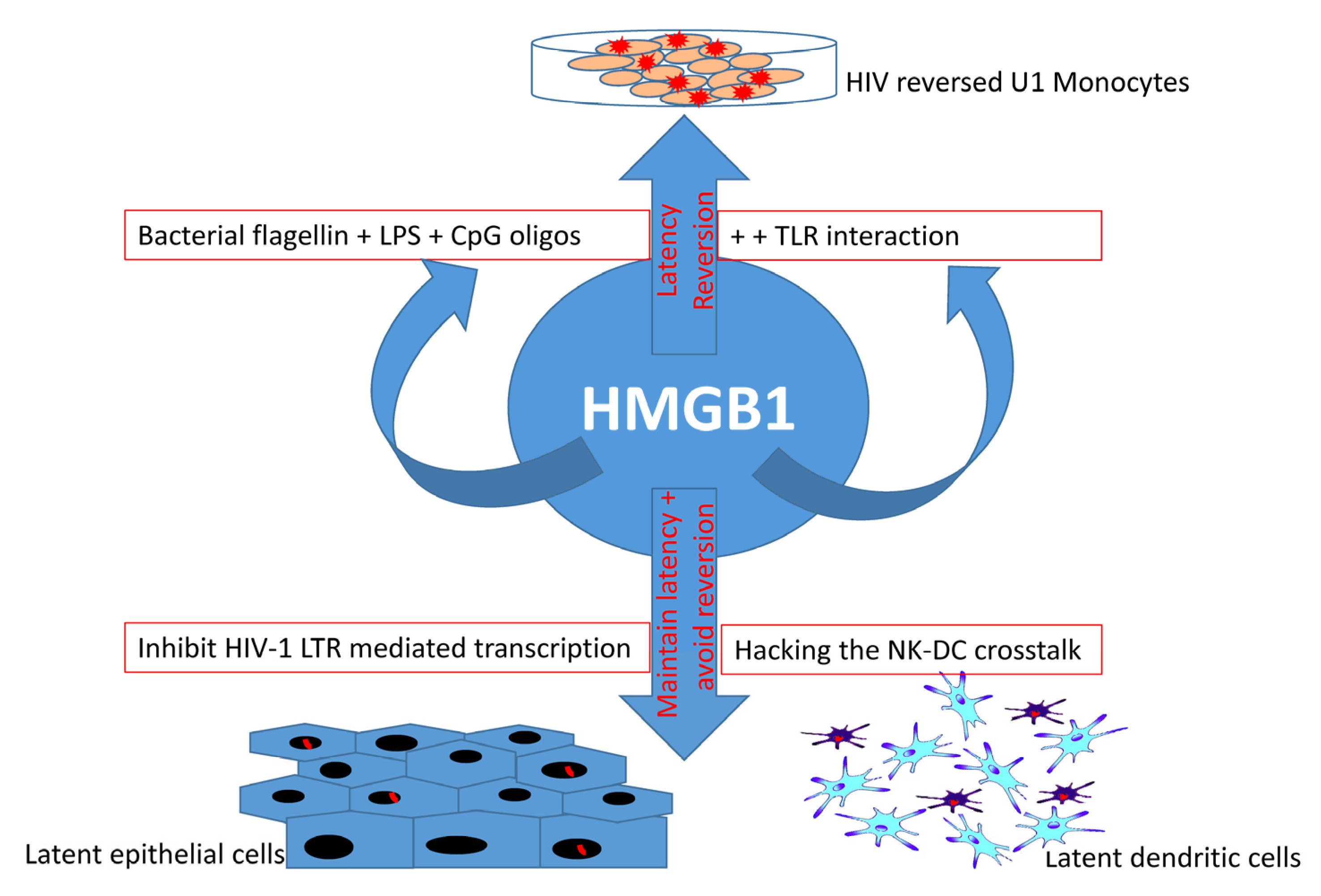{kind=link}
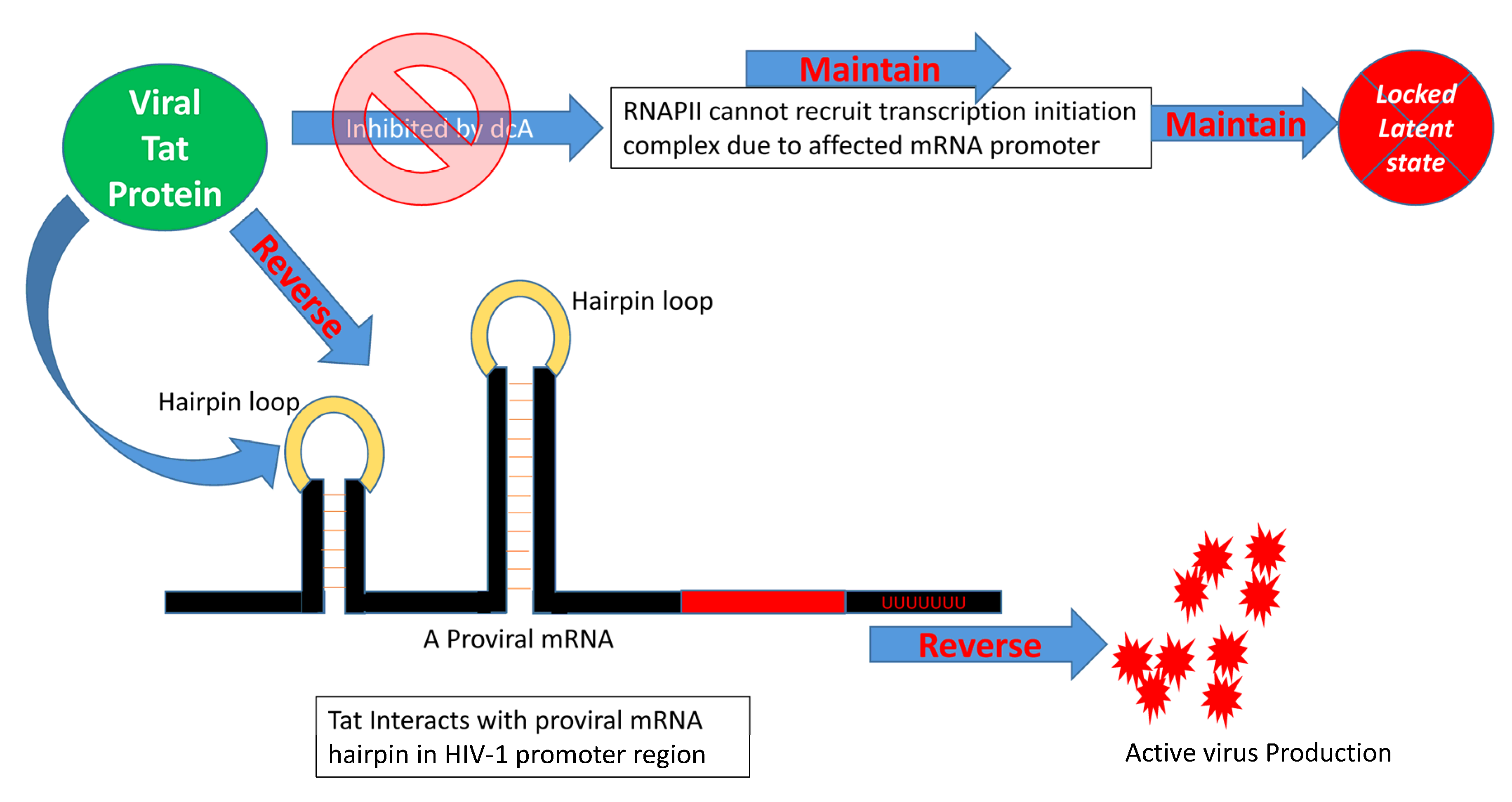{kind=link}
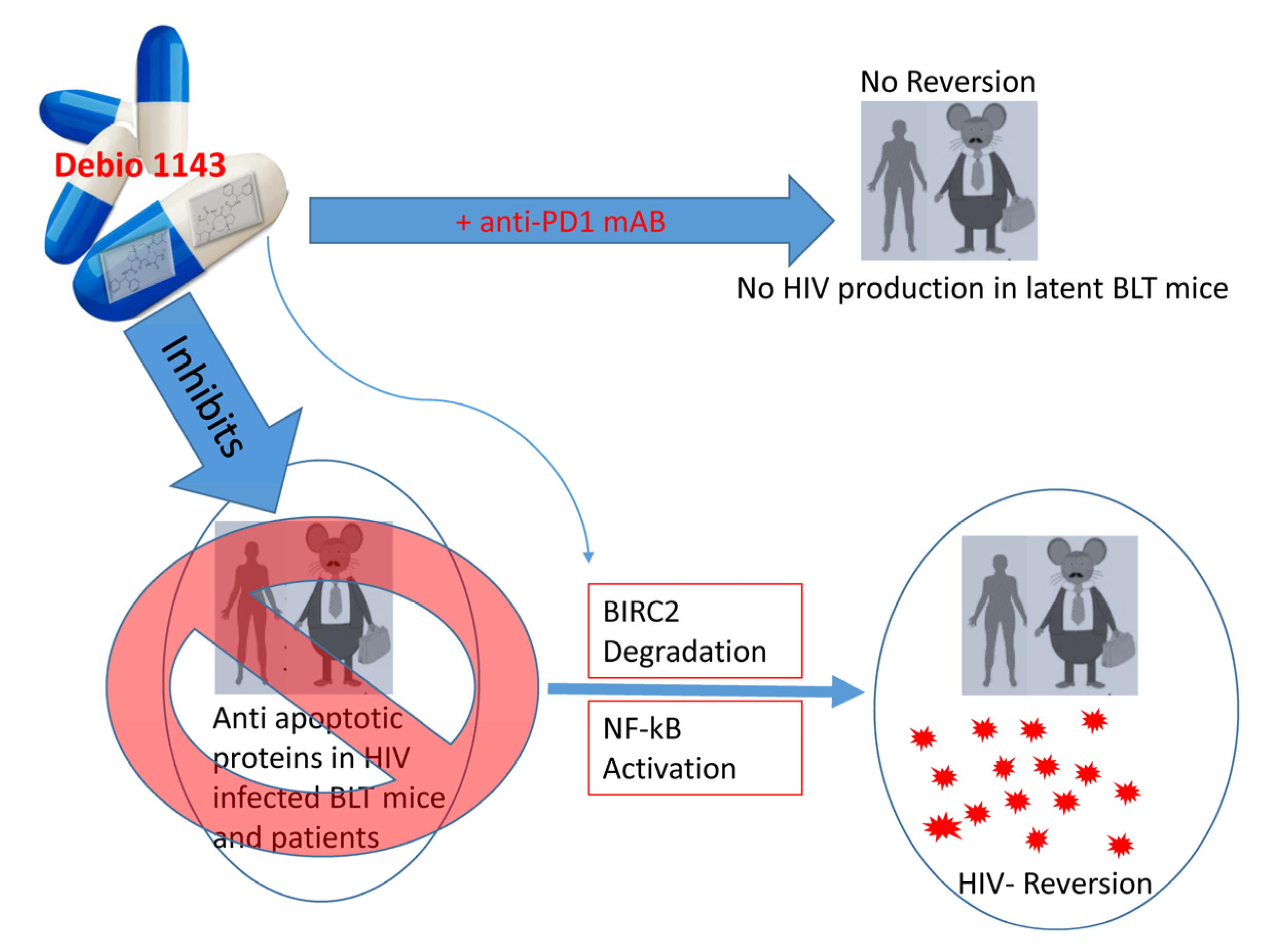{kind=link}
Abstract
:1. Introduction
1.1. HIV Reservoirs, Latency Maintenance, and Clinical Complications
1.2. HIV Latency and Reversal Approaches
2. HIV Latency and Potential Agents for Reversal
2.1. TOX in HIV Latency
2.2. Protein Kinases in HIV Latency
2.3. JAK/STAT Pathway in HIV Latency
2.4. Apoptotic Proteins in HIV Latency
2.5. Transcriptional and Genetic Factors in HIV Latency
3. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ART | Anti-retroviral therapy |
| HIV | Human immunodeficiency virus |
| HSCs | Hematopoietic stem cells |
| LRA | Latency-reversing agents |
| HMG | High-mobility group |
| TCR | T cell receptor |
| LncRNA | Long noncoding RNA |
| BLT | Bone marrow, liver, and thymus |
| MDCs | Monocyte-derived dendritic cells |
| TLRs | Toll-like receptors |
| NLS | Nuclear localization signal |
| NHEJ | Nonhomologous end joining |
| PIR | Pro-Trp-Trp-Pro motif interacting region |
| TOX | Thymocyte selection-associated high-mobility group box |
| HMGB1 | High-mobility group box 1 |
| LPS | lipopolysaccharides |
| mTOR | Mammalian target of Rapamycin |
| JAK | Janus kinase |
| STAT | Signal transducer and activator of transcription |
| PD-1 | Programmed cell death-1 |
| QUECEL | Quiescent effector cell latency |
| LTR | Long terminal repeats |
| RNAPII | RNA polymerase II |
| SNP | Single nucleotide polymorphisms |
| TREM1 | Triggering receptor expressed on myeloid cells 1 |
| mAb | Monoclonal antibody |
| dCA | didehydro-Cortistatin A |
| NK | Natural killer |
References
- Siliciano, R.F.; Greene, W.C. HIV Latency. Cold Spring Harb. Perspect. Med. 2011, 1, a007096. [Google Scholar] [CrossRef] [Green Version]
- Vanhamel, J.; Bruggemans, A.; Debyser, Z. Establishment of latent HIV-1 reservoirs: What do we really know? J. Virus Erad. 2019, 5, 3–9. [Google Scholar] [CrossRef]
- Ananworanich, J.; Dubé, K.; Chomont, N. How does the timing of antiretroviral therapy initiation in acute infection affect HIV reservoirs? Curr. Opin. HIV AIDS 2015, 10, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Leite, T.F.; Delatorre, E.; Côrtes, F.H.; Ferreira, A.C.G.; Cardoso, S.W.; Grinsztejn, B.; De Andrade, M.M.; Veloso, V.G.; Morgado, M.G.; Guimarães, M.L. Reduction of HIV-1 Reservoir Size and Diversity After 1 Year of cART Among Brazilian Individuals Starting Treatment During Early Stages of Acute Infection. Front. Microbiol. 2019, 10, 145. [Google Scholar] [CrossRef]
- Luo, L.; Wang, N.; Yue, Y.; Han, Y.; Lv, W.; Liu, Z.; Qiu, Z.; Lu, H.; Tang, X.; Zhang, T.; et al. The effects of antiretroviral therapy initiation time on HIV reservoir size in Chinese chronically HIV infected patients: A prospective, multi-site cohort study. BMC Infect. Dis. 2019, 19, 257. [Google Scholar] [CrossRef]
- Brodin, J.; Zanini, F.; Thebo, L.; Lanz, C.; Bratt, G.; Neher, R.A.; Albert, J. Establishment and stability of the latent HIV-1 DNA reservoir. eLife 2016, 5, e18889. [Google Scholar] [CrossRef] [PubMed]
- Pankau, M.D.; Reeves, D.B.; Harkins, E.; Ronen, K.; Jaoko, W.; Mandaliya, K.; Graham, S.M.; McClelland, R.S.; Iv, F.A.M.; Schiffer, J.T.; et al. Dynamics of HIV DNA reservoir seeding in a cohort of superinfected Kenyan women. PLoS Pathog. 2020, 16, e1008286. [Google Scholar] [CrossRef]
- Abrahams, M.-R.; Joseph, S.B.; Garrett, N.; Tyers, L.; Moeser, M.; Archin, N.; Council, O.D.; Matten, D.; Zhou, S.; Doolabh, D.; et al. The replication-competent HIV-1 latent reservoir is primarily established near the time of therapy initiation. Sci. Transl. Med. 2019, 11, eaaw5589. [Google Scholar] [CrossRef]
- Kuo, H.-H.; Lichterfeld, M. Recent progress in understanding HIV reservoirs. Curr. Opin. HIV AIDS 2018, 13, 137–142. [Google Scholar] [CrossRef]
- Sadowski, I.; Hashemi, F.B. Strategies to eradicate HIV from infected patients: Elimination of latent provirus reservoirs. Cell. Mol. Life Sci. 2019, 76, 3583–3600. [Google Scholar] [CrossRef] [Green Version]
- Pantaleo, G.; Graziosi, C.; Demarest, J.F.; Butini, L.; Montroni, M.; Fox, C.H.; Orenstein, J.M.; Kotler, D.P.; Fauci, A.S. HIV infection is active and progressive in lymphoid tissue during the clinically latent stage of disease. Nat. Cell Biol. 1993, 362, 355–358. [Google Scholar] [CrossRef]
- Pantaleo, G.; Graziosi, C.; Butini, L.; Pizzo, P.A.; Schnittman, S.M.; Kotler, D.P.; Fauci, A.S. Lymphoid organs function as major reservoirs for human immunodeficiency virus. Proc. Natl. Acad. Sci. USA 1991, 88, 9838–9842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantaleo, G.; Graziosi, C.; Fauci, A.S. The role of lymphoid organs in the pathogenesis of HIV infection. Semin. Immunol. 1993, 5, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Josefsson, L.; Palmer, S.; Faria, N.R.; Lemey, P.; Casazza, J.; Ambrozak, D.; Kearney, M.; Shao, W.; Kottilil, S.; Sneller, M.; et al. Single Cell Analysis of Lymph Node Tissue from HIV-1 Infected Patients Reveals that the Majority of CD4+ T-cells Contain One HIV-1 DNA Molecule. PLoS Pathog. 2013, 9, e1003432. [Google Scholar] [CrossRef] [Green Version]
- Pantaleo, G.; Graziosi, C.; Demarest, J.F.; Cohen, O.J.; Vaccarezza, M.; Gantt, K.; Muro-Cacho, C.; Fauci, A.S. Role of Lymphoid Organs in the Pathogenesis of Human Immunodeficiency Virus (HIV) Infection. Immunol. Rev. 1994, 140, 105–130. [Google Scholar] [CrossRef]
- Wong, J.K.; Yukl, S.A. Tissue reservoirs of HIV. Curr. Opin. HIV AIDS 2016, 11, 362–370. [Google Scholar] [CrossRef]
- Chinnapaiyan, S.; Parira, T.; Dutta, R.; Agudelo, M.; Morris, A.; Nair, M.; Unwalla, H.J. HIV Infects Bronchial Epithelium and Suppresses Components of the Mucociliary Clearance Apparatus. PLoS ONE 2017, 12, e0169161. [Google Scholar] [CrossRef] [Green Version]
- Winston, J.A.; Bruggeman, L.A.; Ross, M.D.; Jacobson, J.; Ross, L.; D’Agati, V.D.; Klotman, P.E.; Klotman, M.E. Nephropathy and Establishment of a Renal Reservoir of HIV Type 1 during Primary Infection. N. Engl. J. Med. 2001, 344, 1979–1984. [Google Scholar] [CrossRef] [PubMed]
- Lutgen, V.; Narasipura, S.D.; Barbian, H.J.; Richards, M.; Wallace, J.; Razmpour, R.; Buzhdygan, T.; Ramirez, S.H.; Prevedel, L.; Eugenin, E.A.; et al. HIV infects astrocytes in vivo and egresses from the brain to the periphery. PLoS Pathog. 2020, 16, e1008381. [Google Scholar] [CrossRef]
- Darcis, G.; Berkhout, B.; Pasternak, A.O. The Quest for Cellular Markers of HIV Reservoirs: Any Color You Like. Front. Immunol. 2019, 10, 2251. [Google Scholar] [CrossRef]
- Descours, B.; Petitjean, G.; López-Zaragoza, J.-L.; Bruel, T.; Raffel, R.; Psomas, C.; Reynes, C.P.J.; Lacabaratz, C.; Levy, Y.; Schwartz, T.B.O.; et al. CD32a is a marker of a CD4 T-cell HIV reservoir harbouring replication-competent proviruses. Nat. Cell Biol. 2017, 543, 564–567. [Google Scholar] [CrossRef]
- Abdel-Mohsen, M.; Kuri-Cervantes, L.; Grau-Exposito, J.; Spivak, A.M.; Nell, R.A.; Tomescu, C.; Vadrevu, S.K.; Giron, L.B.; Serra-Peinado, C.; Genescà, M.; et al. CD32 is expressed on cells with transcriptionally active HIV but does not enrich for HIV DNA in resting T cells. Sci. Transl. Med. 2018, 10, eaar6759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galvani, A.P.; Pandey, A.; Fitzpatrick, M.C.; Medlock, J.; Gray, E.G. Defining control of HIV epidemics. Lancet HIV 2018, 5, e667–e670. [Google Scholar] [CrossRef]
- Tyagi, M.; Bukrinsky, M. Human Immunodeficiency Virus (HIV) Latency: The Major Hurdle in HIV Eradication. Mol. Med. 2012, 18, 1096–1108. [Google Scholar] [CrossRef] [PubMed]
- Dash, P.K.; Kaminski, R.; Bella, R.; Su, H.; Mathews, S.; Ahooyi, T.M.; Chen, C.; Mancuso, P.; Sariyer, R.; Ferrante, P.; et al. Sequential LASER ART and CRISPR Treatments Eliminate HIV-1 in a Subset of Infected Humanized Mice. Nat. Commun. 2019, 10, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Chun, T.-W.; Justement, J.S.; Murray, D.; Hallahan, C.W.; Maenza, J.; Collier, A.C.; Sheth, P.M.; Kaul, R.; Ostrowski, M.; Moir, S.; et al. Rebound of plasma viremia following cessation of antiretroviral therapy despite profoundly low levels of HIV reservoir: Implications for eradication. AIDS 2010, 24, 2803–2808. [Google Scholar] [CrossRef] [Green Version]
- Deeks, S.G.; International AIDS Society Towards a Cure Working Group International AIDS Society Towards a Cure Working Group; Lewin, S.R.; Ross, A.L.; Ananworanich, J.; Benkirane, M.; Cannon, P.; Chomont, N.; Douek, D.; Lifson, J.D.; et al. International AIDS Society global scientific strategy: Towards an HIV cure. Nat. Med. 2016, 22, 839–850. [Google Scholar] [CrossRef]
- Zhao, J.; Nguyen, L.N.T.; Dang, X.; Cao, D.; Khanal, S.; Schank, M.; Thakuri, B.K.C.; Ogbu, S.C.; Morrison, Z.D.; Wu, X.Y.; et al. ATM Deficiency Accelerates DNA Damage, Telomere Erosion, and Premature T Cell Aging in HIV-Infected Individuals on Antiretroviral Therapy. Front. Immunol. 2019, 10, 2531. [Google Scholar] [CrossRef] [Green Version]
- Khanal, S.; Tang, Q.; Cao, D.; Zhao, J.; Nguyen, L.N.; Oyedeji, O.S.; Dang, X.; Schank, M.; Thakuri, B.K.C.; Ogbu, C.; et al. Telomere and ATM Dynamics in CD4 T-Cell Depletion in Active and Virus-Suppressed HIV Infections. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Bestilny, L.J.; Gill, M.J.; Mody, C.H.; Riabowol, K.T. Accelerated replicative senescence of the peripheral immune system induced by HIV infection. AIDS 2000, 14, 771–780. [Google Scholar] [CrossRef]
- Gonzalez-Serna, A.; Ajaykumar, A.; Gadawski, I.; Muñoz-Fernández, M.A.; Hayashi, K.; Harrigan, P.R.; Côté, H.C.F. Rapid Decrease in Peripheral Blood Mononucleated Cell Telomere Length After HIV Seroconversion, but Not HCV Seroconversion. JAIDS J. Acquir. Immune Defic. Syndr. 2017, 76, e29–e32. [Google Scholar] [CrossRef]
- Malaspina, A.; Moir, S.; Orsega, S.M.; Vasquez, J.; Miller, N.J.; Donoghue, E.T.; Kottilil, S.; Gezmu, M.; Follmann, D.; Vodeiko, G.M.; et al. Compromised B Cell Responses to Influenza Vaccination in HIV-Infected Individuals. J. Infect. Dis. 2005, 191, 1442–1450. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Barradas, M.C.; Alexandraki, I.; Nazir, T.; Foltzer, M.; Musher, D.M.; Brown, S.; Thornby, J. Response of Human Immunodeficiency Virus-Infected Patients Receiving Highly Active Antiretroviral Therapy to Vaccination with 23-Valent Pneumococcal Polysaccharide Vaccine. Clin. Infect. Dis. 2003, 37, 438–447. [Google Scholar] [CrossRef] [Green Version]
- Patterson, S.B.; Landrum, M.L.; Okulicz, J.F. Delayed-type hypersensitivity and hepatitis B vaccine responses, in vivo markers of cellular and humoral immune function, and the risk of AIDS or death. Vaccine 2014, 32, 3341–3344. [Google Scholar] [CrossRef] [Green Version]
- Balado, M.D.M.D.P.; Leal, M.; Lagares, G.M.; Mata, R.C.; López-Cortés, L.F.; Viciana, P.; Pacheco, Y.M. Increased Regulatory T Cell Counts in HIV-Infected Nonresponders to Hepatitis B Virus Vaccine. J. Infect. Dis. 2010, 202, 362–369. [Google Scholar] [CrossRef] [Green Version]
- Piekna-Przybylska, D.; Sharma, G.; Maggirwar, S.B.; Bambara, R.A. Deficiency in DNA damage response, a new characteristic of cells infected with latent HIV. Cell Cycle 2017, 16, 968–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Watt, G. Mitochondrial dysfunction and human immunodeficiency virus infection. J. Endocrinol. Metab. Diabetes S. Afr. 2011, 16, 94–100. [Google Scholar] [CrossRef] [Green Version]
- Piekna-Przybylska, D.; Maggirwar, S.B. CD4+ memory T cells infected with latent HIV-1 are susceptible to drugs targeting telomeres. Cell Cycle 2018, 17, 2187–2203. [Google Scholar] [CrossRef] [Green Version]
- Ji, Y.; Dang, X.; Nguyen, L.N.T.; Nguyen, L.N.; Zhao, J.; Cao, D.; Khanal, S.; Schank, M.; Wu, X.Y.; Morrison, Z.D.; et al. Topological DNA damage, telomere attrition and T cell senescence during chronic viral infections. Immun. Ageing 2019, 16, 1–15. [Google Scholar] [CrossRef]
- Cao, D.; Zhao, J.; Nguyan, L.N.; Nguyen, L.N.T.; Khanal, S.; Dang, X.; Schank, M.; Thakuri, B.K.C.; Wu, X.Y.; Morrison, Z.D.; et al. Disruption of Telomere Integrity and DNA Repair Machineries by KML001 Induces T Cell Senescence, Apoptosis, and Cellular Dysfunctions. Front. Immunol. 2019, 10, 1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco, J.-R.; Jarrin, I.; Martinez, A.; Siles, E.; Larrayoz, I.M.; Cañuelo, A.; Gutierrez, F.; Gonzalez-Garcia, J.; Vidal, F.; Moreno, S. Shorter Telomere Length Predicts Poorer Immunological Recovery in Virologically Suppressed HIV-1–Infected Patients Treated With Combined Antiretroviral Therapy. JAIDS J. Acquir. Immune Defic. Syndr. 2015, 68, 21–29. [Google Scholar] [CrossRef]
- Leung, J.M.; Fishbane, N.; Jones, M.; Morin, A.; Xu, S.; Liu, J.C.; MacIsaac, J.; Milloy, M.-J.; Hayashi, K.; Montaner, J.; et al. Longitudinal study of surrogate aging measures during human immunodeficiency virus seroconversion. Aging 2017, 9, 687–705. [Google Scholar] [CrossRef] [Green Version]
- Pathai, S.; Lawn, S.D.; Gilbert, C.E.; McGuinness, D.; McGlynn, L.; Weiss, H.A.; Port, J.; Christ, T.; Barclay, K.; Wood, R.; et al. Accelerated biological ageing in HIV-infected individuals in South Africa. AIDS 2013, 27, 2375–2384. [Google Scholar] [CrossRef] [Green Version]
- Kirkwood, T.B. Understanding the Odd Science of Aging. Cell 2005, 120, 437–447. [Google Scholar] [CrossRef] [Green Version]
- Tedone, E.; Huang, E.; O’Hara, R.; Batten, K.; Ludlow, A.T.; Lai, T.-P.; Arosio, B.; Mari, D.; Wright, W.E.; Shay, J.W. Telomere length and telomerase activity in T cells are biomarkers of high-performing centenarians. Aging Cell 2018, 18, e12859. [Google Scholar] [CrossRef] [PubMed]
- Boyle, S.M.; Fehr, K.; Deering, C.; Raza, A.; Harhay, M.N.; Malat, G.; Ranganna, K.; Lee, D.H. Barriers to kidney transplant evaluation in HIV-positive patients with advanced kidney disease: A single-center study. Transpl. Infect. Dis. 2020, e13253. [Google Scholar] [CrossRef]
- Ekat, M.H.; Diafouka, M. Sp269antiretroviral Therapy-Related Nephrotoxicity in Hiv Infected Patients With Low Body Mass Index Outpatient Follow-Up In Brazzaville, Congo. Nephrol. Dial. Transplant. 2015, 30, iii468. [Google Scholar] [CrossRef] [Green Version]
- Ekat, M.H.; Ndour, C.T.; Ibara, R.B.O.; Diafouka, M.; Boumandoki, P.; Doukaga, T.A.; Aloumba, G.A.; Mahambou-Nsonde, D.; Nzounza, P.R.; Obengui, P.; et al. Faible indice de masse corporelle et impact des antirétroviraux sur la néphrotoxicité, la maladie rénale chronique chez les patients infectés par le VIH à Brazzaville, Congo. Néphrologie Thérapeutique 2020, 16, 97–104. [Google Scholar] [CrossRef]
- McHugh, G.; Rehman, A.M.; Simms, V.; Gonzalez-Martinez, C.; Bandason, T.; Dauya, E.; Moyo, B.; Mujuru, H.; Rylance, J.; Sovershaeva, E.; et al. Chronic lung disease in children and adolescents with HIV: A case—Control study. Trop. Med. Int. Heal. 2020, 25, 590–599. [Google Scholar] [CrossRef]
- Toribio, M.; Neilan, T.G.; Zanni, M.V. Heart Failure among People with HIV: Evolving Risks, Mechanisms, and Preventive Considerations. Curr. HIV/AIDS Rep. 2019, 16, 371–380. [Google Scholar] [CrossRef]
- Barceló, C.; Guidi, M.; Thorball, C.W.; Hammer, C.; Chaouch, A.; Scherrer, A.U.; Hasse, B.; Cavassini, M.; Furrer, H.; Calmy, A.; et al. Impact of Genetic and Nongenetic Factors on Body Mass Index and Waist-Hip Ratio Change in HIV-Infected Individuals Initiating Antiretroviral Therapy. Open Forum Infect. Dis. 2020, 7, ofz464. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Afaaf, L.; Shiau, S.; Strehlau, R.; Pierson, S.; Patel, F.; Wang, L.; Burke, M.; Violari, A.; Coovadia, A.; et al. Mitochondrial Impairment in Well-Suppressed Children with Perinatal HIV-Infection on Antiretroviral Therapy. AIDS Res. Hum. Retroviruses 2020, 36, 27–38. [Google Scholar] [CrossRef]
- Erlandson, K.M.; Bradford, Y.; Samuels, D.C.; Brown, T.T.; Sun, J.; Wu, K.; Tassiopoulos, K.; Ritchie, M.D.; Haas, D.W.; Hulgan, T.; et al. Mitochondrial DNA Haplogroups and Frailty in Adults Living with HIV. AIDS Res. Hum. Retrovir. 2020, 36, 214–219. [Google Scholar] [CrossRef] [Green Version]
- Chwiki, S.; Campos, M.M.; McLaughlin, M.E.; Kleiner, D.E.; Kovacs, J.A.; Morse, C.G.; Abu-Asab, M.S. Adverse effects of antiretroviral therapy on liver hepatocytes and endothelium in HIV patients: An ultrastructural perspective. Ultrastruct. Pathol. 2017, 41, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Hao, Y.; Zhao, H.; Xiao, J.; Han, N.; Zhang, Y.; Dai, G.; Chong, X.; Zeng, H.; Zhang, F. Distinct Mitochondrial Disturbance in CD4+T and CD8+T Cells From HIV-Infected Patients. JAIDS J. Acquir. Immune Defic. Syndr. 2017, 74, 206–212. [Google Scholar] [CrossRef]
- Honnapurmath, V.K.; Patil, V.W. Antiretroviral therapy-induced insulin resistance and oxidative deoxy nucleic acid damage in human immunodeficiency virus-1 patients. Indian J. Endocrinol. Metab. 2017, 21, 316–321. [Google Scholar] [CrossRef]
- Wang, G.P.; Ciuffi, A.; Leipzig, J.; Berry, C.C.; Bushman, F.D. HIV integration site selection: Analysis by massively parallel pyrosequencing reveals association with epigenetic modifications. Genome Res. 2007, 17, 1186–1194. [Google Scholar] [CrossRef] [Green Version]
- Cohn, L.B.; Silva, I.T.; Oliveira, T.Y.; Rosales, R.A.; Parrish, E.H.; Learn, G.H.; Hahn, B.H.; Czartoski, J.L.; McElrath, M.J.; Lehmann, C.; et al. HIV-1 Integration Landscape during Latent and Active Infection. Cell 2015, 160, 420–432. [Google Scholar] [CrossRef] [Green Version]
- Symons, J.; Chopra, A.; Malantinkova, E.; De Spiegelaere, W.; Leary, S.; Cooper, D.; Abana, C.O.; Rhodes, A.; Rezaei, S.D.; Vandekerckhove, L.; et al. HIV integration sites in latently infected cell lines: Evidence of ongoing replication. Retrovirology 2017, 14, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Brady, T.; Agosto, L.M.; Malani, N.; Berry, C.C.; O’Doherty, U.; Bushman, F. HIV integration site distributions in resting and activated CD4 + T cells infected in culture. AIDS 2009, 23, 1461–1471. [Google Scholar] [CrossRef] [Green Version]
- Saleh, S.; Lu, H.K.; Evans, V.; Harisson, D.; Zhou, J.; Jaworowski, A.; Sallmann, G.; Cheong, K.Y.; Mota, T.M.; Tennakoon, S.; et al. HIV integration and the establishment of latency in CCL19-treated resting CD4+ T cells require activation of NF-κB. Retrovirology 2016, 13, 49. [Google Scholar] [CrossRef] [Green Version]
- Einkauf, K.B.; Lee, G.Q.; Gao, C.; Sharaf, R.; Sun, X.; Hua, S.; Chen, S.M.; Jiang, C.; Lian, X.; Chowdhury, F.Z.; et al. Intact HIV-1 proviruses accumulate at distinct chromosomal positions during prolonged antiretroviral therapy. J. Clin. Investig. 2019, 129, 988–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Anderson, J.L.; Lewin, S.R. Getting the “Kill” into “Shock and Kill”: Strategies to Eliminate Latent HIV. Cell Host Microbe 2018, 23, 14–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cameron, P.U.; Saleh, S.; Sallmann, G.; Solomon, A.; Wightman, F.; Evans, V.A.; Boucher, G.; Haddad, E.K.; Sekaly, R.-P.; Harman, A.N.; et al. Establishment of HIV-1 latency in resting CD4+ T cells depends on chemokine-induced changes in the actin cytoskeleton. Proc. Natl. Acad. Sci. USA 2010, 107, 16934–16939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, L.J.; Bonczkowski, P.; Spivak, A.M.; De Spiegelaere, W.; Novis, C.L.; DePaula-Silva, A.B.; Malatinkova, E.; Trypsteen, W.; Bosque, A.; Vanderkerckhove, L.; et al. Modeling HIV-1 Latency in Primary T Cells Using a Replication-Competent Virus. AIDS Res. Hum. Retrovir. 2016, 32, 187–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosque, A.; Planelles, V. Induction of HIV-1 latency and reactivation in primary memory CD4+ T cells. Blood 2009, 113, 58–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kula, A.; Delacourt, N.; Bouchat, S.; Darcis, G.; Avettand-Fenoel, V.; Verdikt, R.; Corazza, F.; Necsoi, C.; VanHulle, C.; Bendoumou, M.; et al. Heterogeneous HIV-1 Reactivation Patterns of Disulfiram and Combined Disulfiram+Romidepsin Treatments. JAIDS J. Acquir. Immune Defic. Syndr. 2019, 80, 605–613. [Google Scholar] [CrossRef] [Green Version]
- Cary, D.C.; Peterlin, B.M. Procyanidin trimer C1 reactivates latent HIV as a triple combination therapy with kansui and JQ. PLoS ONE 2018, 13, e0208055. [Google Scholar] [CrossRef]
- Scheller, C.; Ullrich, A.; McPherson, K.; Hefele, B.; Knöferle, J.; Lamla, S.; Olbrich, A.R.; Stocker, H.; Arasteh, K.; Ter Meulen, V.; et al. CpG Oligodeoxynucleotides Activate HIV Replication in Latently Infected Human T Cells. J. Biol. Chem. 2004, 279, 21897–21902. [Google Scholar] [CrossRef] [Green Version]
- Huan, C.; Li, Z.; Ning, S.; Wang, H.; Yu, X.-F.; Zhang, W. Long Noncoding RNA uc002yug.2 Activates HIV-1 Latency through Regulation of mRNA Levels of Various RUNX1 Isoforms and Increased Tat Expression. J. Virol. 2018, 92, e01844-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McBrien, J.B.; Mavigner, M.; Franchitti, L.; Smith, S.A.; White, E.; Tharp, G.K.; Walum, H.; Busman-Sahay, K.; Aguilera-Sandoval, C.R.; Thayer, W.O.; et al. Robust and persistent reactivation of SIV and HIV by N-803 and depletion of CD8+ cells. Nat. Cell Biol. 2020, 578, 154–159. [Google Scholar] [CrossRef]
- Nixon, C.C.; Mavigner, M.; Sampey, G.C.; Brooks, A.D.; Spagnuolo, R.A.; Irlbeck, D.M.; Mattingly, C.; Ho, P.T.; Schoof, N.; Cammon, C.G.; et al. Systemic HIV and SIV latency reversal via non-canonical NF-κB signalling in vivo. Nat. Cell Biol. 2020, 578, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Perreau, M.; Savoye, A.-L.; De Crignis, E.; Corpataux, J.-M.; Cubas, R.; Haddad, E.K.; De Leval, L.; Graziosi, C.; Pantaleo, G. Follicular helper T cells serve as the major CD4 T cell compartment for HIV-1 infection, replication, and production. J. Exp. Med. 2013, 210, 143–156. [Google Scholar] [CrossRef]
- Banga, R.; Procopio, F.A.; Noto, A.; Pollakis, G.; Cavassini, M.; Ohmiti, K.; Corpataux, J.-M.; De Leval, L.; Pantaleo, G.; Perreau, M. PD-1+ and follicular helper T cells are responsible for persistent HIV-1 transcription in treated aviremic individuals. Nat. Med. 2016, 22, 754–761. [Google Scholar] [CrossRef] [PubMed]
- Noto, A.; Procopio, F.A.; Banga, R.; Suffiotti, M.; Corpataux, J.M.; Cavassini, M.; Riva, A.; Fenwick, C.; Gottardo, R.; Perreau, M.; et al. CD32+ and PD-1+ lymph node CD4 T cells support persistent HIV-1 transcription in treated aviremic individuals. J. Virol. 2018, 92, e00901-18. [Google Scholar] [CrossRef] [Green Version]
- Fromentin, R.; DaFonseca, S.; Costiniuk, C.T.; El-Far, M.; Procopio, F.A.; Hecht, F.M.; Hoh, R.; Deeks, S.G.; Hazuda, D.J.; Lewin, S.R.; et al. PD-1 blockade potentiates HIV latency reversal ex vivo in CD4+ T cells from ART-suppressed individuals. Nat. Commun. 2019, 10, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kristoff, J.; Palma, M.L.; Garcia-Bates, T.M.; Shen, C.; Sluis-Cremer, N.; Gupta, P.; Rinaldo, C.R.; Mailliard, R.B. Type 1-programmed dendritic cells drive antigen-specific latency reversal and immune elimination of persistent HIV. EBioMedicine 2019, 43, 295–306. [Google Scholar] [CrossRef] [Green Version]
- Rezaei, S.D.; Lu, H.K.; Chang, J.J.; Rhodes, A.; Lewin, S.R.; Cameron, P.U. The Pathway To Establishing HIV Latency Is Critical to How Latency Is Maintained and Reversed. J. Virol. 2018, 92, e02225-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Huertas, M.R.; Morín, M.; Madrid-Elena, N.; Gutiérrez, C.; Jiménez-Tormo, L.; Santoyo, J.; Sanz-Rodríguez, F.; Pelayo, M.; Ángel, M.; Bermejo, L.G.; et al. Selective miRNA Modulation Fails to Activate HIV Replication in In Vitro Latency Models. Mol. Ther. Nucleic Acids 2019, 17, 323–336. [Google Scholar] [CrossRef] [Green Version]
- Meås, H.Z.; Haug, M.; Beckwith, M.S.; Louet, C.; Ryan, L.; Hu, Z.; Landskron, J.; Nordbø, S.A.; Taskén, K.; Yin, H.; et al. Sensing of HIV-1 by TLR8 activates human T cells and reverses latency. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef]
- Grau-Expósito, J.; Luque-Ballesteros, L.; Navarro, J.; Curran, A.; Burgos, J.; Ribera, E.; Torrella, A.; Planas, B.; Badía, R.; Martin-Castillo, M.; et al. Latency reversal agents affect differently the latent reservoir present in distinct CD4+ T subpopulations. PLoS Pathog. 2019, 15, e1007991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serra-Peinado, C.; Grau-Expósito, J.; Luque-Ballesteros, L.; Astorga-Gamaza, A.; Navarro, J.; Gallego-Rodriguez, J.; Martin, M.; Curran, A.; Burgos, J.; Ribera, E.; et al. Expression of CD20 after viral reactivation renders HIV-reservoir cells susceptible to Rituximab. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkinson, B.; Chen, J.Y.-F.; Han, P.; Rufner, K.M.; Goularte, O.D.; Kaye, J. TOX: An HMG box protein implicated in the regulation of thymocyte selection. Nat. Immunol. 2002, 3, 272–280. [Google Scholar] [CrossRef]
- Yun, S.; Lee, S.H.; Yoon, S.-R.; Kim, M.S.; Piao, Z.-H.; Myung, P.-K.; Kim, T.-D.; Jung, H.; Choi, I. TOX regulates the differentiation of human natural killer cells from hematopoietic stem cells in vitro. Immunol. Lett. 2011, 136, 29–36. [Google Scholar] [CrossRef]
- Aliahmad, P.; Kaye, J. Development of all CD4 T lineages requires nuclear factor TOX. J. Exp. Med. 2008, 205, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Li, Z. TOX gene: A novel target for human cancer gene therapy. Am. J. Cancer Res. 2015, 5, 3516–3524. [Google Scholar]
- Dittmer, S.; Kovacs, Z.; Yuan, S.H.; Siszler, G.; Kögl, M.; Summer, H.; Geerts, A.; Golz, S.; Shioda, T.; Methner, A. TOX3 is a neuronal survival factor that induces transcription depending on the presence of CITED1 or phosphorylated CREB in the transcriptionally active complex. J. Cell Sci. 2010, 124, 252–260. [Google Scholar] [CrossRef] [Green Version]
- Du Puch, C.B.M.; Barbier, E.; Kraut, A.; Coute, Y.; Fuchs, J.; Buhot, A.; Livache, T.; Sève, M.; Favier, A.; Douki, T.; et al. TOX4 and its binding partners recognize DNA adducts generated by platinum anticancer drugs. Arch. Biochem. Biophys. 2011, 507, 296–303. [Google Scholar] [CrossRef]
- Vong, Q.P.; Leung, W.-H.; Houston, J.; Li, Y.; Rooney, B.; Holladay, M.; Oostendorp, R.A.J.; Leung, W. TOX2 regulates human natural killer cell development by controlling T-BET expression. Blood 2014, 124, 3905–3913. [Google Scholar] [CrossRef]
- Seo, H.; Chen, J.; González-Avalos, E.; Samaniego-Castruita, D.; Das, A.; Wang, Y.H.; López-Moyado, I.F.; Georges, R.O.; Zhang, W.; Onodera, A.; et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8+ T cell exhaustion. Proc. Natl. Acad. Sci. USA 2019, 116, 12410–12415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobbardi, R.; Pinder, J.; Martinez-Pastor, B.; Theodorou, M.; Blackburn, J.S.; Abraham, B.J.; Namiki, Y.; Mansour, M.; Abdelfattah, N.S.; Molodtsov, A.; et al. TOX Regulates Growth, DNA Repair, and Genomic Instability in T-cell Acute Lymphoblastic Leukemia. Cancer Discov. 2017, 7, 1336–1353. [Google Scholar] [CrossRef] [Green Version]
- Morchikh, M.; Naughtin, M.; Di Nunzio, F.; Xavier, J.; Charneau, P.; Jacob, Y.; Lavigne, M. TOX4 and NOVA1 Proteins Are Partners of the LEDGF PWWP Domain and Affect HIV-1 Replication. PLoS ONE 2013, 8, e81217. [Google Scholar] [CrossRef]
- Gougeon, M.-L.; Melki, M.-T.; Saïdi, H. HMGB1, an alarmin promoting HIV dissemination and latency in dendritic cells. Cell Death Differ. 2011, 19, 96–106. [Google Scholar] [CrossRef]
- Naghavi, M.H.; Nowak, P.; Andersson, J.; Sönnerborg, A.; Yang, H.; Tracey, K.J.; Vahlne, A. Intracellular high mobility group B1 protein (HMGB1) represses HIV-1 LTR-directed transcription in a promoter- and cell-specific manner. Virology 2003, 314, 179–189. [Google Scholar] [CrossRef] [Green Version]
- Nowak, P.; Abdurahman, S.; Lindkvist, A.; Troseid, M.; Sönnerborg, A. Impact of HMGB1/TLR Ligand Complexes on HIV-1 Replication: Possible Role for Flagellin during HIV-1 Infection. Int. J. Microbiol. 2012, 2012, 1–10. [Google Scholar] [CrossRef] [PubMed]
- O’Flaherty, E.; Kaye, J. TOX defines a conserved subfamily of HMG-box proteins. BMC Genom. 2003, 4, 13. [Google Scholar] [CrossRef] [Green Version]
- Khan, O.; Giles, J.R.; McDonald, S.; Manne, S.; Ngiow, S.F.; Patel, K.P.; Werner, M.T.; Huang, A.C.; Alexander, K.A.; Wu, J.E.; et al. TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature 2019, 571, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, V.; Su, M.; Huang, Y.; Hsing, M.; Cherkasov, A.; Zhou, Y. Computer-Aided Discovery of Small Molecule Inhibitors of Thymocyte Selection-Associated High Mobility Group Box Protein (TOX) as Potential Therapeutics for Cutaneous T-Cell Lymphomas. Molecules 2019, 24, 3459. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Wang, H.; Andersson, U. Targeting Inflammation Driven by HMGB. Front. Immunol. 2020, 11, 484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Wang, H.; Ju, Z.; Ragab, A.A.; Lundbäck, P.; Long, W.; Valdés-Ferrer, S.I.; He, M.; Pribis, J.P.; Li, J.; et al. MD-2 is required for disulfide HMGB1&ndash, dependent TLR4 signaling. J. Exp. Med. 2015, 212, 5–14. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Li, S.; Yang, Q.; Shi, Y.; Zheng, M.; Liu, Y.; Chen, F.; Song, G.; Xu, H.; Wan, T.; et al. Resveratrol Reduces the Proinflammatory Effects and Lipopolysaccharide- Induced Expression of HMGB1 and TLR4 in RAW264.7 Cells. Cell. Physiol. Biochem. 2014, 33, 1283–1292. [Google Scholar] [CrossRef]
- Vanpatten, S.; Al-Abed, Y. High Mobility Group Box-1 (HMGb1): Current Wisdom and Advancement as a Potential Drug Target. J. Med. Chem. 2018, 61, 5093–5107. [Google Scholar] [CrossRef] [Green Version]
- András, I.E.; Garcia-Contreras, M.; Yanick, C.; Perez, P.; Sewell, B.; Durand, L.; Toborek, M. Extracellular vesicle-mediated amyloid transfer to neural progenitor cells: Implications for RAGE and HIV infection. Mol. Brain 2020, 13, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Yu, J.; Li, L.; Zhang, B.; Liu, L.; Wu, C.-H.; Jong, A.; Mao, D.-A.; Huang, S.-H. Alpha7 nicotinic acetylcholine receptor is required for amyloid pathology in brain endothelial cells induced by Glycoprotein 120, methamphetamine and nicotine. Sci. Rep. 2017, 7, 40467. [Google Scholar] [CrossRef] [Green Version]
- Yuan, S.; Liu, Z.; Xu, Z.; Liu, J.; Zhang, J. High mobility group box 1 (HMGB1): A pivotal regulator of hematopoietic malignancies. J. Hematol. Oncol. 2020, 13, 1–19. [Google Scholar] [CrossRef]
- Stoszko, M.; Ne, E.; Abner, E.; Mahmoudi, T. A broad drug arsenal to attack a strenuous latent HIV reservoir. Curr. Opin. Virol. 2019, 38, 37–53. [Google Scholar] [CrossRef]
- Vargas, B.; Giacobbi, N.S.; Sanyal, A.; Venkatachari, N.J.; Han, F.; Gupta, P.; Sluis-Cremer, N. Inhibitors of Signaling Pathways That Block Reversal of HIV-1 Latency. Antimicrob. Agents Chemother. 2018, 63, e01744-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Vidal, E.; Badia, R.; Pujantell, M.; Castellví, M.; Felip, E.; Clotet, B.; Riveira-Muñoz, E.; Ballana, E.; Esté, J.A. Dual effect of the broad spectrum kinase inhibitor midostaurin in acute and latent HIV-1 infection. Antivir. Res. 2019, 168, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Chougui, G.; Margottin-Goguet, F. HUSH, a Link Between Intrinsic Immunity and HIV Latency. Front. Microbiol. 2019, 10, 224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duverger, A.; Wolschendorf, F.; Anderson, J.C.; Wagner, F.; Bosque, A.; Shishido, T.; Jones, J.; Planelles, V.; Willey, C.; Cron, R.Q.; et al. Kinase Control of Latent HIV-1 Infection: PIM-1 Kinase as a Major Contributor to HIV-1 Reactivation. J. Virol. 2013, 88, 364–376. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, K.; Kobayakawa, T.; Tsuchiya, K.; Hattori, S.-I.H.; Nomura, W.; Gatanaga, H.; Yoshimura, K.; Oka, S.; Endo, Y.; Tamamura, H.; et al. Benzolactam-related compounds promote apoptosis of HIV-infected human cells via protein kinase C–induced HIV latency reversal. J. Biol. Chem. 2019, 294, 116–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Montfort, T.; Van Der Sluis, R.; Darcis, G.; Beaty, D.; Groen, K.; Pasternak, A.O.; Pollakis, G.; Vink, M.; Westerhout, E.M.; Hamdi, M.; et al. Dendritic cells potently purge latent HIV-1 beyond TCR-stimulation, activating the PI3K-Akt-mTOR pathway. EBioMedicine 2019, 42, 97–108. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Mousseau, G.; Valente, S.T. Tat inhibition by didehydro-Cortistatin A promotes heterochromatin formation at the HIV-1 long terminal repeat. Epigenetics Chromatin 2019, 12, 1–17. [Google Scholar] [CrossRef]
- Besnard, E.; Hakre, S.; Kampmann, M.; Lim, H.W.; Hosmane, N.N.; Martin, A.; Bassik, M.C.; Verschueren, E.; Battivelli, E.; Chan, J.; et al. The mTOR Complex Controls HIV Latency. Cell Host Microbe 2016, 20, 785–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulinski, T.; Olejniczak, M.; Huthoff, H.; Bielecki, L.; Pachulska-Wieczorek, K.; Das, A.T.; Berkhout, B.; Adamiak, R.W. The Apical Loop of the HIV-1 TAR RNA Hairpin Is Stabilized by a Cross-loop Base Pair. J. Biol. Chem. 2003, 278, 38892–38901. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Kadakkuzha, B.M.; Zhao, L.; Fan, M.; Qi, X.; Xia, T. Dynamic Ensemble View of the Conformational Landscape of HIV-1 TAR RNA and Allosteric Recognition. Biochemistry 2011, 50, 5042–5057. [Google Scholar] [CrossRef]
- Wei, P.; Garber, E.M.; Fang, S.-M.; Fischer, W.H.; Jones, A.K. A Novel CDK9-Associated C-Type Cyclin Interacts Directly with HIV-1 Tat and Mediates Its High-Affinity, Loop-Specific Binding to TAR RNA. Cell 1998, 92, 451–462. [Google Scholar] [CrossRef] [Green Version]
- Garber, M.E.; Mayall, T.P.; Suess, E.M.; Meisenhelder, J.; Thompson, N.E.; Jones, K.A. CDK9 Autophosphorylation Regulates High-Affinity Binding of the Human Immunodeficiency Virus Type 1 Tat–P-TEFb Complex to TAR RNA. Mol. Cell. Biol. 2000, 20, 6958–6969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salerno, D.; Hasham, M.G.; Marshall, R.; Garriga, J.; Tsygankov, A.Y.; Graña, X. Direct inhibition of CDK9 blocks HIV-1 replication without preventing T-cell activation in primary human peripheral blood lymphocytes. Gene 2007, 405, 65–78. [Google Scholar] [CrossRef] [Green Version]
- Mancebo, H.S.; Lee, G.; Flygare, J.; Tomassini, J.; Luu, P.; Zhu, Y.; Peng, J.; Blau, C.; Hazuda, D.; Price, D.; et al. P-TEFb kinase is required for HIV Tat transcriptional activation in vivo and in vitro. Genes Dev. 1997, 11, 2633–2644. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Herrmann, C.H.; Rice, A.P. The human immunodeficiency virus Tat proteins specifically associate with TAK in vivo and require the carboxyl-terminal domain of RNA polymerase II for function. J. Virol. 1996, 70, 4576–4584. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Pe’Ery, T.; Peng, J.; Ramanathan, Y.; Marshall, N.; Marshall, T.; Amendt, B.; Mathews, M.B.; Price, D.H. Transcription elongation factor P-TEFb is required for HIV-1 Tat transactivation in vitro. Genes Dev. 1997, 11, 2622–2632. [Google Scholar] [CrossRef] [Green Version]
- Budhiraja, S.; Famiglietti, M.; Bosque, A.; Planelles, V.; Rice, A.P. Cyclin T1 and CDK9 T-Loop Phosphorylation Are Downregulated during Establishment of HIV-1 Latency in Primary Resting Memory CD4+ T Cells. J. Virol. 2013, 87, 1211–1220. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Song, L.; Zhou, T.; Zeng, C.; Jia, Y.; Zhao, Y. A computational study of Tat–CDK9–Cyclin binding dynamics and its implication in transcription-dependent HIV latency. Phys. Chem. Chem. Phys. 2020, 22, 25474–25482. [Google Scholar] [CrossRef] [PubMed]
- Morton, E.L.; Forst, C.V.; Zheng, Y.; DePaula-Silva, A.B.; Ramirez, N.-G.P.; Planelles, V.; D’Orso, I. Transcriptional Circuit Fragility Influences HIV Proviral Fate. Cell Rep. 2019, 27, 154–171.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seif, F.; Khoshmirsafa, M.; Aazami, H.; Mohsenzadegan, M.; Sedighi, G.; Bahar, M. The role of JAK-STAT signaling pathway and its regulators in the fate of T helper cells. Cell Commun. Signal. 2017, 15, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Shea, J.J.; Plenge, R. JAK and STAT Signaling Molecules in Immunoregulation and Immune-Mediated Disease. Immunity 2012, 36, 542–550. [Google Scholar] [CrossRef] [Green Version]
- Rose, N.R. Autoimmune Diseases. Int. Encycl. Public Health 2017, 12, 192–195. [Google Scholar] [CrossRef]
- Darnell, J.E.D., Jr. STATs and Gene Regulation. Science 1997, 277, 1630–1635. [Google Scholar] [CrossRef]
- Quan, Y.; Xu, H.; Han, Y.; Mesplède, T.; Wainberg, M.A. JAK-STAT Signaling Pathways and Inhibitors Affect Reversion of Envelope-Mutated HIV. J. Virol. 2017, 91, e00075-17. [Google Scholar] [CrossRef] [Green Version]
- Gavegnano, C.; Brehm, J.H.; Dupuy, F.P.; Talla, A.; Ribeiro, S.P.; Kulpa, D.A.; Cameron, C.; Santos, S.; Hurwitz, S.J.; Marconi, V.C.; et al. Novel mechanisms to inhibit HIV reservoir seeding using Jak inhibitors. PLoS Pathog. 2017, 13, e1006740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gavegnano, C.; Detorio, M.; Montero, C.; Bosque, A.; Planelles, V.; Schinazi, R.F. Ruxolitinib and Tofacitinib Are Potent and Selective Inhibitors of HIV-1 Replication and Virus Reactivation In Vitro. Antimicrob. Agents Chemother. 2014, 58, 1977–1986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatachari, N.J.; Zerbato, J.M.; Jain, S.; Mancini, A.E.; Chattopadhyay, A.; Sluis-Cremer, N.; Bar-Joseph, Z.; Ayyavoo, V. Temporal transcriptional response to latency reversing agents identifies specific factors regulating HIV-1 viral transcriptional switch. Retrovirology 2015, 12, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosque, A.; Nilson, K.A.; Macedo, A.B.; Spivak, A.M.; Archin, N.M.; Van Wagoner, R.M.; Martins, L.J.; Novis, C.L.; Szaniawski, M.A.; Ireland, C.M.; et al. Benzotriazoles Reactivate Latent HIV-1 through Inactivation of STAT5 SUMOylation. Cell Rep. 2017, 18, 1324–1334. [Google Scholar] [CrossRef]
- Selliah, N.; Zhang, M.; DeSimone, D.; Kim, H.; Brunner, M.; Ittenbach, R.F.; Rui, H.; Cron, R.Q.; Finkel, T.H. The γc-cytokine regulated transcription factor, STAT5, increases HIV-1 production in primary CD4 T cells. Virology 2006, 344, 283–291. [Google Scholar] [CrossRef] [Green Version]
- Della Chiara, G.; Crotti, A.; Liboi, E.; Giacca, M.; Poli, G.; Lusic, M. Negative Regulation of HIV-1 Transcription by a Heterodimeric NF-κB1/p50 and C-Terminally Truncated STAT5 Complex. J. Mol. Biol. 2011, 410, 933–943. [Google Scholar] [CrossRef]
- Quan, Y.; Xu, H.; Kramer, V.G.; Han, Y.; Sloan, R.D.; Wainberg, M.A. Identification of an env-defective HIV-1 mutant capable of spontaneous reversion to a wild-type phenotype in certain T-cell lines. Virol. J. 2014, 11, 177. [Google Scholar] [CrossRef] [Green Version]
- Chaudhuri, A.; Yang, B.; Gendelman, H.E.; Persidsky, Y.; Kanmogne, G.D. STAT1 signaling modulates HIV-1–induced inflammatory responses and leukocyte transmigration across the blood-brain barrier. Blood 2008, 111, 2062–2072. [Google Scholar] [CrossRef]
- Van Der Sluis, R.M.; Zerbato, J.M.; Rhodes, J.W.; Pascoe, R.D.; Solomon, A.; Kumar, N.A.; Dantanarayana, A.I.; Tennakoon, S.; Dufloo, J.; McMahon, J.; et al. Diverse effects of interferon alpha on the establishment and reversal of HIV latency. PLoS Pathog. 2020, 16, e1008151. [Google Scholar] [CrossRef]
- Gargan, S.; Ahmed, S.; Mahony, R.; Bannan, C.; Napoletano, S.; O’Farrelly, C.; Borrow, P.; Bergin, C.; Stevenson, N.J. HIV-1 Promotes the Degradation of Components of the Type 1 IFN JAK/STAT Pathway and Blocks Anti-viral ISG Induction. EBioMedicine 2018, 30, 203–216. [Google Scholar] [CrossRef] [Green Version]
- Shytaj, I.L.; Lucic, B.; Forcato, M.; Penzo, C.; Billingsley, J.; Laketa, V.; Bosinger, S.; Stanic, M.; Gregoretti, F.; Antonelli, L.; et al. Alterations of redox and iron metabolism accompany the development of HIV latency. EMBO J. 2020, 39, e102209. [Google Scholar] [CrossRef] [PubMed]
- Diaz, R.S.; Shytaj, I.L.; Giron, L.B.; Obermaier, B.; Della Libera, E.; Galinskas, J.; Dias, D.; Hunter, J.; Janini, M.; Gosuen, G.; et al. Potential impact of the antirheumatic agent auranofin on proviral HIV-1 DNA in individuals under intensified antiretroviral therapy: Results from a randomised clinical trial. Int. J. Antimicrob. Agents 2019, 54, 592–600. [Google Scholar] [CrossRef]
- Valle-Casuso, J.C.; Angin, M.; Volant, S.; Passaes, C.; Monceaux, V.; Mikhailova, A.; Bourdic, K.; Avettand-Fenoel, V.; Boufassa, F.; Sitbon, M.; et al. Cellular Metabolism Is a Major Determinant of HIV-1 Reservoir Seeding in CD4+ T Cells and Offers an Opportunity to Tackle Infection. Cell Metab. 2019, 29, 611–626.e5. [Google Scholar] [CrossRef] [Green Version]
- Castellano, P.; Prevedel, L.; Valdebenito, S.; Eugenin, E.A. HIV infection and latency induce a unique metabolic signature in human macrophages. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Thaker, S.K.; Ch’Ng, J.; Christofk, H.R. Viral hijacking of cellular metabolism. BMC Biol. 2019, 17, 1–15. [Google Scholar] [CrossRef]
- Eisenreich, W.; Rudel, T.; Heesemann, J.; Goebel, W. How Viral and Intracellular Bacterial Pathogens Reprogram the Metabolism of Host Cells to Allow Their Intracellular Replication. Front. Cell. Infect. Microbiol. 2019, 9, 42. [Google Scholar] [CrossRef] [Green Version]
- Timilsina, U.; Gaur, R. Modulation of apoptosis and viral latency—an axis to be well understood for successful cure of human immunodeficiency virus. J. Gen. Virol. 2016, 97, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Bobardt, M.; Kuo, J.; Chatterji, U.; Chanda, S.; Little, S.J.; Wiedemann, N.; Vuagniaux, G.; Gallay, P.A. The inhibitor apoptosis protein antagonist Debio 1143 Is an attractive HIV-1 latency reversal candidate. PLoS ONE 2019, 14, e0211746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bobardt, M.; Kuo, J.; Chatterji, U.; Wiedemann, N.; Vuagniaux, G.; Gallay, P. The inhibitor of apoptosis proteins antagonist Debio 1143 promotes the PD-1 blockade-mediated HIV load reduction in blood and tissues of humanized mice. PLoS ONE 2020, 15, e0227715. [Google Scholar] [CrossRef] [PubMed]
- Kuo, H.-H.; Ahmad, R.; Lee, G.Q.; Gao, C.; Chen, H.-R.; Ouyang, Z.; Szucs, M.J.; Kim, D.; Tsibris, A.; Chun, T.-W.; et al. Anti-apoptotic Protein BIRC5 Maintains Survival of HIV-1-Infected CD4+ T Cells. Immunity 2018, 48, 1183–1194.e5. [Google Scholar] [CrossRef] [Green Version]
- Chugh, P.; Bradel-Tretheway, B.; Monteiro-Filho, C.M.; Planelles, V.; Maggirwar, S.B.; Dewhurst, S.; Kim, B. Akt inhibitors as an HIV-1 infected macrophage-specific anti-viral therapy. Retrovirology 2008, 5, 11. [Google Scholar] [CrossRef] [Green Version]
- Wolf, D.; Witte, V.; Laffert, B.; Blume, K.; Stromer, E.; Trapp, S.; D’Aloja, P.; Schürmann, A.; Baur, A.S. HIV-1 Nef associated PAK and PI3-Kinases stimulate Akt-independent Bad-phosphorylation to induce anti-apoptotic signals. Nat. Med. 2001, 7, 1217–1224. [Google Scholar] [CrossRef]
- She, Q.-B.; Halilovic, E.; Ye, Q.; Zhen, W.; Shirasawa, S.; Sasazuki, T.; Solit, D.B.; Rosen, N. 4E-BP1 Is a Key Effector of the Oncogenic Activation of the AKT and ERK Signaling Pathways that Integrates Their Function in Tumors. Cancer Cell 2010, 18, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Rodrik-Outmezguine, V.S.; Chandarlapaty, S.; Pagano, N.C.; Poulikakos, P.I.; Scaltriti, M.; Moskatel, E.; Baselga, J.; Guichard, S.; Rosen, N. mTOR Kinase Inhibition Causes Feedback-Dependent Biphasic Regulation of AKT Signaling. Cancer Discov. 2011, 1, 248–259. [Google Scholar] [CrossRef] [Green Version]
- Campbell, G.R.; To, R.K.; Spector, S.A. TREM-1 Protects HIV-1-Infected Macrophages from Apoptosis through Maintenance of Mitochondrial Function. mBio 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Chandrasekar, A.P.; Cummins, N.W.; Badley, A.D. The Role of the BCL-2 Family of Proteins in HIV-1 Pathogenesis and Persistence. Clin. Microbiol. Rev. 2019, 33. [Google Scholar] [CrossRef]
- Doitsh, G.; Galloway, N.L.K.; Geng, X.; Yang, Z.; Monroe, K.M.; Zepeda, O.; Hunt, P.W.; Hatano, H.; Sowinski, S.; Muñoz-Arias, I.; et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nat. Cell Biol. 2014, 505, 509–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, G.; Luk, B.T.; Wei, X.; Campbell, G.R.; Fang, R.H.; Zhang, L.; Spector, S.A. Selective cell death of latently HIV-infected CD4+ T cells mediated by autosis inducing nanopeptides. Cell Death Dis. 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, J.Y.; Dixon, S.J. Mechanisms of ferroptosis. Cell. Mol. Life Sci. 2016, 73, 2195–2209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, R.N.; Reed, J.C.; Chanda, S.K. HIV-1 protease cleaves the serine-threonine kinases RIPK1 and RIPK. Retrovirology 2015, 12, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Fletcher-Etherington, A.; Nobre, L.; Nightingale, K.; Antrobus, R.; Nichols, J.; Davison, A.J.; Stanton, R.J.; Weekes, M.P. Human cytomegalovirus protein pUL36: A dual cell death pathway inhibitor. Proc. Natl. Acad. Sci. USA 2020, 117, 18771–18779. [Google Scholar] [CrossRef]
- Mehrbod, P.; Ande, S.R.; Alizadeh, J.; Rahimizadeh, S.; Shariati, A.; Malek, H.; Hashemi, M.; Glover, K.K.M.; Sher, A.A.; Coombs, K.M.; et al. The roles of apoptosis, autophagy and unfolded protein response in arbovirus, influenza virus, and HIV infections. Virulence 2019, 10, 376–413. [Google Scholar] [CrossRef] [Green Version]
- Swadling, L.; Pallett, L.J.; Diniz, M.O.; Baker, J.M.; Amin, O.E.; Stegmann, K.A.; Burton, A.R.; Schmidt, N.M.; Jeffery-Smith, A.; Zakeri, N.; et al. Human Liver Memory CD8+ T Cells Use Autophagy for Tissue Residence. Cell Rep. 2020, 30, 687–698.e6. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Yang, W.; Zeng, Z.; Wei, Y.; Gao, J.; Zhang, B.; Li, L.; Liu, L.; Wan, Y.; Zeng, Q.; et al. NLRP3-dependent pyroptosis is required for HIV-1 gp120-induced neuropathology. Cell. Mol. Immunol. 2020, 17, 283–299. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.R.; Pinto, C.M.; Tavares, J.N. Maintenance of the latent reservoir by pyroptosis and superinfection in a fractional order HIV transmission model. Int. J. Optim. Control. Theor. Appl. 2019, 9, 69–75. [Google Scholar] [CrossRef] [Green Version]
- Narayanan, A.; Iordanskiy, S.; Das, R.; Van Duyne, R.; Santos, S.; Jaworski, E.; Guendel, I.; Sampey, G.; Dalby, E.; Iglesias-Ussel, M.; et al. Exosomes Derived from HIV-1-infected Cells Contain Trans-activation Response Element RNA. J. Biol. Chem. 2013, 288, 20014–20033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampey, G.C.; Saifuddin, M.; Schwab, A.; Barclay, R.; Punya, S.; Chung, M.-C.; Hakami, R.M.; Zadeh, M.A.; Lepene, B.; Klase, Z.A.; et al. Exosomes from HIV-1-infected Cells Stimulate Production of Pro-inflammatory Cytokines through Trans-activating Response (TAR) RNA. J. Biol. Chem. 2016, 291, 1251–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, L.J.; Johnson, T.P.; Smith, B.R.; Reoma, L.B.; Santamaria, U.A.; Bachani, M.; DeMarino, C.; Barclay, R.A.; Snow, J.; Sacktor, N.; et al. Presence of Tat and transactivation response element in spinal fluid despite antiretroviral therapy. AIDS 2019, 33, S145–S157. [Google Scholar] [CrossRef]
- Chen, L.; Feng, Z.; Yuan, G.; Emerson, C.C.; Stewart, P.L.; Ye, F.; Jin, G. Human Immunodeficiency Virus-Associated Exosomes Promote Kaposi’s Sarcoma-Associated Herpesvirus Infection via the Epidermal Growth Factor Receptor. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [Green Version]
- Barclay, R.A.; Schwab, A.; DeMarino, C.; Akpamagbo, Y.; Lepene, B.; Kassaye, S.; Iordanskiy, S.; Kashanchi, F. Exosomes from uninfected cells activate transcription of latent HIV. J. Biol. Chem. 2017, 292, 11682–11701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barclay, R.A.; Mensah, G.A.; Cowen, M.; DeMarino, C.; Kim, Y.; Pinto, D.O.; Erickson, J.; Kashanchi, F. Extracellular Vesicle Activation of Latent HIV-1 Is Driven by EV-Associated c-Src and Cellular SRC-1 via the PI3K/AKT/mTOR Pathway. Viruses 2020, 12, 665. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Feng, Z.; Yue, H.; Bazdar, D.; Mbonye, U.; Zender, C.; Harding, C.V.; Bruggeman, L.; Karn, J.; Sieg, S.F.; et al. Exosomes derived from HIV-1-infected cells promote growth and progression of cancer via HIV TAR RNA. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeMarino, C.; Pleet, M.L.; Cowen, M.; Barclay, R.A.; Akpamagbo, Y.; Erickson, J.; Ndembi, N.; Charurat, M.; Jumare, J.; Bwala, S.; et al. Antiretroviral Drugs Alter the Content of Extracellular Vesicles from HIV-1-Infected Cells. Sci. Rep. 2018, 8, 1–20. [Google Scholar] [CrossRef] [PubMed]
- DeMarino, C.; Cowen, M.; Pleet, M.L.; Pinto, D.O.; Khatkar, P.; Erickson, J.; Docken, S.S.; Russell, N.; Reichmuth, B.; Phan, T.; et al. Differences in Transcriptional Dynamics Between T-cells and Macrophages as Determined by a Three-State Mathematical Model. Sci. Rep. 2020, 10, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Sampey, G.C.; Iordanskiy, S.; Pleet, M.L.; DeMarino, C.; Romerio, F.; Mahieux, R.; Kashanchi, F. Identification of Modulators of HIV-1 Proviral Transcription from a Library of FDA-Approved Pharmaceuticals. Viruses 2020, 12, 1067. [Google Scholar] [CrossRef] [PubMed]
- Dobrowolski, C.; Valadkhan, S.; Graham, A.C.; Shukla, M.; Ciuffi, A.; Telenti, A.; Karn, J.; Ott, M.; Henderson, A.; Spina, C. Entry of Polarized Effector Cells into Quiescence Forces HIV Latency. mBio 2019, 10, e00337-19. [Google Scholar] [CrossRef] [Green Version]
- Theron, A.J.; Anderson, R.; Rossouw, T.M.; Steel, H.C. The Role of Transforming Growth Factor Beta-1 in the Progression of HIV/AIDS and Development of Non-AIDS-Defining Fibrotic Disorders. Front. Immunol. 2017, 8, 1461. [Google Scholar] [CrossRef]
- Mbonye, U.; Wang, B.; Gokulrangan, G.; Shi, W.; Yang, S.; Karn, J. Cyclin-dependent kinase 7 (CDK7)-mediated phosphorylation of the CDK9 activation loop promotes P-TEFb assembly with Tat and proviral HIV reactivation. J. Biol. Chem. 2018, 293, 10009–10025. [Google Scholar] [CrossRef] [Green Version]
- Giffin, M.J.; Stroud, J.C.; Bates, D.L.; Von Koenig, K.D.; Hardin, J.W.; Chen, L. Structure of NFAT1 bound as a dimer to the HIV-1 LTR κB element. Nat. Struct. Mol. Biol. 2003, 10, 800–806. [Google Scholar] [CrossRef]
- Chan, J.K.; Bhattacharyya, D.; Lassen, K.G.; Ruelas, D.; Greene, W.C. Calcium/Calcineurin Synergizes with Prostratin to Promote NF-κB Dependent Activation of Latent HIV. PLoS ONE 2013, 8, e77749. [Google Scholar] [CrossRef] [PubMed]
- Gerritsen, M.E.; Williams, A.J.; Neish, A.S.; Moore, S.; Shi, Y.; Collins, T. CREB-binding protein/p300 are transcriptional coactivators of p65. Proc. Natl. Acad. Sci. USA 1997, 94, 2927–2932. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, S.; Su, L.; Amano, M.; Timmerman, A.L.; Kaneshima, H.; Nolan, G.P. The T Cell Activation Factor NF-ATc Positively Regulates HIV-1 Replication and Gene Expression in T Cells. Immunity 1997, 6, 235–244. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Chen, Y.; Gabuzda, D. ERK MAP Kinase Links Cytokine Signals to Activation of Latent HIV-1 Infection by Stimulating a Cooperative Interaction of AP-1 and NF-κB. J. Biol. Chem. 1999, 274, 27981–27988. [Google Scholar] [CrossRef] [Green Version]
- Nabel, G.; Baltimore, D. An inducible transcription factor activates expression of human immunodeficiency virus in T cells. Nat. Cell Biol. 1987, 326, 711–713. [Google Scholar] [CrossRef] [PubMed]
- Mbonye, U.; Karn, J. The Molecular Basis for Human Immunodeficiency Virus Latency. Annu. Rev. Virol. 2017, 4, 261–285. [Google Scholar] [CrossRef] [PubMed]
- Cullen, B.R.; Lomedico, P.T.; Ju, G. Transcriptional interference in avian retroviruses—Implications for the promoter insertion model of leukaemogenesis. Nat. Cell Biol. 1984, 307, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Lenasi, T.; Contreras, X.; Peterlin, B.M. Transcriptional Interference Antagonizes Proviral Gene Expression to Promote HIV Latency. Cell Host Microbe 2008, 4, 123–133. [Google Scholar] [CrossRef] [Green Version]
- Colin, L.; Van Lint, C. Molecular control of HIV-1 postintegration latency: Implications for the development of new therapeutic strategies. Retrovirology 2009, 6, 1–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greger, I.H. Transcriptional interference perturbs the binding of Sp1 to the HIV-1 promoter. Nucleic Acids Res. 1998, 26, 1294–1301. [Google Scholar] [CrossRef] [Green Version]
- Renner, D.B.; Yamaguchi, Y.; Wada, T.; Handa, H.; Price, D.H. A Highly Purified RNA Polymerase II Elongation Control System. J. Biol. Chem. 2001, 276, 42601–42609. [Google Scholar] [CrossRef] [Green Version]
- Pagano, J.M.; Kwak, H.; Waters, C.T.; Sprouse, R.O.; White, B.S.; Ozer, A.; Szeto, K.; Shalloway, D.; Craighead, H.G.; Lis, J.T. Defining NELF-E RNA Binding in HIV-1 and Promoter-Proximal Pause Regions. PLoS Genet. 2014, 10, e1004090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, S.; Lied, A.; Kulkarni, V.; Rucevic, M.; Martin, M.P.; Walker-Sperling, V.; Anderson, S.K.; Ewy, R.; Singh, S.; Nguyen, H.; et al. CCR5AS lncRNA variation differentially regulates CCR5, influencing HIV disease outcome. Nat. Immunol. 2019, 20, 824–834. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.; Amorim, R.; Niu, M.; Breton, Y.; Tremblay, M.J.; Mouland, A.J. Host mRNA decay proteins influence HIV-1 replication and viral gene expression in primary monocyte-derived macrophages. Retrovirology 2019, 16, 1–15. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khanal, S.; Schank, M.; El Gazzar, M.; Moorman, J.P.; Yao, Z.Q. HIV-1 Latency and Viral Reservoirs: Existing Reversal Approaches and Potential Technologies, Targets, and Pathways Involved in HIV Latency Studies. Cells 2021, 10, 475. https://doi.org/10.3390/cells10020475
Khanal S, Schank M, El Gazzar M, Moorman JP, Yao ZQ. HIV-1 Latency and Viral Reservoirs: Existing Reversal Approaches and Potential Technologies, Targets, and Pathways Involved in HIV Latency Studies. Cells. 2021; 10(2):475. https://doi.org/10.3390/cells10020475
Chicago/Turabian StyleKhanal, Sushant, Madison Schank, Mohamed El Gazzar, Jonathan P. Moorman, and Zhi Q. Yao. 2021. "HIV-1 Latency and Viral Reservoirs: Existing Reversal Approaches and Potential Technologies, Targets, and Pathways Involved in HIV Latency Studies" Cells 10, no. 2: 475. https://doi.org/10.3390/cells10020475
APA StyleKhanal, S., Schank, M., El Gazzar, M., Moorman, J. P., & Yao, Z. Q. (2021). HIV-1 Latency and Viral Reservoirs: Existing Reversal Approaches and Potential Technologies, Targets, and Pathways Involved in HIV Latency Studies. Cells, 10(2), 475. https://doi.org/10.3390/cells10020475