
IQGAP1 Is a Scaffold of the Core Proteins of the Hippo Pathway and Negatively Regulates the Pro-Apoptotic Signal Mediated by This Pathway

 , ,
, , 
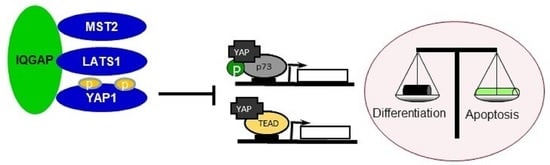
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Constructs and siRNA
2.2. Cell Culture and Transfections
2.3. Immunoprecipitation and Immunoblotting
2.4. Antibodies
2.5. Luciferase Reporter Assays
2.6. Real Time PCR (rtPCR)
2.7. Cell Cycle and Apoptosis Assays
3. Results
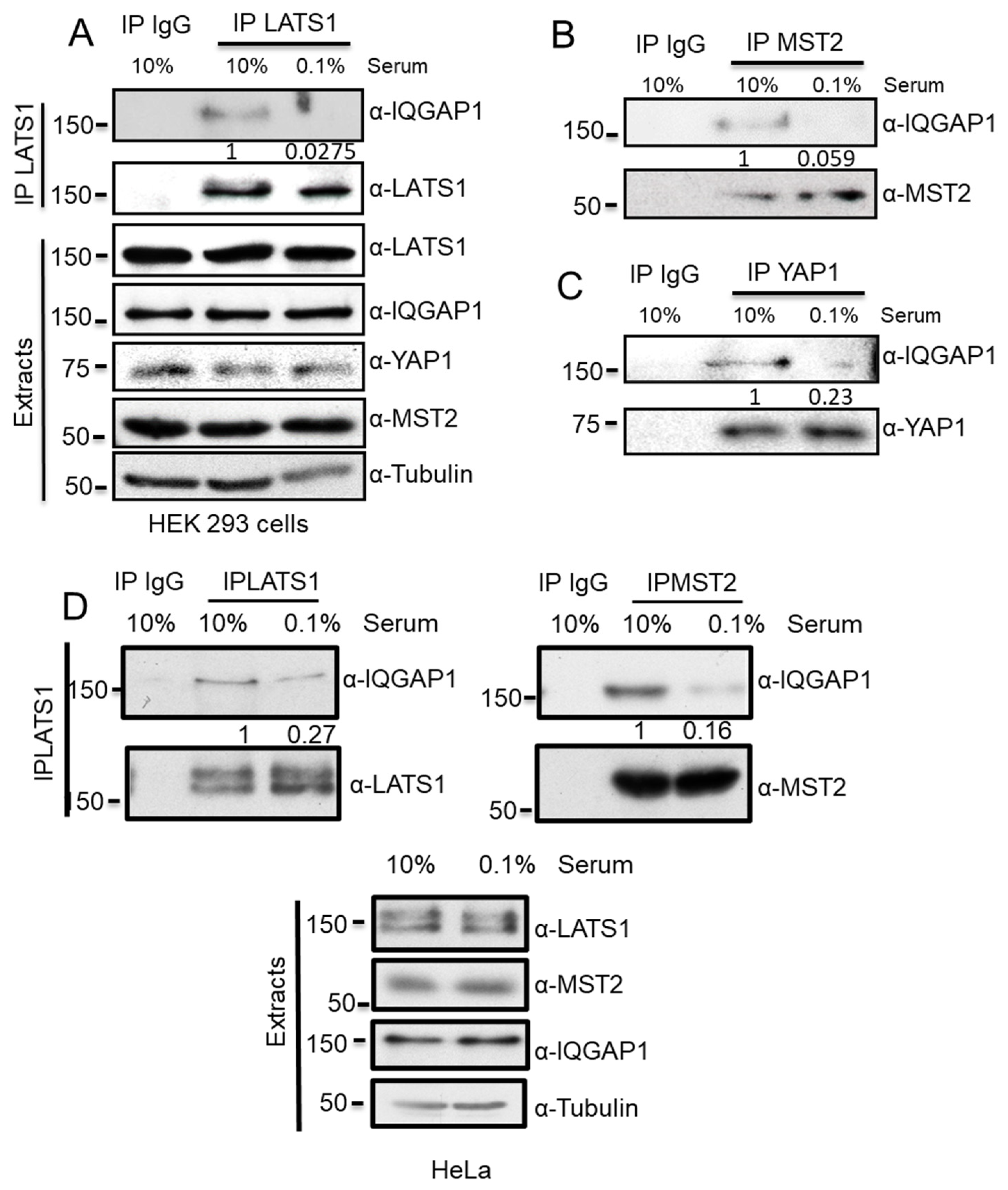
3.1. IQGAP1 Specifically Interact with the Core Proteins of the Hippo Pathway
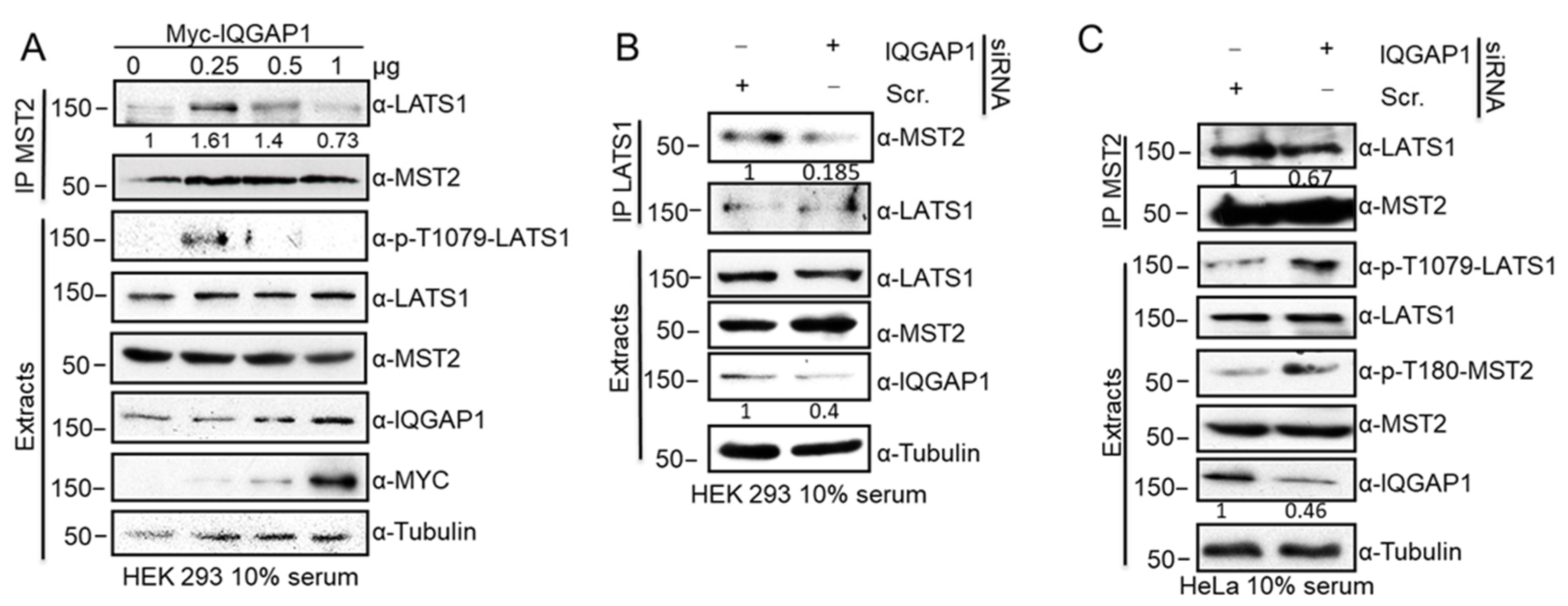
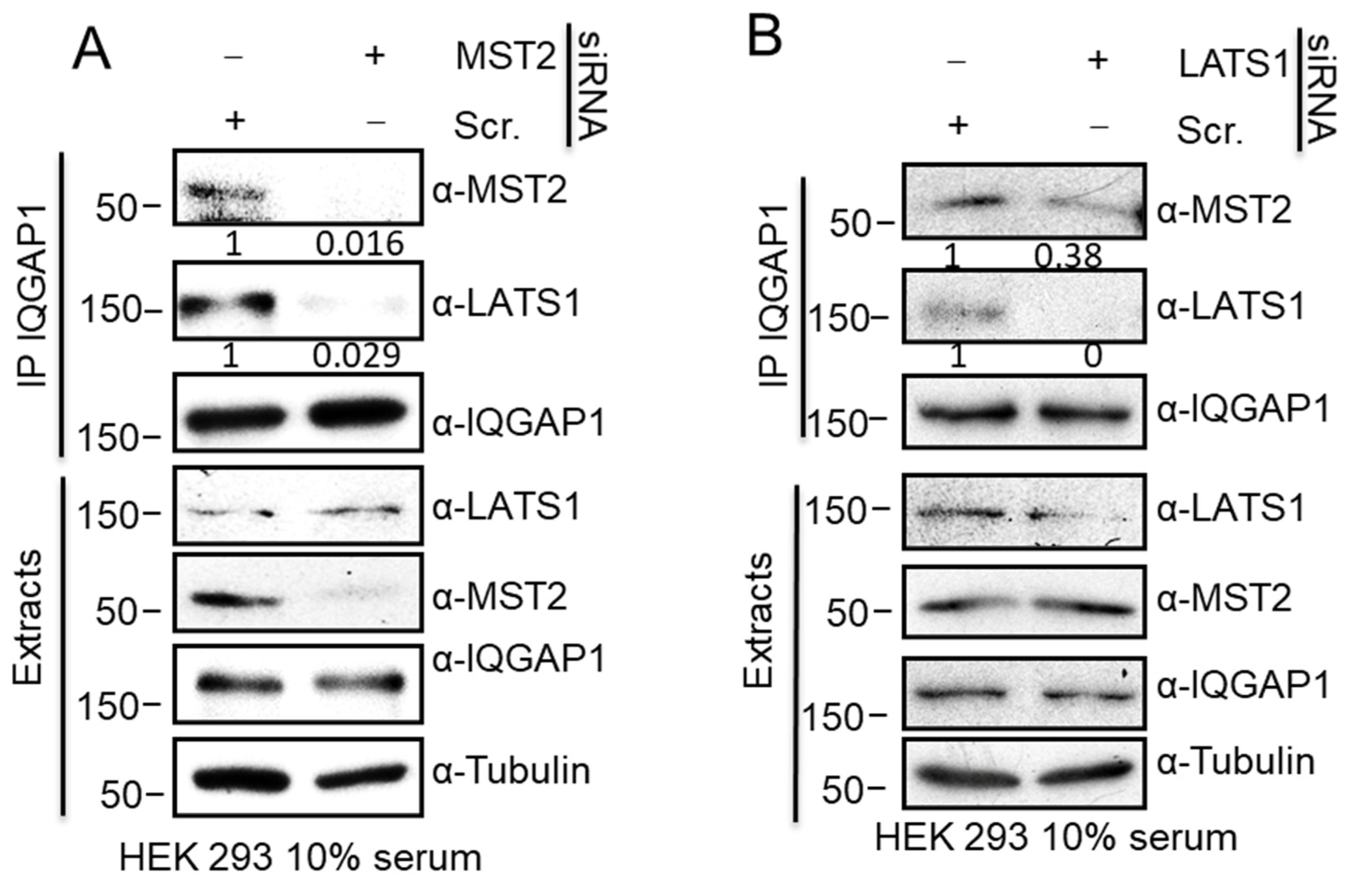
3.2. IQGAP1 Scaffolds the MST2-LATS1 Interaction and Regulates Their Activation
3.3. MST2 and LATS1 Bind to the IQ Domain of IQGAP1
3.4. MST2 and LATS1 Cooperate to Bind to IQGAP1
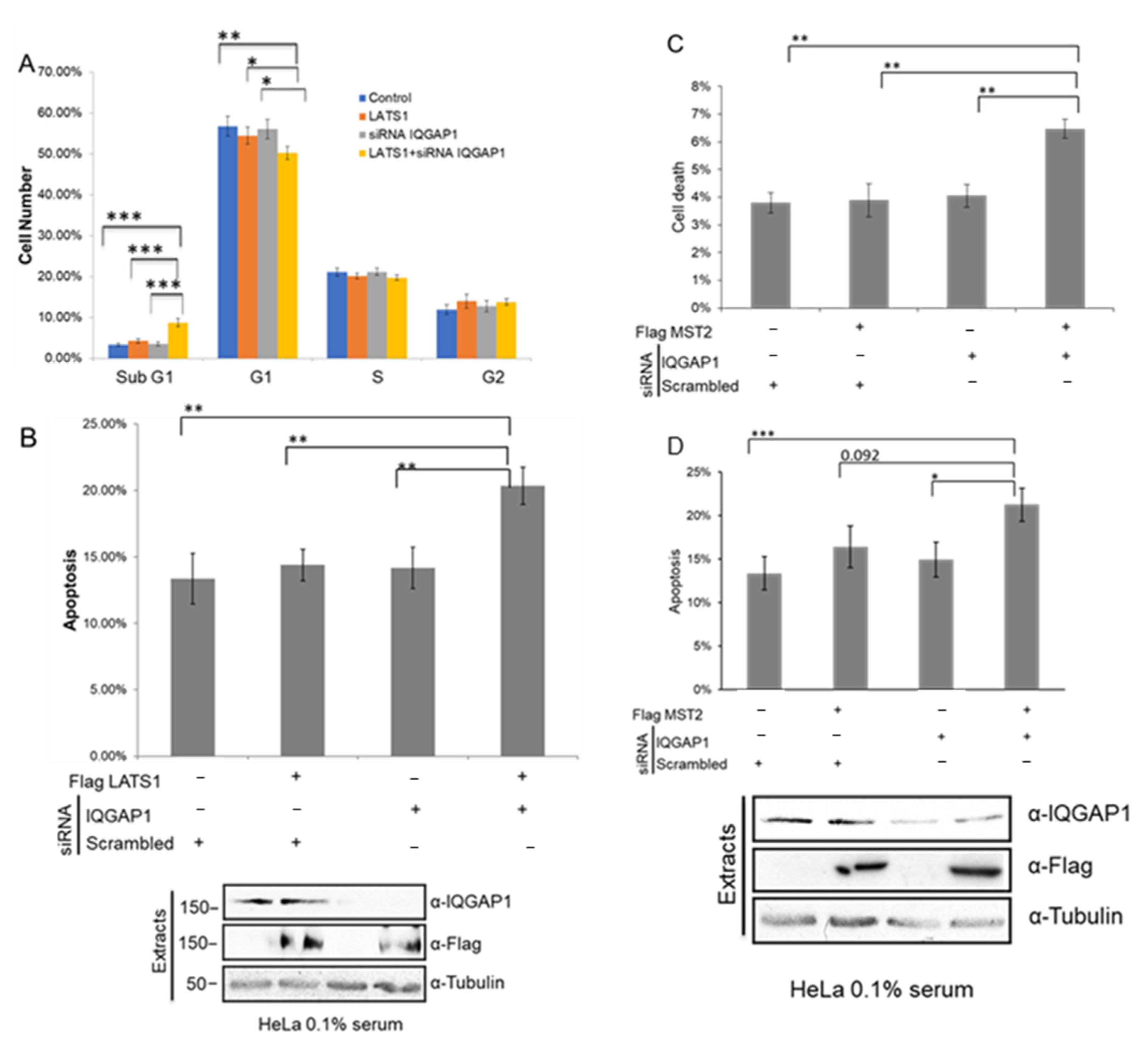
3.5. IQGAP1 Regulates MST2-LATS1-Dependent Apoptosis
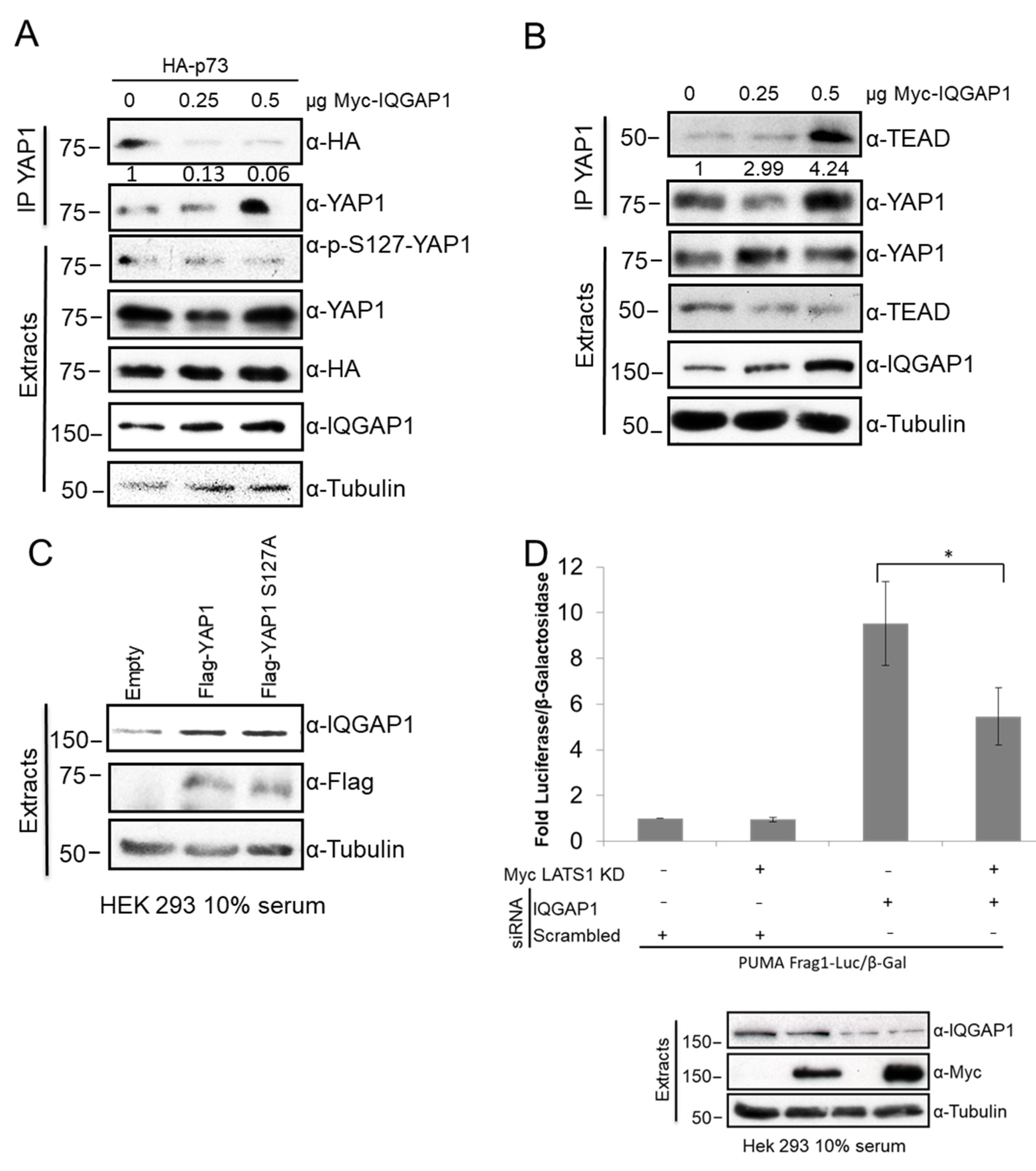
3.6. IQGAP1 Regulates YAP-p73 Interaction and Transcriptional Activity
3.7. The IQGAP1-Hippo Module Is Regulated by CDCA in Hepatocellular Cells
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fallahi, E.; O’Driscoll, N.A.; Matallanas, D. The MST/Hippo Pathway and Cell Death: A Non-Canonical Affair. Genes 2016, 7, 28. [Google Scholar] [CrossRef] [PubMed]
- Pan, D. The hippo signaling pathway in development and cancer. Dev. Cell 2010, 19, 491–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matallanas, D.; Romano, D.; Hamilton, G.; Kolch, W.; O’Neill, E. A Hippo in the ointment: MST signalling beyond the fly. Cell Cycle 2008, 7, 879–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romano, D.; Matallanas, D.; Frederick, D.T.; Flaherty, K.T.; Kolch, W. One Hippo and many masters: Differential regulation of the Hippo pathway in cancer. Biochem. Soc. Trans. 2014, 42, 816–821. [Google Scholar] [CrossRef]
- Matallanas, D.; Romano, D.; Yee, K.; Meissl, K.; Kucerova, L.; Piazzolla, D.; Baccarini, M.; Vass, J.K.; Kolch, W.; O’Neill, E. RASSF1A elicits apoptosis through an MST2 pathway directing proapoptotic transcription by the p73 tumor suppressor protein. Mol. Cell 2007, 27, 962–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gundogdu, R.; Hergovich, A. MOB (Mps one Binder) Proteins in the Hippo Pathway and Cancer. Cells 2019, 8, 569. [Google Scholar] [CrossRef] [Green Version]
- Hong, L.; Cai, Y.; Jiang, M.; Zhou, D.; Chen, L. The Hippo signaling pathway in liver regeneration and tumorigenesis. Acta Biochim. Biophys. Sin. (Shanghai) 2015, 47, 46–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donninger, H.; Schmidt, M.L.; Mezzanotte, J.; Barnoud, T.; Clark, G.J. Ras signaling through RASSF proteins. Semin. Cell Dev. Biol. 2016, 58, 86–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Gutierrez, L.; McKenna, S.; Kolch, W.; Matallanas, D. RASSF1A Tumour Suppressor: Target the Network for Effective Cancer Therapy. Cancers 2020, 12, 229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matallanas, D.; Romano, D.; Al-Mulla, F.; O’Neill, E.; Al-Ali, W.; Crespo, P.; Doyle, B.; Nixon, C.; Sansom, O.; Drosten, M.; et al. Mutant K-Ras activation of the proapoptotic MST2 pathway is antagonized by wild-type K-Ras. Mol. Cell 2011, 44, 893–906. [Google Scholar] [CrossRef] [Green Version]
- Romano, D.; Maccario, H.; Doherty, C.; Quinn, N.P.; Kolch, W.; Matallanas, D. The differential effects of wild-type and mutated K-Ras on MST2 signaling are determined by K-Ras activation kinetics. Mol. Cell Biol. 2013, 33, 1859–1868. [Google Scholar] [CrossRef] [Green Version]
- Strano, S.; Monti, O.; Pediconi, N.; Baccarini, A.; Fontemaggi, G.; Lapi, E.; Mantovani, F.; Damalas, A.; Citro, G.; Sacchi, A.; et al. The transcriptional coactivator Yes-associated protein drives p73 gene-target specificity in response to DNA Damage. Mol. Cell 2005, 18, 447–459. [Google Scholar] [CrossRef]
- Oka, T.; Mazack, V.; Sudol, M. Mst2 and Lats Kinases Regulate Apoptotic Function of Yes Kinase-associated Protein (YAP). J. Biol. Chem. 2008, 283, 27534–27546. [Google Scholar] [CrossRef] [Green Version]
- Kwan, J.; Sczaniecka, A.; Arash, E.H.; Nguyen, L.; Chen, C.C.; Ratkovic, S.; Klezovitch, O.; Attisano, L.; McNeill, H.; Emili, A.; et al. DLG5 connects cell polarity and Hippo signaling protein networks by linking PAR-1 with MST1/2. Genes Dev. 2016, 30, 2696–2709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hergovich, A. MOB control: Reviewing a conserved family of kinase regulators. Cell Signal. 2011, 23, 1433–1440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hergovich, A. Mammalian Hippo signalling: A kinase network regulated by protein-protein interactions. Biochem. Soc. Trans. 2012, 40, 124–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, M.; Sharma, M.; Henderson, B.R. IQGAP1 regulation and roles in cancer. Cell Signal. 2009, 21, 1471–1478. [Google Scholar] [CrossRef] [PubMed]
- Malarkannan, S.; Awasthi, A.; Rajasekaran, K.; Kumar, P.; Schuldt, K.M.; Bartoszek, A.; Manoharan, N.; Goldner, N.K.; Umhoefer, C.M.; Thakar, M.S. IQGAP1: A regulator of intracellular spacetime relativity. J. Immunol. 2012, 188, 2057–2063. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.M.; Hedman, A.C.; Sacks, D.B. IQGAPs choreograph cellular signaling from the membrane to the nucleus. Trends Cell Biol. 2015, 25, 171–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayedyahossein, S.; Li, Z.; Hedman, A.C.; Morgan, C.J.; Sacks, D.B. IQGAP1 Binds to Yes-associated Protein (YAP) and Modulates Its Transcriptional Activity. J. Biol. Chem. 2016, 291, 19261–19273. [Google Scholar] [CrossRef] [Green Version]
- Anakk, S.; Bhosale, M.; Schmidt, V.A.; Johnson, R.L.; Finegold, M.J.; Moore, D.D. Bile acids activate YAP to promote liver carcinogenesis. Cell Rep. 2013, 5, 1060–1069. [Google Scholar] [CrossRef] [Green Version]
- Gulfo, J.; Rotondo, F.; de Leon, C.G.A.; Cornide-Petronio, M.E.; Fuster, C.; Gracia-Sancho, J.; Jimenez-Castro, M.B.; Peralta, C. FGF15 improves outcomes after brain dead donor liver transplantation with steatotic and non-steatotic grafts in rats. J. Hepatol. 2020, 5, 1131–1143. [Google Scholar] [CrossRef]
- Ji, S.; Liu, Q.; Zhang, S.; Chen, Q.; Wang, C.; Zhang, W.; Xiao, C.; Li, Y.; Nian, C.; Li, J.; et al. FGF15 Activates Hippo Signaling to Suppress Bile Acid Metabolism and Liver Tumorigenesis. Dev. Cell 2019, 48, 460–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Wei, L.; Fan, F.; Ji, S.; Zhang, S.; Geng, J.; Hong, L.; Fan, X.; Chen, Q.; Tian, J.; et al. Integration of Hippo signalling and the unfolded protein response to restrain liver overgrowth and tumorigenesis. Nat. Commun. 2015, 6, 6239. [Google Scholar] [CrossRef] [Green Version]
- Roy, M.; Li, Z.; Sacks, D.B. IQGAP1 is a scaffold for mitogen-activated protein kinase signaling. Mol. Cell Biol. 2005, 25, 7940–7952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romano, D.; Matallanas, D.; Weitsman, G.; Preisinger, C.; Ng, T.; Kolch, W. Proapoptotic kinase MST2 coordinates signaling crosstalk between RASSF1A, Raf-1, and Akt. Cancer Res. 2010, 70, 1195–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, Y.; Chun, A.; Cheung, K.; Rashidi, B.; Yang, X. Tumor suppressor LATS1 is a negative regulator of oncogene YAP. J. Biol. Chem. 2008, 283, 5496–5509. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Zhang, L.; Hwang, P.M.; Kinzler, K.W.; Vogelstein, B. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol. Cell 2001, 7, 673–682. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Papaspyropoulos, A.; Bradley, L.; Thapa, A.; Leung, C.Y.; Toskas, K.; Koennig, D.; Pefani, D.E.; Raso, C.; Grou, C.; Hamilton, G.; et al. RASSF1A uncouples Wnt from Hippo signalling and promotes YAP mediated differentiation via p73. Nat. Commun. 2018, 9, 424. [Google Scholar] [CrossRef]
- Sun, C.; De Mello, V.; Mohamed, A.; Ortuste Quiroga, H.P.; Garcia-Munoz, A.; Al Bloshi, A.; Tremblay, A.M.; von Kriegsheim, A.; Collie-Duguid, E.; Vargesson, N.; et al. Common and Distinctive Functions of the Hippo Effectors Taz and Yap in Skeletal Muscle Stem Cell Function. Stem Cells 2017, 35, 1958–1972. [Google Scholar] [CrossRef] [Green Version]
- Zeke, A.; Lukacs, M.; Lim, W.A.; Remenyi, A. Scaffolds: Interaction platforms for cellular signalling circuits. Trends Cell Biol. 2009, 19, 364–374. [Google Scholar] [CrossRef] [Green Version]
- Good, M.C.; Zalatan, J.G.; Lim, W.A. Scaffold proteins: Hubs for controlling the flow of cellular information. Science 2011, 332, 680–686. [Google Scholar] [CrossRef] [Green Version]
- Roy, M.; Li, Z.; Sacks, D.B. IQGAP1 binds ERK2 and modulates its activity. J. Biol. Chem. 2004, 279, 17329–17337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, H.; Qi, H.; Li, Y.; Pei, J.; Barton, J.; Blackstad, M.; Xu, T.; Tao, W. LATS1 tumor suppressor regulates G2/M transition and apoptosis. Oncogene 2002, 21, 1233–1241. [Google Scholar] [CrossRef] [Green Version]
- Xia, F.D.; Wang, Z.L.; Chen, H.X.; Huang, Y.; Li, J.D.; Wang, Z.M.; Li, X.Y. Differential expression of IQGAP1/2 in Hepatocellular carcinoma and its relationship with clinical outcomes. Asian Pac. J. Cancer Prev. 2014, 15, 4951–4956. [Google Scholar] [CrossRef] [Green Version]
- Miao, J.; Xiao, Z.; Kanamaluru, D.; Min, G.; Yau, P.M.; Veenstra, T.D.; Ellis, E.; Strom, S.; Suino-Powell, K.; Xu, H.E.; et al. Bile acid signaling pathways increase stability of Small Heterodimer Partner (SHP) by inhibiting ubiquitin-proteasomal degradation. Genes Dev. 2009, 23, 986–996. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Lei, Q.Y.; Guan, K.L. The Hippo-YAP pathway: New connections between regulation of organ size and cancer. Curr. Opin. Cell Biol. 2008, 20, 638–646. [Google Scholar] [CrossRef] [Green Version]
- Levchenko, A.; Bruck, J.; Sternberg, P.W. Scaffold proteins may biphasically affect the levels of mitogen-activated protein kinase signaling and reduce its threshold properties. Proc. Natl. Acad. Sci. USA 2000, 97, 5818–5823. [Google Scholar] [CrossRef] [Green Version]
- Kolch, W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat. Rev. Mol. Cell Biol. 2005, 6, 827–837. [Google Scholar] [CrossRef] [PubMed]
- Romano, D.; Nguyen, L.K.; Matallanas, D.; Halasz, M.; Doherty, C.; Kholodenko, B.N.; Kolch, W. Protein interaction switches coordinate Raf-1 and MST2/Hippo signalling. Nat. Cell Biol. 2014, 16, 673–684. [Google Scholar] [CrossRef]
- O’Neill, E.; Rushworth, L.; Baccarini, M.; Kolch, W. Role of the kinase MST2 in suppression of apoptosis by the proto-oncogene product Raf-1. Science 2004, 306, 2267–2270. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.; Kim, W.; Kim, S.; Kim, Y.; Song, Y.; Bilousov, O.; Kim, J.; Lee, T.; Cha, B.; Kim, M.; et al. Phosphorylation by NLK inhibits YAP-14-3-3-interactions and induces its nuclear localization. EMBO Rep. 2017, 18, 61–71. [Google Scholar] [CrossRef]
- Hong, A.W.; Meng, Z.; Yuan, H.X.; Plouffe, S.W.; Moon, S.; Kim, W.; Jho, E.H.; Guan, K.L. Osmotic stress-induced phosphorylation by NLK at Ser128 activates YAP. EMBO Rep. 2017, 18, 72–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, F.; Zhu, H.H.; Zhou, L.F.; Wu, S.S.; Wang, J.; Chen, Z. IQGAP1 is overexpressed in hepatocellular carcinoma and promotes cell proliferation by Akt activation. Exp. Mol. Med. 2010, 42, 477–483. [Google Scholar] [CrossRef]
- Song, H.; Mak, K.K.; Topol, L.; Yun, K.; Hu, J.; Garrett, L.; Chen, Y.; Park, O.; Chang, J.; Simpson, R.M.; et al. Mammalian Mst1 and Mst2 kinases play essential roles in organ size control and tumor suppression. Proc. Natl. Acad. Sci. USA 2010, 107, 1431–1436. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, V.A.; Chiariello, C.S.; Capilla, E.; Miller, F.; Bahou, W.F. Development of hepatocellular carcinoma in Iqgap2-deficient mice is IQGAP1 dependent. Mol. Cell Biol. 2008, 28, 1489–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, C.D.; Khurana, H.; Gnatenko, D.V.; Li, Z.; Odze, R.D.; Sacks, D.B.; Schmidt, V.A. IQGAP1 and IQGAP2 are reciprocally altered in hepatocellular carcinoma. BMC Gastroenterol. 2010, 10, 125. [Google Scholar] [CrossRef] [Green Version]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quinn, N.P.; García-Gutiérrez, L.; Doherty, C.; von Kriegsheim, A.; Fallahi, E.; Sacks, D.B.; Matallanas, D. IQGAP1 Is a Scaffold of the Core Proteins of the Hippo Pathway and Negatively Regulates the Pro-Apoptotic Signal Mediated by This Pathway. Cells 2021, 10, 478. https://doi.org/10.3390/cells10020478
Quinn NP, García-Gutiérrez L, Doherty C, von Kriegsheim A, Fallahi E, Sacks DB, Matallanas D. IQGAP1 Is a Scaffold of the Core Proteins of the Hippo Pathway and Negatively Regulates the Pro-Apoptotic Signal Mediated by This Pathway. Cells. 2021; 10(2):478. https://doi.org/10.3390/cells10020478
Chicago/Turabian StyleQuinn, Niall P., Lucía García-Gutiérrez, Carolanne Doherty, Alexander von Kriegsheim, Emma Fallahi, David B. Sacks, and David Matallanas. 2021. "IQGAP1 Is a Scaffold of the Core Proteins of the Hippo Pathway and Negatively Regulates the Pro-Apoptotic Signal Mediated by This Pathway" Cells 10, no. 2: 478. https://doi.org/10.3390/cells10020478
APA StyleQuinn, N. P., García-Gutiérrez, L., Doherty, C., von Kriegsheim, A., Fallahi, E., Sacks, D. B., & Matallanas, D. (2021). IQGAP1 Is a Scaffold of the Core Proteins of the Hippo Pathway and Negatively Regulates the Pro-Apoptotic Signal Mediated by This Pathway. Cells, 10(2), 478. https://doi.org/10.3390/cells10020478