
Muscle Enriched Lamin Interacting Protein (Mlip) Binds Chromatin and Is Required for Myoblast Differentiation


Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
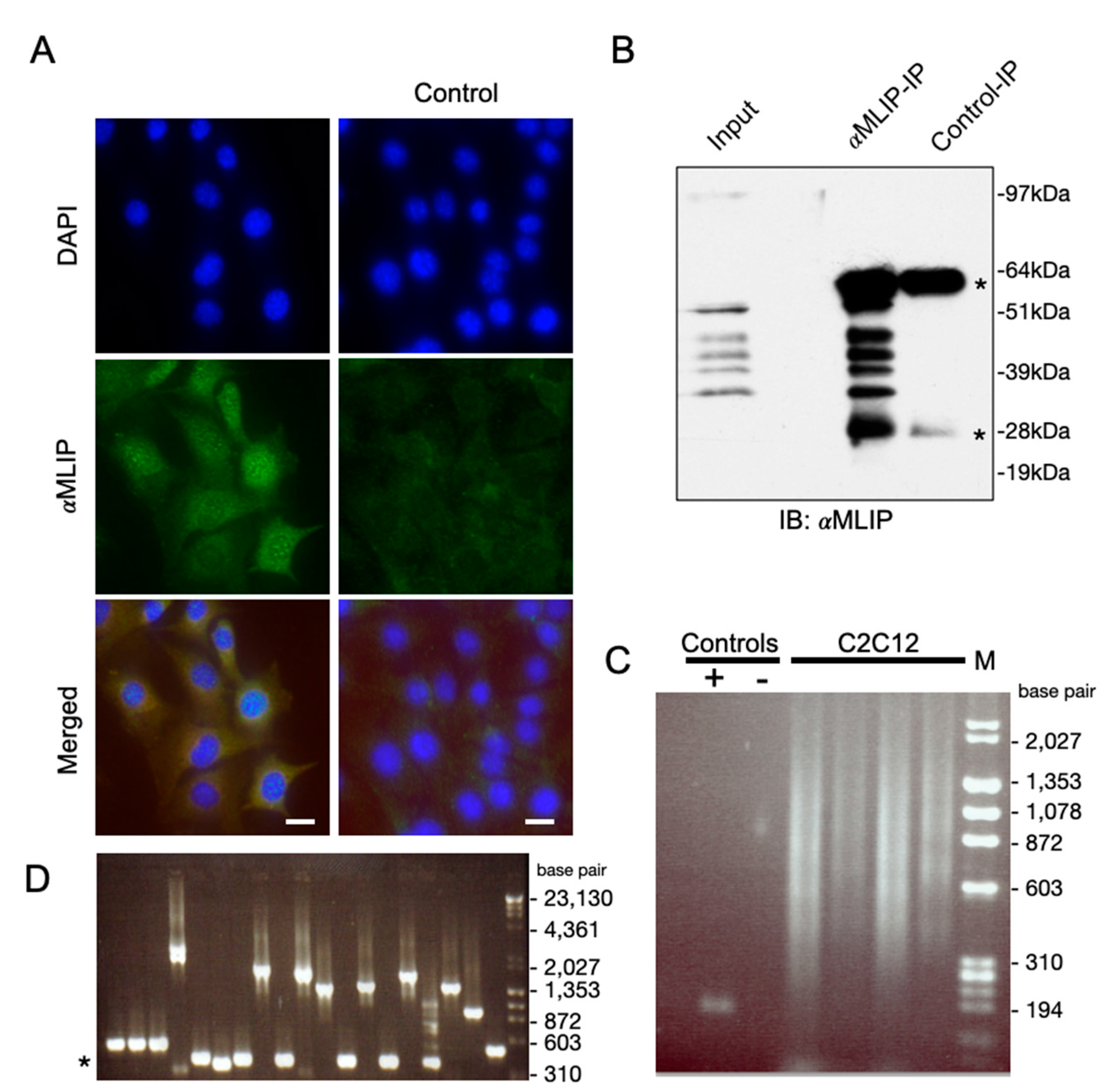
2.2. Whole Cell Extract Preparation for Immunoprecipitation and Western Blotting
2.3. Immunofluorescence
2.4. Chromatin Immunoprecipitation and ChIP-on-Chip Studies
2.5. Nuclear Extracts and Electrophoretic Mobility Shift Assays (EMSA)
2.6. Reporter Assay
2.7. Transfection and Generation of Stable Mlip Knockdown C2C12 Cell Lines
3. Results
3.1. Mlip Interacts with Chromatin in Areas of Close Proximity to Developmental Genes
3.2. Mlip Contains a Transcriptional Activation Domain
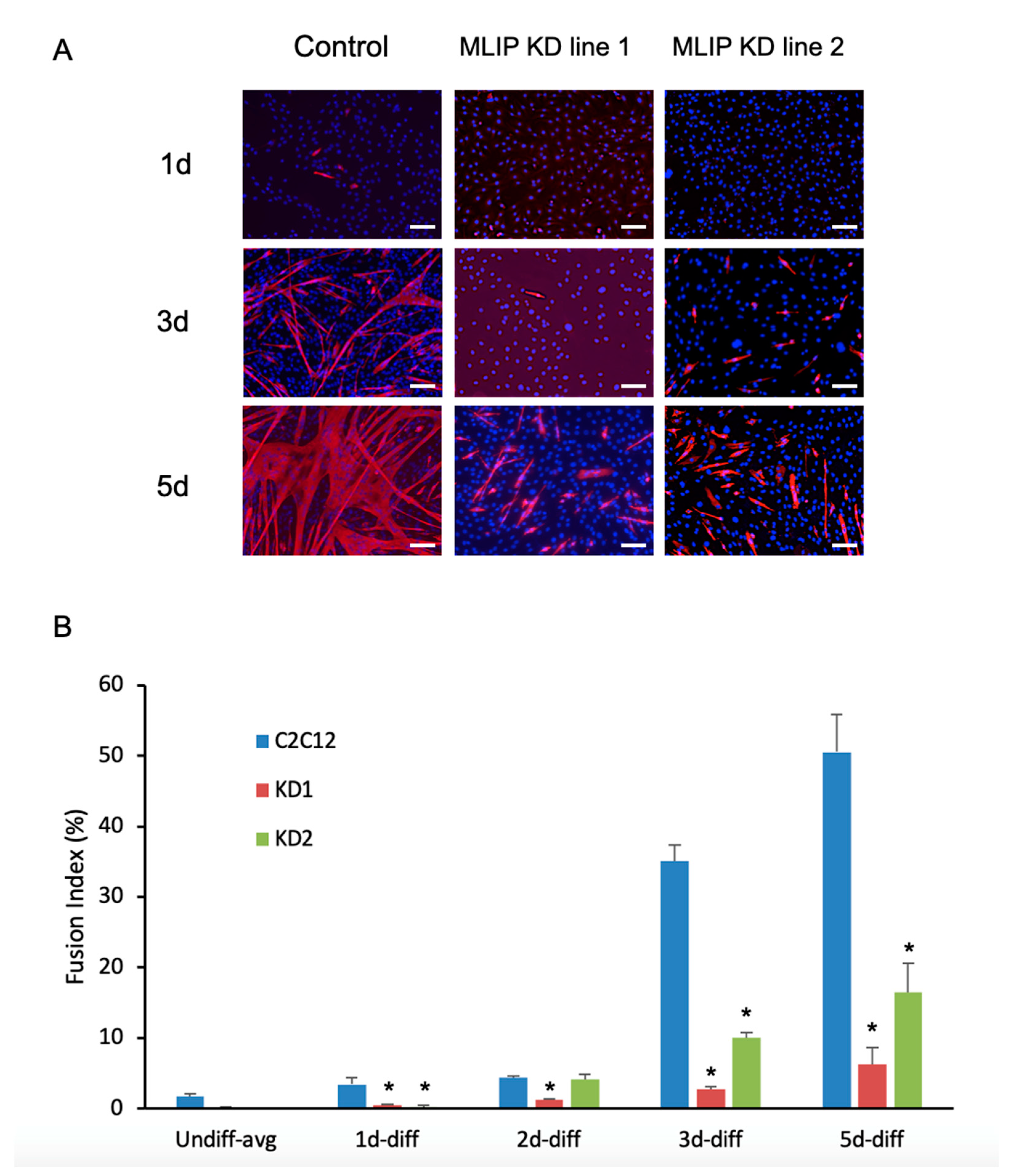
3.3. Impaired Myotube Formation in MLIP-Depleted C2C12 Myoblasts
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Broers, J.L.V.; Ramaekers, F.C.S.; Bonne, G.; Yaou, R.B.; Hutchison, C.J. Nuclear lamins: Laminopathies and their role in premature ageing. Physiol. Rev. 2006, 86, 967–1008. [Google Scholar] [CrossRef]
- Capell, B.C.; Collins, F.S. Human laminopathies: Nuclei gone genetically awry. Nat. Rev. Genet. 2006, 7, 940–952. [Google Scholar] [CrossRef]
- Scaffidi, P.; Misteli, T. Lamin A-dependent nuclear defects in human aging. Science 2006, 312, 1059–1063. [Google Scholar] [CrossRef] [Green Version]
- Worman, H.J.; Courvalin, J.-C. How do mutations in lamins A and C cause disease? J. Clin. Invest. 2004, 113, 349–351. [Google Scholar] [CrossRef] [PubMed]
- Ahmady, E.; Deeke, S.A.; Rabaa, S.; Kouri, L.; Kenney, L.; Stewart, A.F.R.; Burgon, P.G. Identification of a novel muscle A-type lamin-interacting protein (MLIP). J. Biol. Chem. 2011, 286, 19702–19713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cattin, M.-E.; Deeke, S.A.; Dick, S.A.; Verret-Borsos, Z.J.A.; Tennakoon, G.; Gupta, R.; Mak, E.; Roeske, C.L.; Weldrick, J.J.; Megeney, L.A.; et al. Expression of murine muscle-enriched A-type lamin-interacting protein (MLIP) is regulated by tissue-specific alternative transcription start sites. J. Biol. Chem. 2018, 293, 19761–19770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santini, M.P.; Forte, E.; Harvey, R.P.; Kovacic, J.C. Developmental origin and lineage plasticity of endogenous cardiac stem cells. Development 2016, 143, 1242–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moretti, A.; Lam, J.; Evans, S.M.; Laugwitz, K.-L. Biology of Isl1+ cardiac progenitor cells in development and disease. Cell. Mol. Life Sci. 2007, 64. [Google Scholar] [CrossRef]
- Huang, Z.-P.; Young Seok, H.; Zhou, B.; Chen, J.J.-F.; Chen, J.J.-F.; Tao, Y.; Pu, W.T.; Wang, D.-Z. CIP, a cardiac Isl1-interacting protein, represses cardiomyocyte hypertrophy. Circ. Res. 2012, 110, 818–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cattin, M.-E.; Wang, J.; Weldrick, J.J.; Roeske, C.L.; Mak, E.; Thorn, S.L.; DaSilva, J.N.; Wang, Y.; Lusis, A.J.; Burgon, P.G. Deletion of MLIP (Muscle-enriched A-type Lamin-interacting Protein) Leads to Cardiac Hyperactivation of Akt/Mammalian Target of Rapamycin (mTOR) and Impaired Cardiac Adaptation. J. Biol. Chem. 2015, 290, 26699–26714. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.-P.; Kataoka, M.; Chen, J.; Wu, G.; Ding, J.; Nie, M.; Lin, Z.; Liu, J.; Hu, X.; Ma, L.; et al. Cardiomyocyte-enriched protein CIP protects against pathophysiological stresses and regulates cardiac homeostasis. J. Clin. Invest. 2015, 125, 4122–4134. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Huang, Z.P.; Nie, M.; Wang, G.; Silva, W.J.; Yang, Q.; Freire, P.P.; Hu, X.; Chen, H.; Deng, Z.; et al. Regulation of myonuclear positioning and muscle function by the skeletal muscle-specific CIP protein. Proc. Natl. Acad. Sci. USA 2020, 117, 19254–19265. [Google Scholar] [CrossRef]
- Liu, Y.; Chu, A.; Chakroun, I.; Islam, U.; Blais, A. Cooperation between myogenic regulatory factors and SIX family transcription factors is important for myoblast differentiation. Nucleic Acids Res. 2010, 38, 6857–6871. [Google Scholar] [CrossRef] [Green Version]
- Farrance, I.K.; Ordahl, C.P. The role of transcription enhancer factor-1 (TEF-1) related proteins in the formation of M-CAT binding complexes in muscle and non-muscle tissues. J. Biol. Chem. 1996, 271, 8266–8274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueyama, T.; Zhu, C.; Valenzuela, Y.M.; Suzow, J.G.; Stewart, A.F.R. Identification of the Functional Domain in the Transcription Factor RTEF-1 That Mediates 1-Adrenergic Signaling in Hypertrophied Cardiac Myocytes. J. Biol. Chem. 2000, 275, 17476–17480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dechat, T.; Pfleghaar, K.; Sengupta, K.; Shimi, T.; Shumaker, D.K.; Solimando, L.; Goldman, R.D. Nuclear lamins: Major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 2008, 22, 832–853. [Google Scholar] [CrossRef] [Green Version]
- Kandert, S.; Wehnert, M.; Müller, C.R.; Buendia, B.; Dabauvalle, M.-C. Impaired nuclear functions lead to increased senescence and inefficient differentiation in human myoblasts with a dominant p.R545C mutation in the LMNA gene. Eur. J. Cell Biol. 2009, 88, 593–608. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.Y.; Gomez-Sanchez, C.E. Universal TA cloning. Curr. Issues Mol. Biol. 2000, 2, 1–7. [Google Scholar] [PubMed]
- Dennis, G.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4, P3. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2008, 4, 44–57. [Google Scholar] [CrossRef]
- Rubinson, D.A.; Dillon, C.P.; Kwiatkowski, A.V.; Sievers, C.; Yang, L.; Kopinja, J.; Zhang, M.; McManus, M.T.; Gertler, F.B.; Scott, M.L.; et al. A lentivirus-based system to functionally silence genes in primary mammalian cells, stem cells and transgenic mice by RNA interference. Nat. Genet. 2003, 33. [Google Scholar] [CrossRef] [PubMed]
- Kesireddy, V.; Van Der Ven, P.F.M.; Fürst, D.O. Multipurpose modular lentiviral vectors for RNA interference and transgene expression. Mol. Biol. Rep. 2010, 37. [Google Scholar] [CrossRef] [PubMed]
- Rossi, F.; Charlton, C.A.; Blau, H.M. Monitoring protein-protein interactions in intact eukaryotic cells by beta-galactosidase complementation. Proc. Natl. Acad. Sci. USA 1997, 94, 8405–8410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakroun, I.; Yang, D.; Girgis, J.; Gunasekharan, A.; Phenix, H.; Kærn, M.; Blais, A. Genome-wide association between Six4, MyoD, and the histone demethylase Utx during myogenesis. FASEB J. 2015, 29, 4738–4755. [Google Scholar] [CrossRef]
- Liu, Y.; Chakroun, I.; Yang, D.; Horner, E.; Liang, J.; Aziz, A.; Chu, A.; De Repentigny, Y.; Dilworth, F.J.; Kothary, R.; et al. Six1 regulates MyoD expression in adult muscle progenitor cells. PLoS ONE 2013, 8, e67762. [Google Scholar] [CrossRef] [Green Version]
- Giordani, J.; Bajard, L.; Demignon, J.; Daubas, P.; Buckingham, M.; Maire, P. Six proteins regulate the activation of Myf5 expression in embryonic mouse limbs. Proc. Natl. Acad. Sci. USA 2007, 104, 11310–11315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grifone, R.; Demignon, J.; Houbron, C.; Souil, E.; Niro, C.; Seller, M.J.; Hamard, G.; Maire, P. Six1 and Six4 homeoproteins are required for Pax3 and Mrf expression during myogenesis in the mouse embryo. Development 2005, 132, 2235–2249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blau, H.M.; Chiu, C.P.; Webster, C. Cytoplasmic activation of human nuclear genes in stable heterocaryons. Cell 1983, 32, 1171–1180. [Google Scholar] [CrossRef]
- Schneider, M.D.; Olson, E.N. Control of myogenic differentiation by cellular oncogenes. Mol. Neurobiol. 1988, 2, 1–39. [Google Scholar] [CrossRef]
- Olson, E.N. Proto-oncogenes in the regulatory circuit for myogenesis. Semin. Cell Biol. 1992, 3, 127–136. [Google Scholar] [CrossRef]
- Glass, C.A.; Glass, J.R.; Taniura, H.; Hasel, K.W.; Blevitt, J.M.; Gerace, L. The alpha-helical rod domain of human lamins A and C contains a chromatin binding site. EMBO J. 1993, 12, 4413–4424. [Google Scholar] [CrossRef] [PubMed]
- Taniura, H.; Glass, C.; Gerace, L. A chromatin binding site in the tail domain of nuclear lamins that interacts with core histones. J. Cell Biol. 1995, 131, 33–44. [Google Scholar] [CrossRef] [Green Version]
- Markiewicz, E.; Dechat, T.; Foisner, R.; Quinlan, R.A.; Hutchison, C.J. Lamin A/C Binding Protein LAP2alpha Is Required for Nuclear Anchorage of Retinoblastoma Protein. Mol. Biol. Cell 2002, 13, 4401–4413. [Google Scholar] [CrossRef] [Green Version]
- Ozaki, T.; Saijo, M.; Murakami, K.; Enomoto, H.; Taya, Y.; Sakiyama, S. Complex formation between lamin A and the retinoblastoma gene product: Identification of the domain on lamin A required for its interaction. Oncogene 1994, 9, 2649–2653. [Google Scholar]
- Lloyd, D.J.; Trembath, R.C.; Shackleton, S. A novel interaction between lamin A and SREBP1: Implications for partial lipodystrophy and other laminopathies. Hum. Mol. Genet. 2002, 11, 769–777. [Google Scholar] [CrossRef] [Green Version]
- Dreuillet, C.; Tillit, J.; Kress, M.; Ernoult-Lange, M. In vivo and in vitro interaction between human transcription factor MOK2 and nuclear lamin A/C. Nucleic Acids Res. 2002, 30, 4634–4642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laugwitz, K.-L.; Moretti, A.; Lam, J.; Gruber, P.; Chen, Y.; Woodard, S.; Lin, L.-Z.; Cai, C.-L.; Lu, M.M.; Reth, M.; et al. Postnatal isl1+ cardioblasts enter fully differentiated cardiomyocyte lineages. Nature 2005, 433, 647–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Röber, R.A.; Weber, K.; Osborn, M. Differential timing of nuclear lamin A/C expression in the various organs of the mouse embryo and the young animal: A developmental study. Development 1989, 105, 365–378. [Google Scholar] [PubMed]
- Watkins, S.C.; Culien, M.J. A quantitative study of myonuclear and satellite cell nuclear size in Duchenne’s muscular dystrophy, polymyositis and normal human skeletal muscle. Anat. Rec. 1988, 222, 6–11. [Google Scholar] [CrossRef]
- Haslett, J.N.; Sanoudou, D.; Kho, A.T.; Han, M.; Bennett, R.R.; Kohane, I.S.; Beggs, A.H.; Kunkel, L.M. Gene expression profiling of Duchenne muscular dystrophy skeletal muscle. Neurogenetics 2003, 4, 163–171. [Google Scholar] [CrossRef]
- Decostre, V.; Ben Yaou, R.; Bonne, G. Laminopathies affecting skeletal and cardiac muscles: Clinical and pathophysiological aspects. Acta Myol. 2005, 24, 104–109. [Google Scholar]
- Worman, H.J.; Bonne, G. “Laminopathies”: A wide spectrum of human diseases. Exp. Cell Res. 2007, 313, 2121–2133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertrand, A.T.; Chikhaoui, K.; Yaou, R.B.; Bonne, G. Clinical and genetic heterogeneity in laminopathies. Biochem. Soc. Trans. 2011, 39, 1687–1692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakay, M.; Wang, Z.; Melcon, G.; Schiltz, L.; Xuan, J.; Zhao, P.; Sartorelli, V.; Seo, J.; Pegoraro, E.; Angelini, C.; et al. Nuclear envelope dystrophies show a transcriptional fingerprint suggesting disruption of Rb-MyoD pathways in muscle regeneration. Brain 2006, 129, 996–1013. [Google Scholar] [CrossRef] [PubMed]
- Frock, R.L.; Kudlow, B.A.; Evans, A.M.; Jameson, S.A.; Hauschka, S.D.; Kennedy, B.K. Lamin A/C and emerin are critical for skeletal muscle satellite cell differentiation. Genes Dev. 2006, 20, 486–500. [Google Scholar] [CrossRef] [Green Version]
- Melcon, G.; Kozlov, S.; Cutler, D.A.; Sullivan, T.; Hernandez, L.; Zhao, P.; Mitchell, S.; Nader, G.; Bakay, M.; Rottman, J.N.; et al. Loss of emerin at the nuclear envelope disrupts the Rb1/E2F and MyoD pathways during muscle regeneration. Hum. Mol. Genet. 2006, 15, 637–651. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene. | Differentiation | Commitment | Apoptosis | Proliferation | Survival | Growth |
|---|---|---|---|---|---|---|
| Akt2 | x | x | x | x | x | |
| CREM | x | |||||
| SOX5 | x | x | ||||
| PEL1 | ||||||
| KIF5C | x | |||||
| PLC1 | x | x | x | |||
| Met | x | x | x | |||
| MMP3 | x | x | x | |||
| Runx | x | x | x | x | ||
| Nek7 | ||||||
| FLI1 | x | x | x | x | ||
| PP2R3A | x | |||||
| Notch2 | x | x | x | x | x | x |
| Mlip Enriched Chr 11 Genes | All Chr 11 Genes | |
|---|---|---|
| Gene Ontology Term (Biological Process) | p-Value | p-Value |
| 0007275~multicellular organismal development | 0.00004415 | 0.01820829 |
| 0032502~developmental process | 0.00099698 | 0.03170131 |
| 0009987~cellular process | 0.00146013 | 0.00000553 |
| 0016043~cellular component organization & biogenesis | 0.00175770 | 0.00000001 |
| 0048731~system development | 0.00243918 | 0.04057584 |
| 0006665~sphingolipid metabolic process | 0.00257593 | ND |
| 0048856~anatomical structure development | 0.00297968 | 0.02503862 |
| 0006886~intracellular protein transport | 0.00359201 | 0.00028535 |
| 0048513~organ development | 0.00386375 | ND |
| 0043407~negative regulation of MAP kinase activity | 0.00476669 | 0.03816350 |
| 0009888~tissue development | 0.00621520 | ND |
| 0006629~lipid metabolic process | 0.00952032 | ND |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmady, E.; Blais, A.; Burgon, P.G. Muscle Enriched Lamin Interacting Protein (Mlip) Binds Chromatin and Is Required for Myoblast Differentiation. Cells 2021, 10, 615. https://doi.org/10.3390/cells10030615
Ahmady E, Blais A, Burgon PG. Muscle Enriched Lamin Interacting Protein (Mlip) Binds Chromatin and Is Required for Myoblast Differentiation. Cells. 2021; 10(3):615. https://doi.org/10.3390/cells10030615
Chicago/Turabian StyleAhmady, Elmira, Alexandre Blais, and Patrick G. Burgon. 2021. "Muscle Enriched Lamin Interacting Protein (Mlip) Binds Chromatin and Is Required for Myoblast Differentiation" Cells 10, no. 3: 615. https://doi.org/10.3390/cells10030615
APA StyleAhmady, E., Blais, A., & Burgon, P. G. (2021). Muscle Enriched Lamin Interacting Protein (Mlip) Binds Chromatin and Is Required for Myoblast Differentiation. Cells, 10(3), 615. https://doi.org/10.3390/cells10030615