
The G-Protein-Coupled Estrogen Receptor (GPER) Regulates Trimethylation of Histone H3 at Lysine 4 and Represses Migration and Proliferation of Ovarian Cancer Cells In Vitro


 , ,
, ,
Abstract
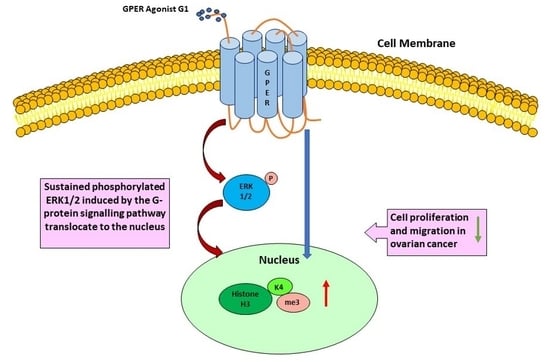
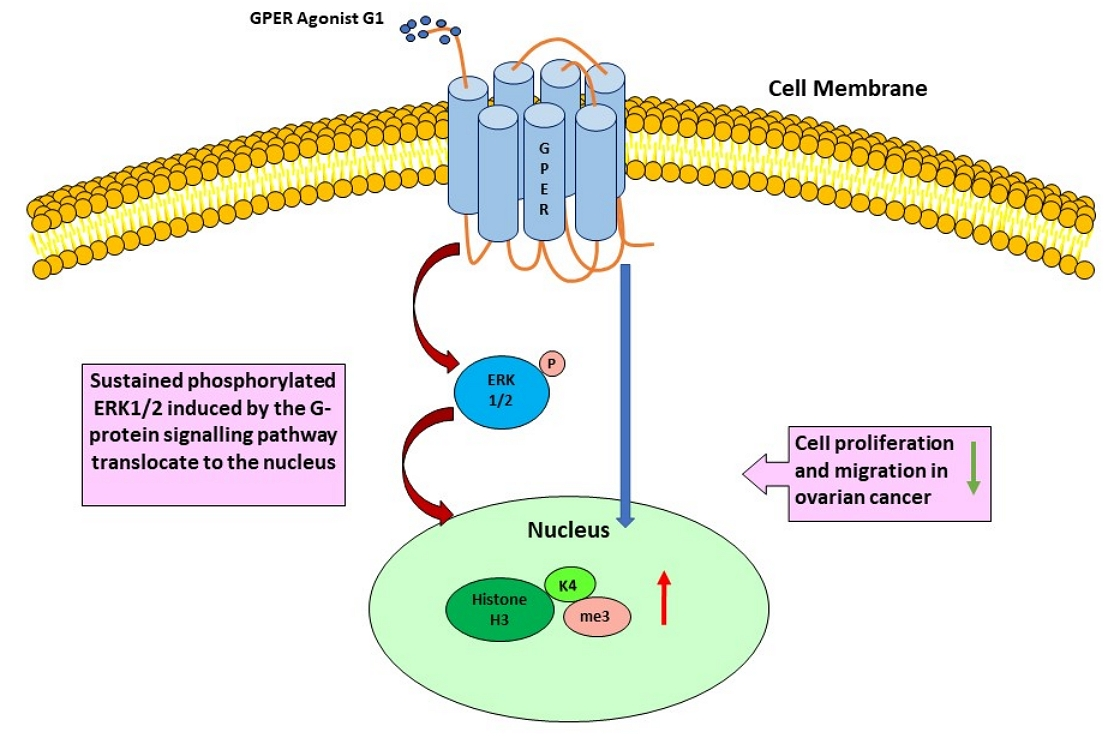
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Ethics Approval
2.2. Patients and Tissue Microarray
2.3. Immunohistochemistry
2.4. Chemicals and Cell Culture
2.5. MTT Assay
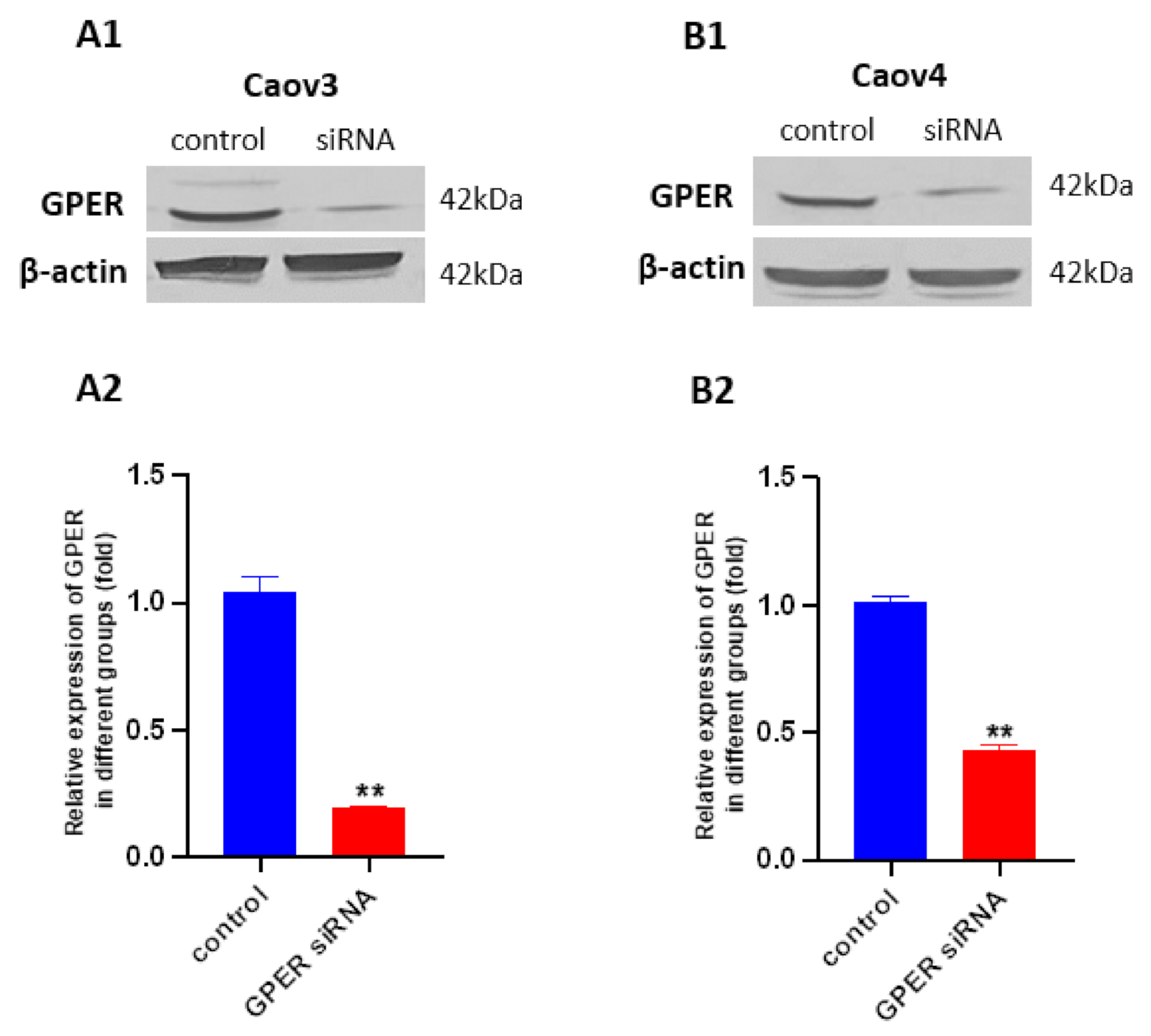
2.6. siRNA Transfection
2.7. Preparation of Cell Lysates and Western Blot
2.8. Wound Healing (Scratch) Assay
2.9. BrdU Assay
2.10. Statistical Analysis
3. Results
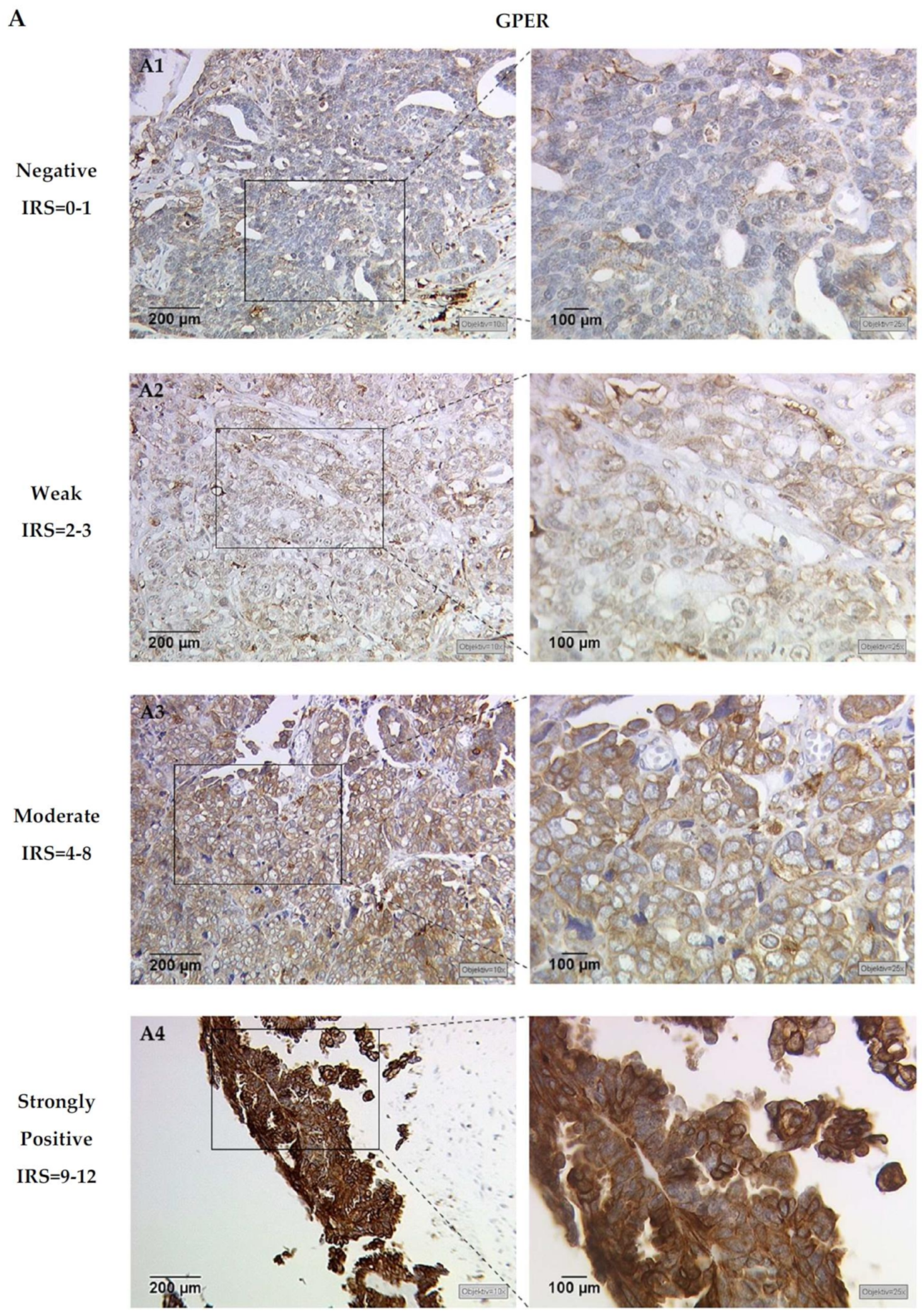
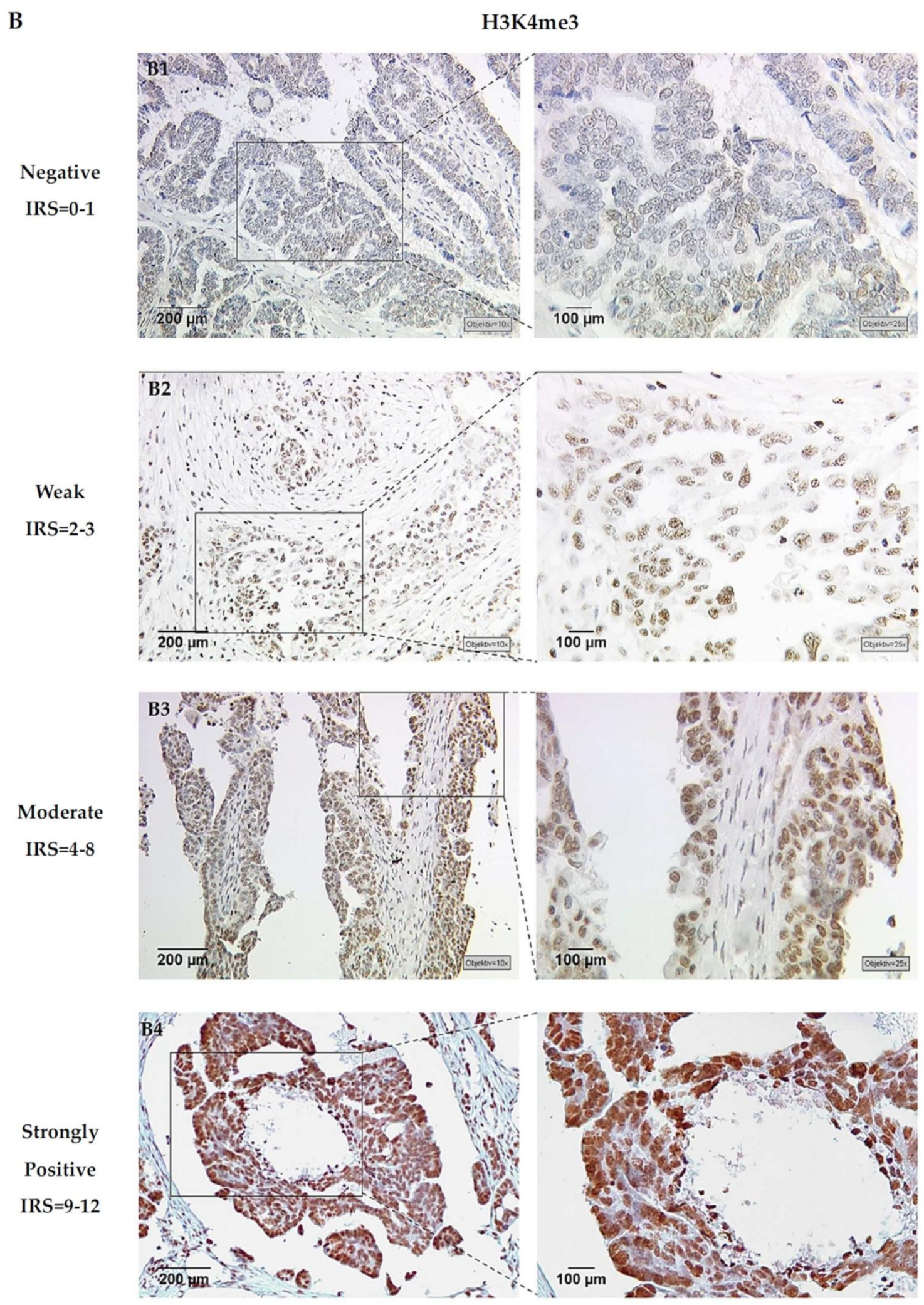
3.1. H3K4me3 Correlates with GPER Expression in EOC
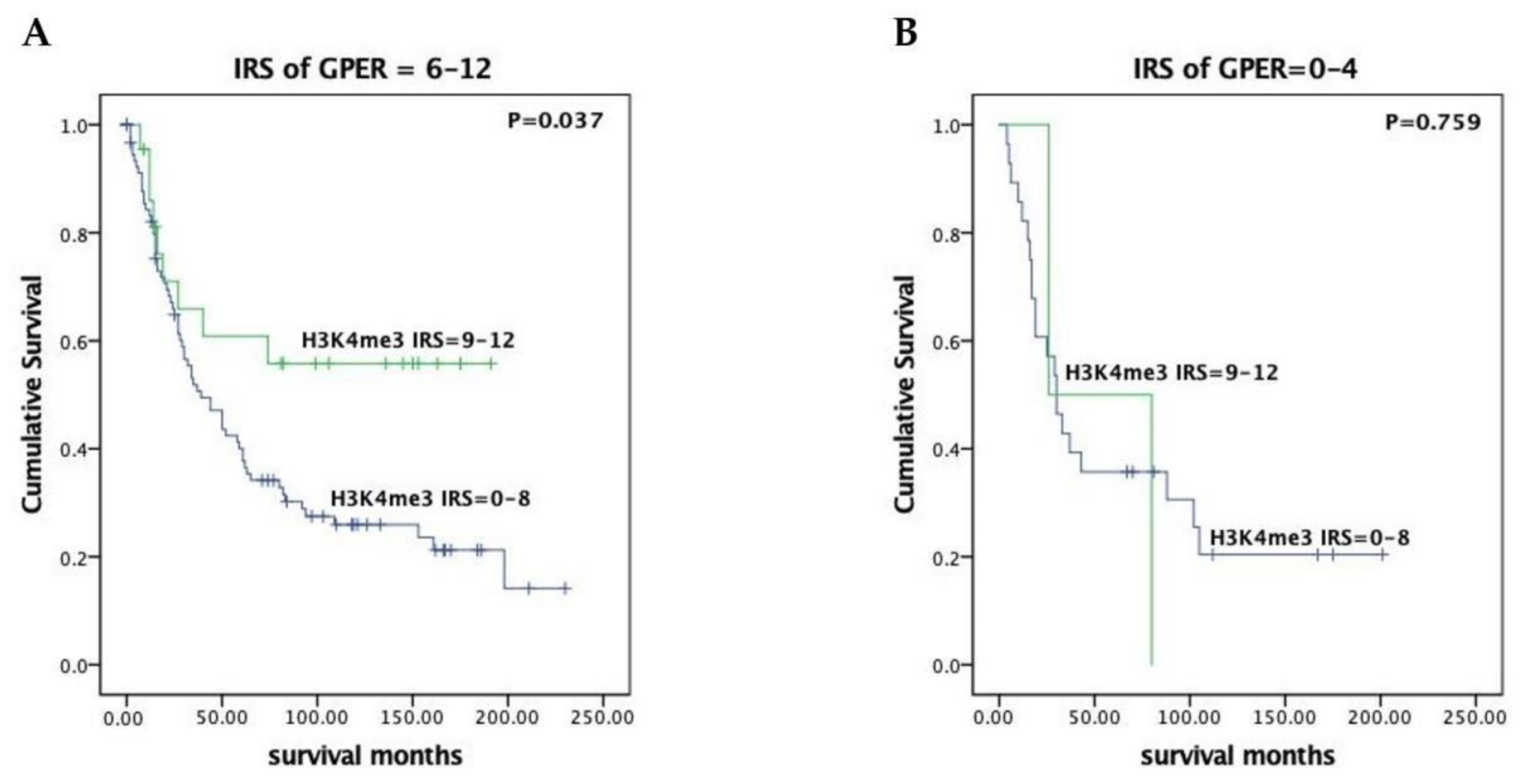
3.2. H3K4me3 Expression Has Prognostic Impact in GPER Positive EOC Patients
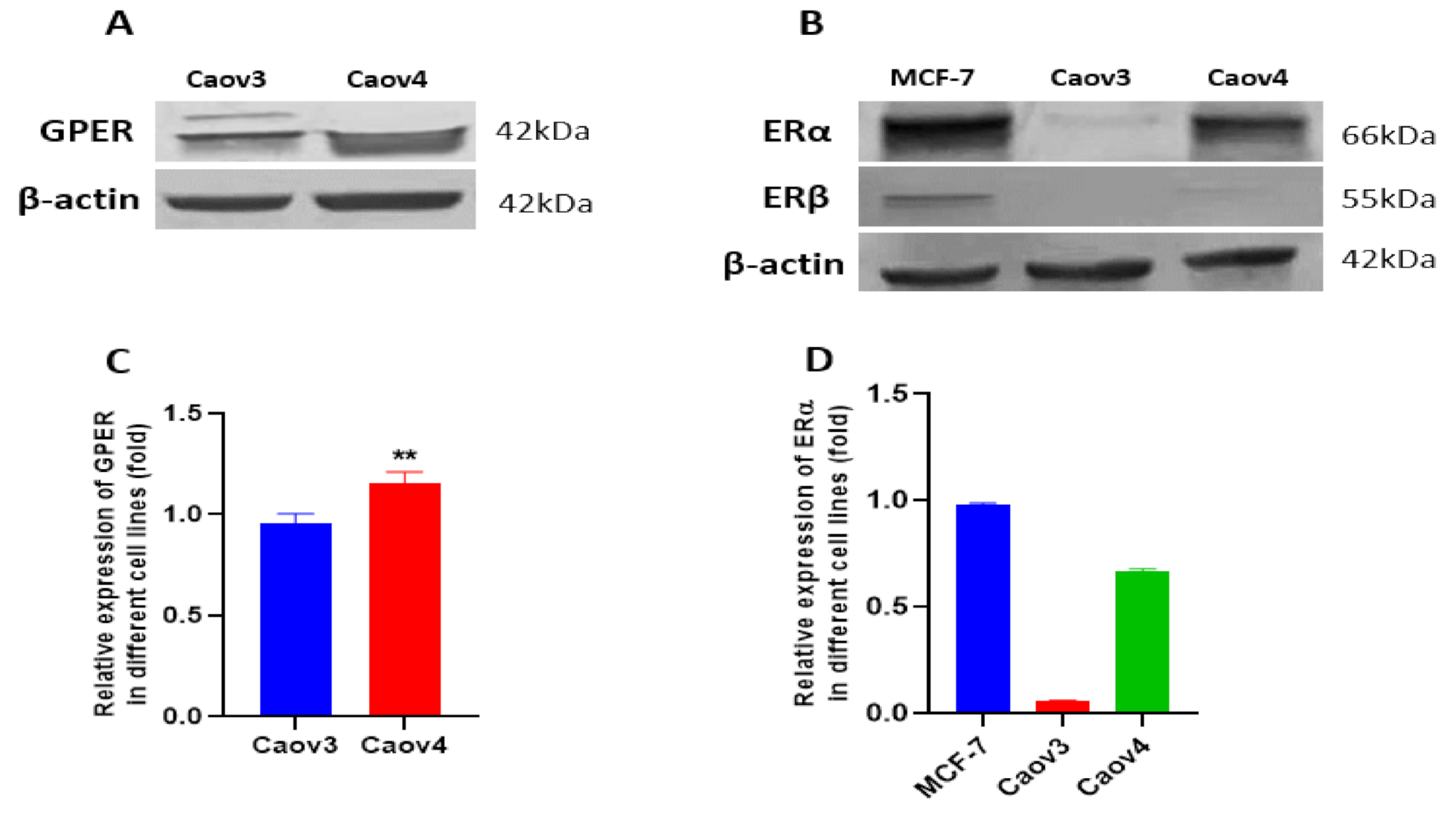
3.3. Expressions of GPER, ERα and ERβ in Caov3 and Caov4 Cell Lines
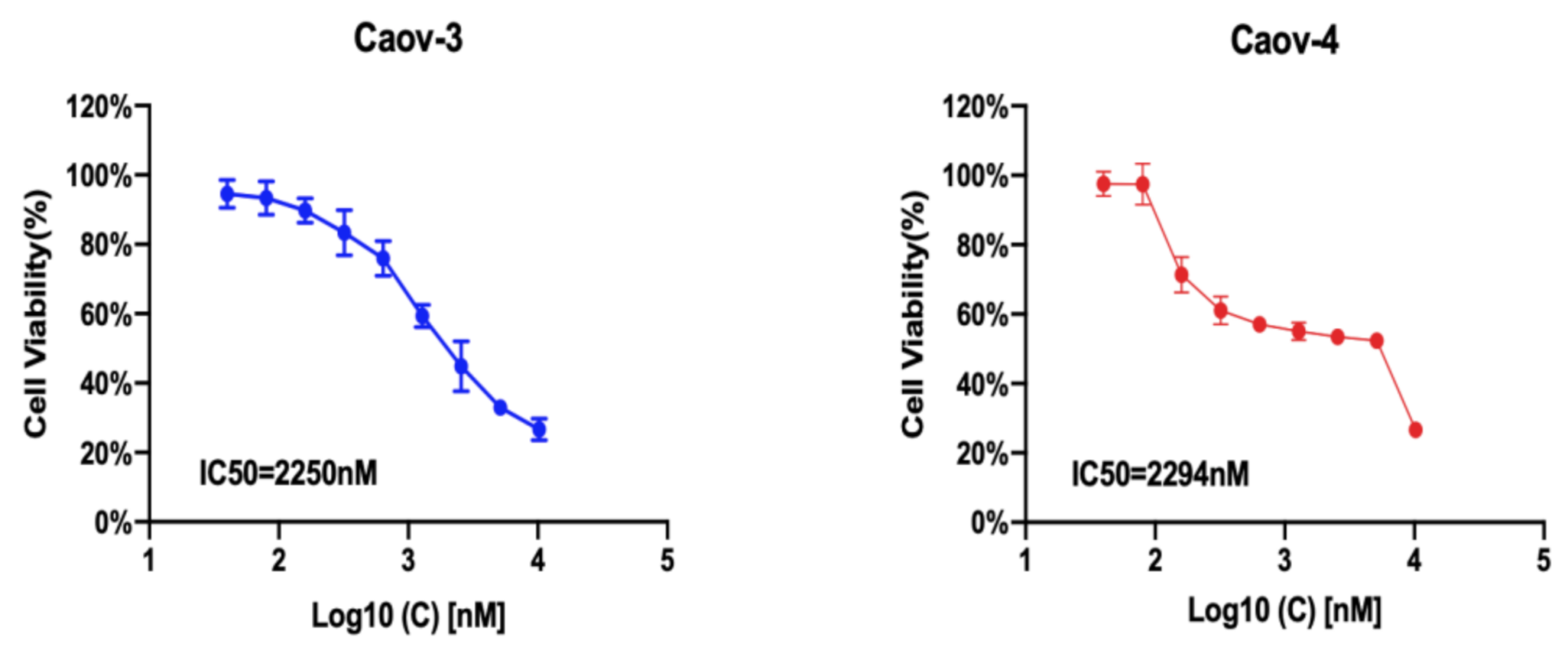
3.4. IC50 of G1 in Ovarian Cancer Cells
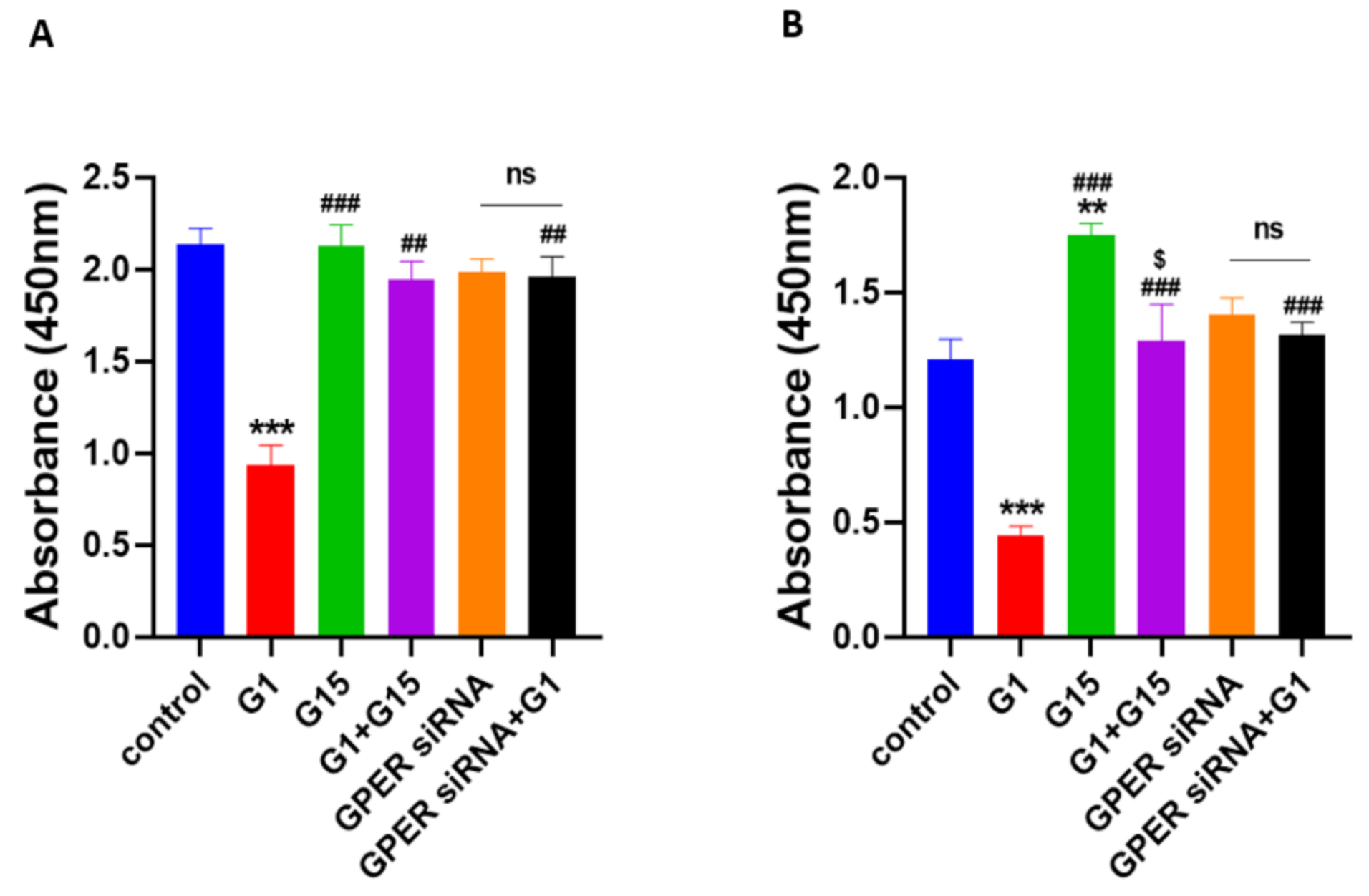
3.5. The Selective GPER Agonist G1 Inhibits Cell Proliferation via Activation of GPER in Caov3 and Caov4 Cell Lines
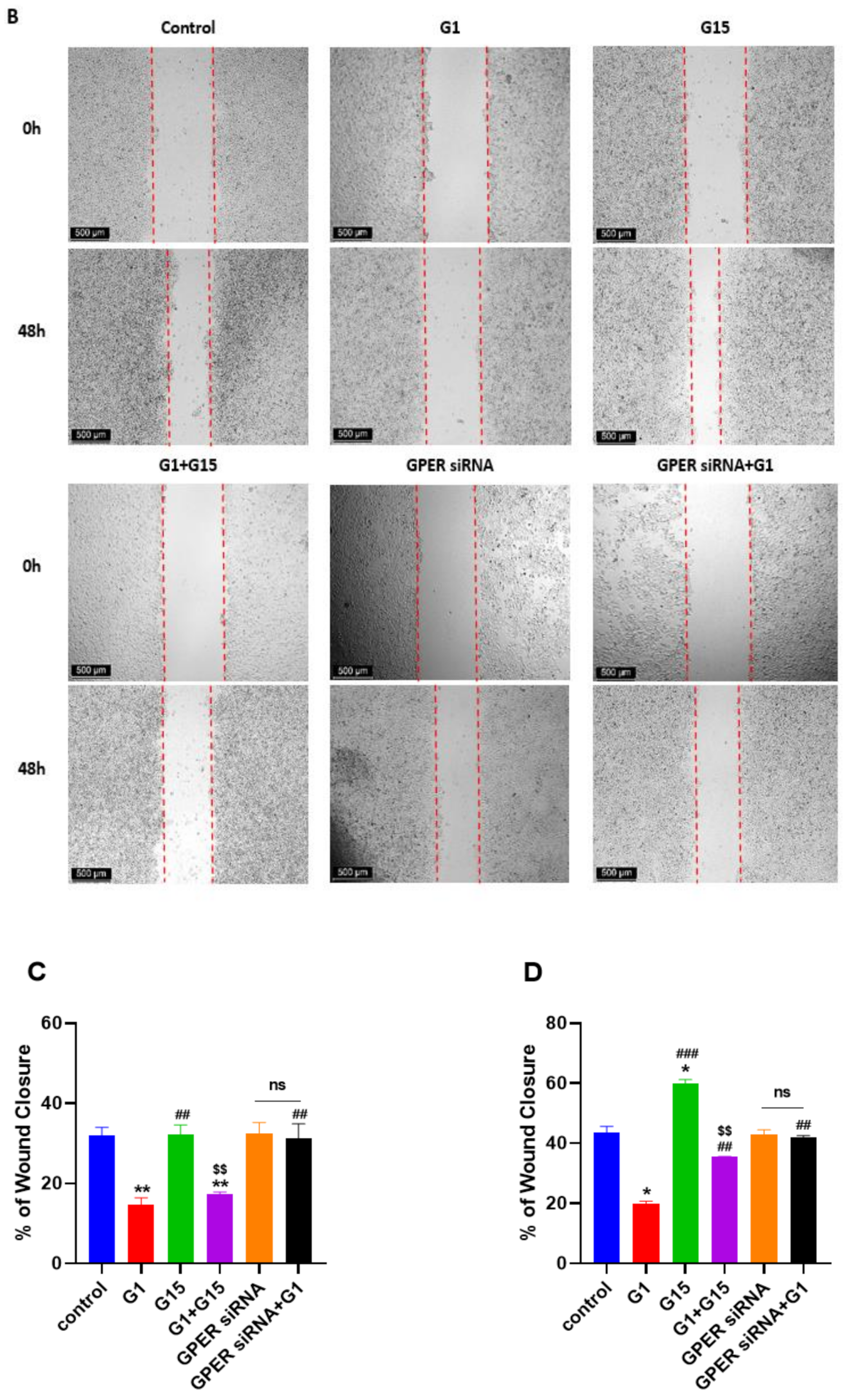
3.6. G1 Suppresses Cell Migration through Activation of GPER in Caov3 and Caov4 Cell Lines
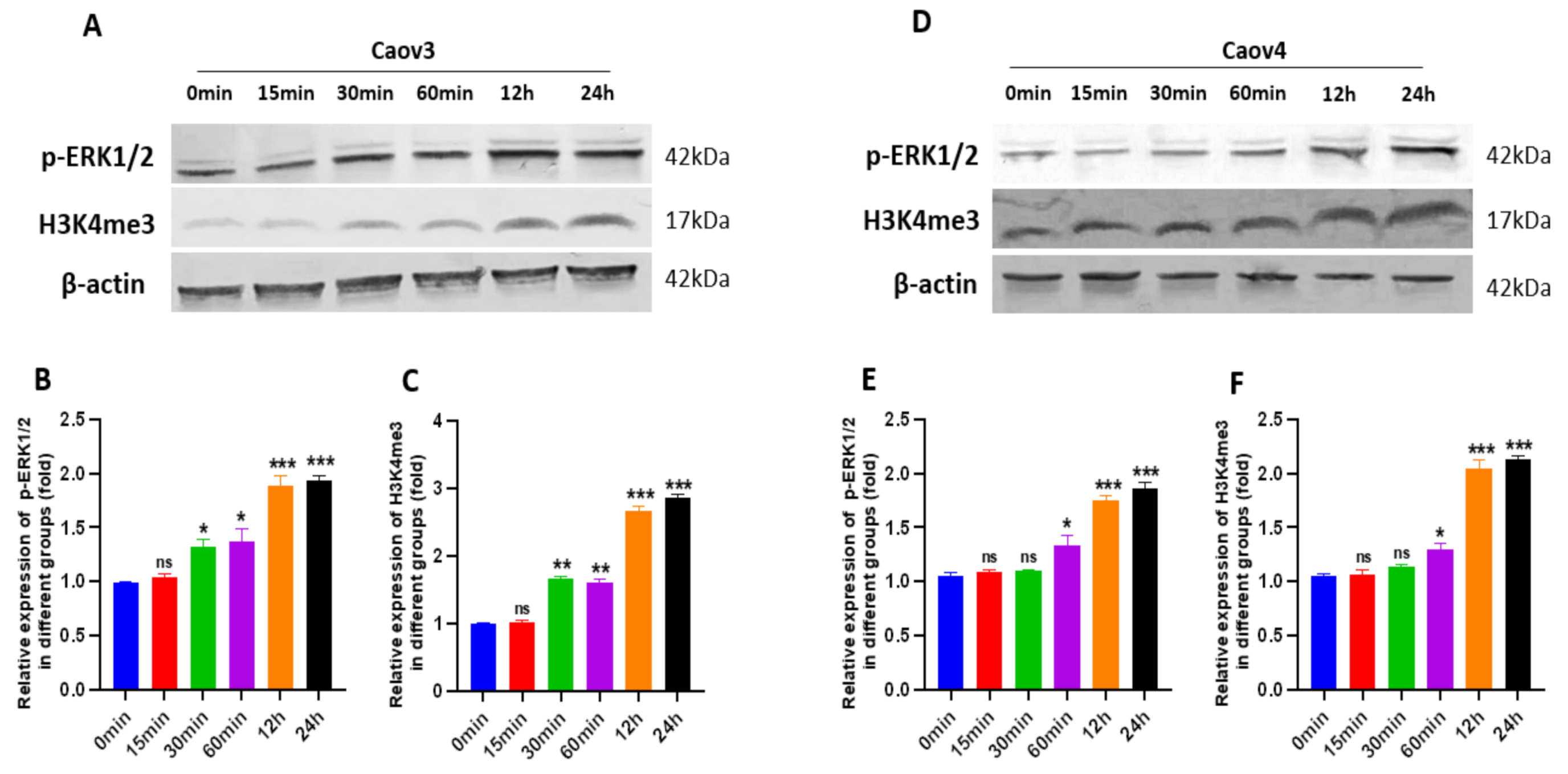
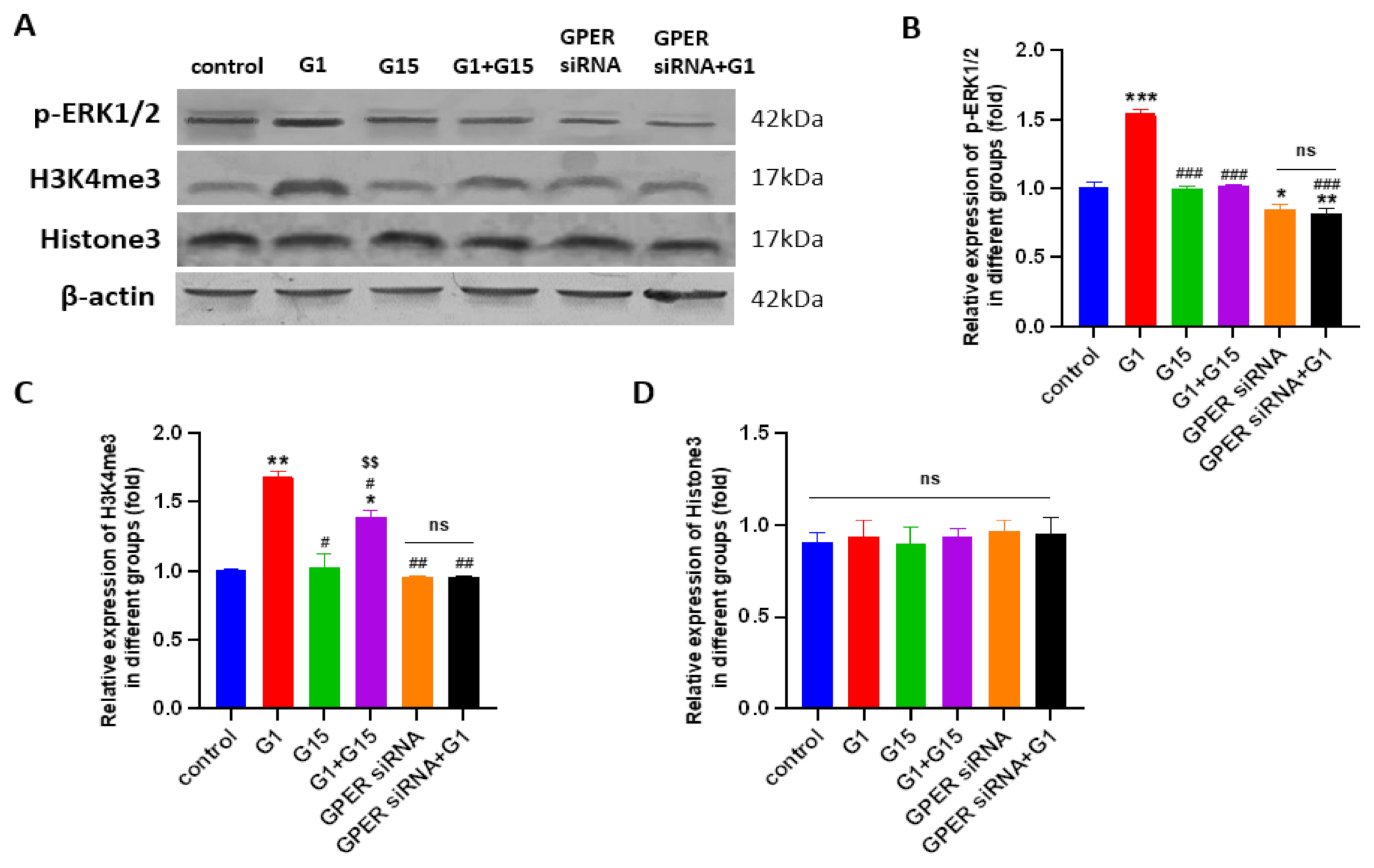
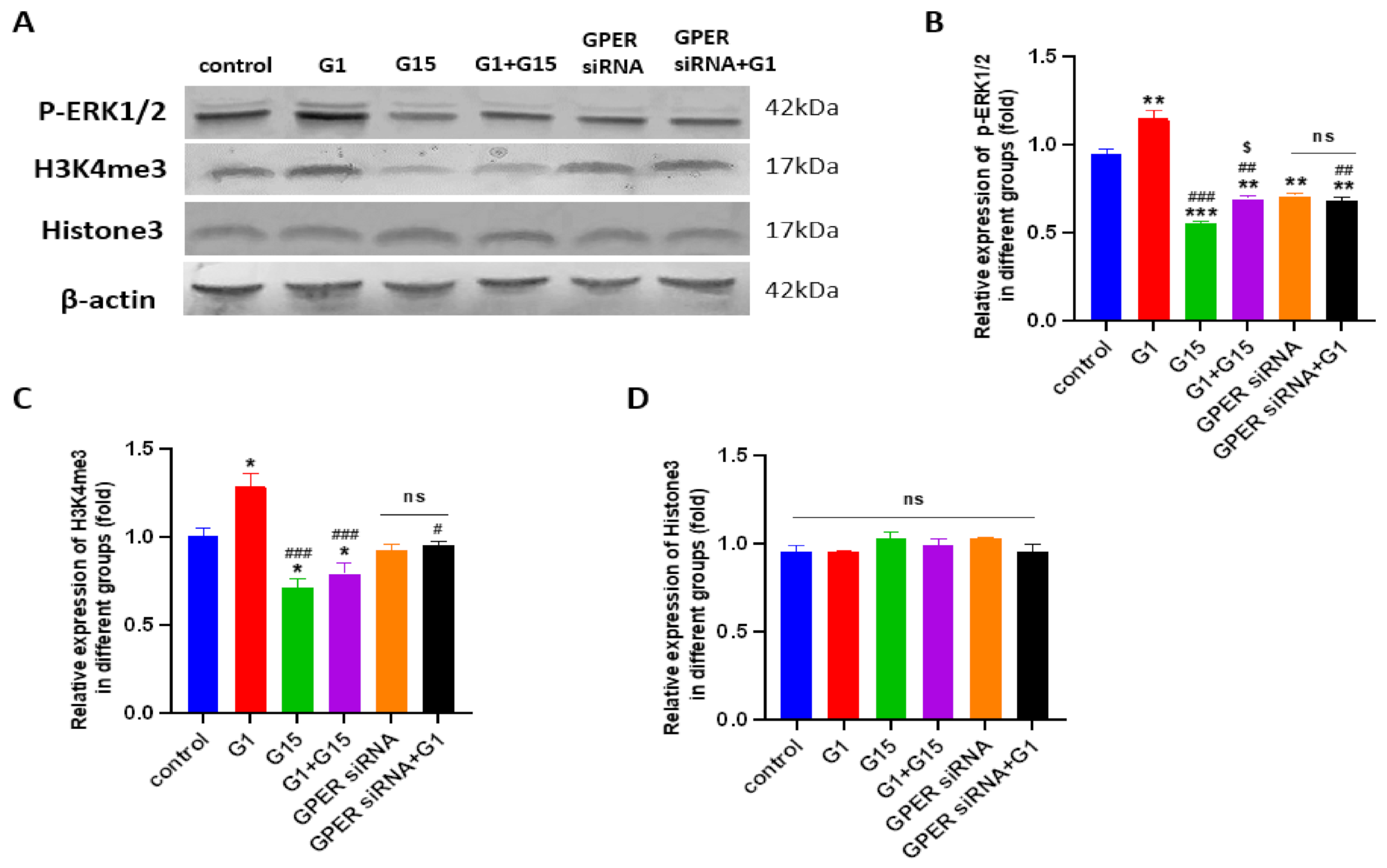
3.7. G1 Treatment Elevatesthe Levels of ERK1/2 Phosphorylation and H3K4me3 via Activation of GPER
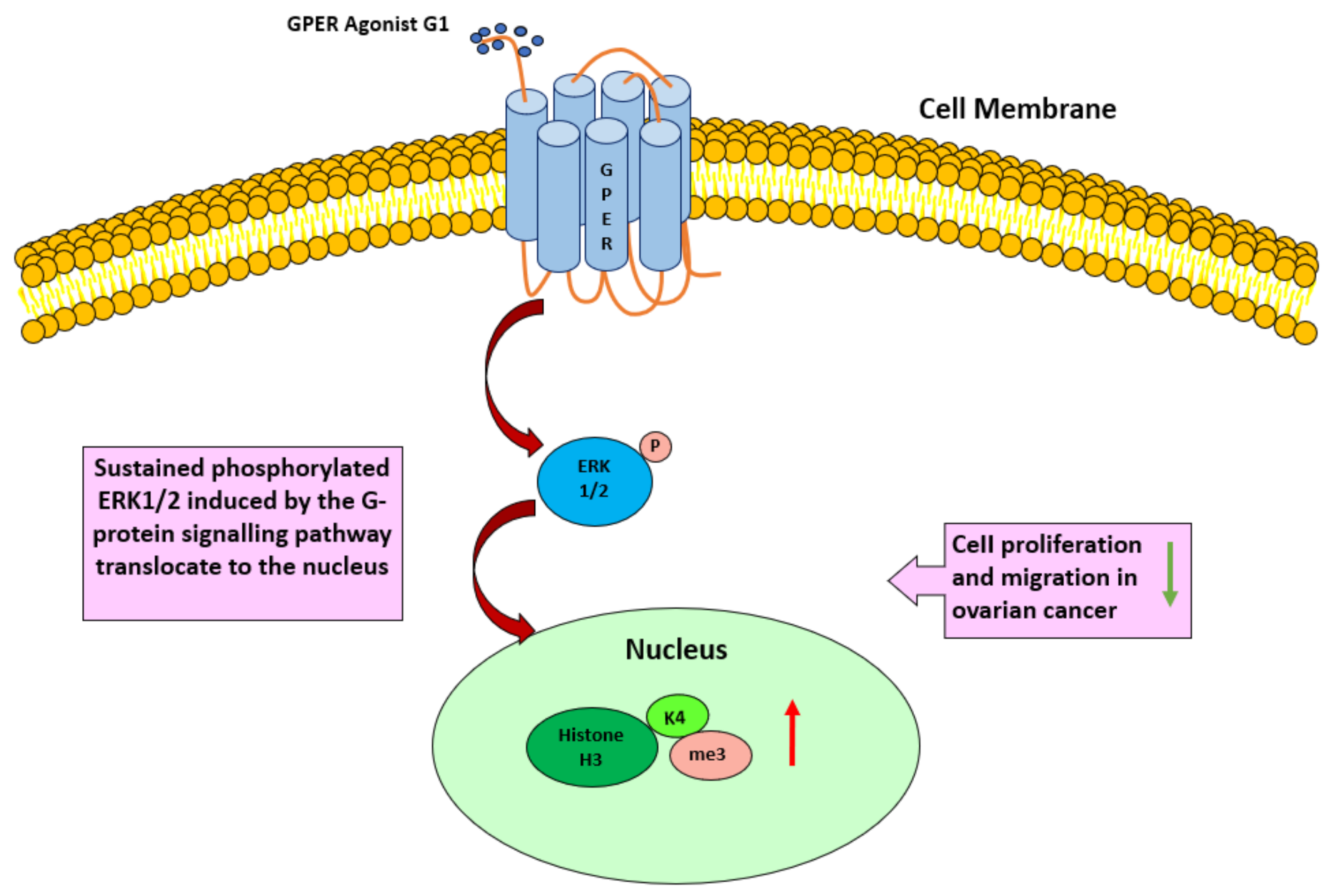
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Jayson, G.C.; Kohn, E.C.; Kitchener, H.C.; Ledermann, J.A. Ovarian cancer. Lancet 2014, 384, 1376–1388. [Google Scholar] [CrossRef]
- Bast, R.C.; Hennessy, B.; Mills, G.B. The biology of ovarian cancer: New opportunities for translation. Nat. Rev. Cancer 2009, 9, 415–428. [Google Scholar] [CrossRef]
- Hunn, J.; Rodriguez, G.C. Ovarian Cancer. Clin. Obstet. Gynecol. 2012, 55, 3–23. [Google Scholar] [CrossRef] [PubMed]
- Persson, I. Estrogens in the causation of breast, endometrial and ovarian cancers-evidence and hypotheses from epidemiological findings. J. Steroid Biochem. Mol. Biol. 2000, 74, 357–364. [Google Scholar] [CrossRef]
- Prossnitz, E.R.; Barton, M. The G-protein-coupled estrogen receptor GPER in health and disease. Nat. Rev. Endocrinol. 2011, 7, 715–726. [Google Scholar] [CrossRef] [Green Version]
- Filardo, E.J.; Quinn, J.A.; Bland, K.I.; Frackelton, A.R. Estrogen-Induced Activation of Erk-1 and Erk-2 Requires the G Protein-Coupled Receptor Homolog, GPR30, and Occurs via Trans-Activation of the Epidermal Growth Factor Receptor through Release of HB-EGF. Mol. Endocrinol. 2000, 14, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Revankar, C.M.; Cimino, D.F.; Sklar, L.A.; Arterburn, J.B.; Prossnitz, E.R. A Transmembrane Intracellular Estrogen Receptor Mediates Rapid Cell Signaling. Science 2005, 307, 1625–1630. [Google Scholar] [CrossRef] [Green Version]
- Prossnitz, E.R.; Barton, M. Signaling, physiological functions and clinical relevance of the G protein-coupled estrogen receptor GPER. Prostaglandins Other Lipid Mediat. 2009, 89, 89–97. [Google Scholar] [CrossRef] [Green Version]
- Prossnitz, E.R.; Barton, M. Estrogen biology: New insights into GPER function and clinical opportunities. Mol. Cell. Endocrinol. 2014, 389, 71–83. [Google Scholar] [CrossRef] [Green Version]
- Albanito, L.; Madeo, A.; Lappano, R.; Vivacqua, A.; Rago, V.; Carpino, A.; Oprea, T.I.; Prossnitz, E.R.; Musti, A.M.; Andò, S.; et al. G Protein–Coupled Receptor 30 (GPR30) Mediates Gene Expression Changes and Growth Response to 17β-Estradiol and Selective GPR30 Ligand G-1 in Ovarian Cancer Cells. Cancer Res. 2007, 67, 1859–1866. [Google Scholar] [CrossRef] [Green Version]
- Barton, M.; Filardo, E.J.; Lolait, S.J.; Thomas, P.; Maggiolini, M.; Prossnitz, E.R. Twenty years of the G protein-coupled estrogen receptor GPER: Historical and personal perspectives. J. Steroid Biochem. Mol. Biol. 2018, 176, 4–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ariazi, E.A.; Brailoiu, E.; Yerrum, S.; Shupp, H.A.; Slifker, M.J.; Cunliffe, H.E.; Black, M.A.; Donato, A.L.; Arterburn, J.B.; Oprea, T.I.; et al. The G Protein–Coupled Receptor GPR30 Inhibits Proliferation of Estrogen Receptor–Positive Breast Cancer Cells. Cancer Res. 2010, 70, 1184–1194. [Google Scholar] [CrossRef] [Green Version]
- Chan, Q.K.; Lam, H.-M.; Ng, C.-F.; Lee, A.Y.Y.; Chan, E.S.; Ng, H.-K.; Ho, S.-M.; Lau, K.-M. Activation of GPR30 inhibits the growth of prostate cancer cells through sustained activation of Erk1/2, c-jun/c-fos-dependent upregulation of p21, and induction of G2 cell-cycle arrest. Cell Death Differ. 2010, 17, 1511–1523. [Google Scholar] [CrossRef]
- Liu, Q.; Chen, Z.; Jiang, G.; Zhou, Y.; Yang, X.; Huang, H.; Liu, H.; Du, J.; Wang, H. Epigenetic down regulation of G protein-coupled estrogen receptor (GPER) functions as a tumor suppressor in colorectal cancer. Mol. Cancer 2017, 16, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Lv, X.; Jiang, C.; Davis, J.S. The putative G-protein coupled estrogen receptor agonist G-1 suppresses prolifer-ation of ovarian and breast cancer cells in a GPER-independent manner. Am. J. Transl. Res. 2012, 4, 390–402. [Google Scholar]
- Yan, Y.; Jiang, X.; Zhao, Y.; Wen, H.; Liu, G. Role of GPER on proliferation, migration and invasion in ligand-independent manner in human ovarian cancer cell line SKOV3. Cell Biochem. Funct. 2015, 33, 552–559. [Google Scholar] [CrossRef]
- Yan, Y.; Liu, H.; Wen, H.; Jiang, X.; Cao, X.; Zhang, G.; Liu, G. The novel estrogen receptor GPER regulates the migration and invasion of ovarian cancer cells. Mol. Cell. Biochem. 2013, 378, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ignatov, T.; Modl, S.; Thulig, M.; Weißenborn, C.; Treeck, O.; Ortmann, O.; Zenclussen, A.; Costa, S.D.; Kalinski, T.; Ignatov, A. GPER-1 acts as a tumor suppressor in ovarian cancer. J. Ovarian Res. 2013, 6, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dennis, M.K.; Burai, R.; Ramesh, C.; Petrie, W.K.; Alcon, S.N.; Nayak, T.K.; Bologa, C.G.; Leitao, A.; Brailoiu, E.; Deliu, E.; et al. In vivo effects of a GPR30 antagonist. Nat. Chem. Biol. 2009, 5, 421–427. [Google Scholar] [CrossRef] [Green Version]
- Dawson, M.A.; Kouzarides, T. Cancer Epigenetics: From Mechanism to Therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.; Kim, G.W.; Kwon, S.H.; Lee, J. Broad domains of histone H3 lysine 4 trimethylation in transcriptional regulation and disease. FEBS J. 2020, 287, 2891–2902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, C.M.; Andrews, R.M.; Flicek, P.; Dillon, S.C.; Karaöz, U.; Clelland, G.K.; Wilcox, S.; Beare, D.M.; Fowler, J.C.; Couttet, P.; et al. The landscape of histone modifications across 1% of the human genome in five human cell lines. Genome Res. 2007, 17, 691–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-Resolution Profiling of Histone Methylations in the Human Genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Chen, Z.; Wu, D.; Zhang, L.; Lin, X.; Su, J.; Rodriguez, B.; Xi, Y.; Xia, Z.; Chen, X.; et al. Broad H3K4me3 is associated with increased transcription elongation and enhancer activity at tumor-suppressor genes. Nat. Genet. 2015, 47, 1149–1157. [Google Scholar] [CrossRef] [PubMed]
- Chapman-Rothe, N.; Curry, E.; Zeller, C.; Liber, D.; Stronach, E.; Gabra, H.; Ghaem-Maghami, S.; Brown, R. Chromatin H3K27me3/H3K4me3 histone marks define gene sets in high-grade serous ovarian cancer that distinguish malignant, tumour-sustaining and chemo-resistant ovarian tumour cells. Oncogene 2012, 32, 4586–4592. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Yang, D.; Sabbatini, M.E.; Colby, A.H.; Grinstaff, M.W.; Oberlies, N.H.; Pearce, C.; Liu, K. Contrasting roles of H3K4me3 and H3K9me3 in regulation of apoptosis and gemcitabine resistance in human pancreatic cancer cells. BMC Cancer 2018, 18, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Chen, L.; Feng, L.; Zhu, M.; Shen, Q.; Fang, Y.; Liu, X.; Zhang, X. NEK2 promotes proliferation, migration and tumor growth of gastric cancer cells via regulating KDM5B/H3K4me3. Am. J. Cancer Res. 2019, 9, 2364–2378. [Google Scholar] [PubMed]
- Dhar, S.S.; Zhao, D.; Lin, T.; Gu, B.; Pal, K.; Wu, S.J.; Alam, H.; Lv, J.; Yun, K.; Gopalakrishnan, V.; et al. MLL4 Is Required to Maintain Broad H3K4me3 Peaks and Super-Enhancers at Tumor Suppressor Genes. Mol. Cell 2018, 70, 825–841.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, N.; Jeschke, U.; Kuhn, C.; Hester, A.; Czogalla, B.; Mahner, S.; Rottmann, M.; Mayr, D.; Schmoeckel, E.; Trillsch, F. H3K4me3 Is a Potential Mediator for Antiproliferative Effects of Calcitriol (1α,25(OH)2D3) in Ovarian Cancer Biology. Int. J. Mol. Sci. 2020, 21, 2151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, M.; Cortez, V.; Vadlamudi, R.K. Epigenetics of Estrogen Receptor Signaling: Role in Hormonal Cancer Progression and Therapy. Cancers 2011, 3, 1691–1707. [Google Scholar] [CrossRef]
- Li, Y.; Sun, L.; Zhang, Y.; Wang, D.; Wang, F.; Liang, J.; Gui, B.; Shang, Y. The Histone Modifications Governing TFF1 Transcription Mediated by Estrogen Receptor. J. Biol. Chem. 2011, 286, 13925–13936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, W.; Katzenellenbogen, B.S. Estrogen Receptor-β Modulation of the ERα-p53 Loop Regulating Gene Expression, Proliferation, and Apoptosis in Breast Cancer. Horm. Cancer 2017, 8, 230–242. [Google Scholar] [CrossRef] [PubMed]
- Templin, M.F.; Stoll, D.; Schrenk, M.; Traub, P.C.; Vöhringer, C.F.; Joos, T.O. Protein microarray technology. Drug Discov. Today 2002, 7, 815–822. [Google Scholar] [CrossRef]
- Heublein, S.; Mayr, D.; Vrekoussis, T.; Friese, K.; Hofmann, S.S.; Jeschke, U.; Lenhard, M. The G-Protein Coupled Estrogen Receptor (GPER/GPR30) is a Gonadotropin Receptor Dependent Positive Prognosticator in Ovarian Carcinoma Patients. PLoS ONE 2013, 8, e71791. [Google Scholar] [CrossRef] [Green Version]
- Friese, K.; Kost, B.; Vattai, A.; Marmé, F.; Kuhn, C.; Mahner, S.; Dannecker, C.; Jeschke, U.; Heublein, S. The G protein-coupled estrogen receptor (GPER/GPR30) may serve as a prognostic marker in early-stage cervical cancer. J. Cancer Res. Clin. Oncol. 2017, 144, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Heublein, S.; Mayr, D.; Friese, K.; Jarrin-Franco, M.C.; Lenhard, M.; Mayerhofer, A.; Jeschke, U. The G-Protein-Coupled Estrogen Receptor (GPER/GPR30) in Ovarian Granulosa Cell Tumors. Int. J. Mol. Sci. 2014, 15, 15161–15172. [Google Scholar] [CrossRef] [Green Version]
- Heublein, S.; Vrekoussis, T.; Kühn, C.; Friese, K.; Makrigiannakis, A.; Mayr, D.; Lenhard, M.; Jeschke, U. Inducers of G-protein coupled estrogen receptor (GPER) in endometriosis: Potential implications for macrophages and follicle maturation. J. Reprod. Immunol. 2013, 97, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Fluss, R.; Faraggi, D.; Reiser, B. Estimation of the Youden Index and its Associated Cutoff Point. Biom. J. 2005, 47, 458–472. [Google Scholar] [CrossRef] [Green Version]
- Perkins, N.J.; Schisterman, E.F. The Inconsistency of “Optimal” Cutpoints Obtained using Two Criteria based on the Receiver Operating Characteristic Curve. Am. J. Epidemiol. 2006, 163, 670–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eishingdrelo, H.; Kongsamut, S. Minireview: Targeting GPCR Activated ERK Pathways for Drug Discovery. Curr. Chem. Genomics Transl. Med. 2013, 7, 9–15. [Google Scholar] [CrossRef] [Green Version]
- Smith, H.O.; Arias-Pulido, H.; Kuo, D.Y.; Howard, T.; Qualls, C.R.; Lee, S.-J.; Verschraegen, C.F.; Hathaway, H.J.; Joste, N.E.; Prossnitz, E.R. GPR30 predicts poor survival for ovarian cancer. Gynecol. Oncol. 2009, 114, 465–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, A.-C.; Heukamp, L.C.; Rogenhofer, S.; Fechner, G.; Bastian, P.J.; Von Ruecker, A.; Müller, S.C.; Ellinger, J. Global histone H4K20 trimethylation predicts cancer-specific survival in patients with muscle-invasive bladder cancer. BJU Int. 2011, 108, E290–E296. [Google Scholar] [CrossRef]
- Ellinger, J.; Kahl, P.; Mertens, C.; Rogenhofer, S.; Hauser, S.; Hartmann, W.; Bastian, P.J.; Büttner, R.; Müller, S.C.; von Ruecker, A. Prognostic relevance of global histone H3 lysine 4 (H3K4) methylation in renal cell carcinoma. Int. J. Cancer 2010, 127, 2360–2366. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Xu, J.; Zhang, J.; Xie, D.; Ye, H.; Xiao, Z.; Cai, M.; Xu, K.; Zeng, Y.; Li, H.; et al. High expression of trimethylated histone H3 lysine 4 is associated with poor prognosis in hepatocellular carcinoma. Hum. Pathol. 2012, 43, 1425–1435. [Google Scholar] [CrossRef] [PubMed]
- Messier, T.L.; Gordon, J.A.R.; Boyd, J.R.; Tye, C.E.; Browne, G.; Stein, J.L.; Lian, J.B.; Stein, G.S. Histone H3 lysine 4 acetylation and methylation dynamics define breast cancer subtypes. Oncotarget 2016, 7, 5094–5109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Li, Y.; Li, J.; Liu, Y.; Cui, B. H3K4me3 and Wdr82 are associated with tumor progression and a favorable prognosis in human colorectal cancer. Oncol. Lett. 2018, 16, 2125–2134. [Google Scholar] [CrossRef]
- Boesch, M.; Zeimet, A.G.; Reimer, D.; Schmidt, S.; Gastl, G.; Parson, W.; Spoeck, F.; Hatina, J.; Wolf, D.; Sopper, S. The side population of ovarian cancer cells defines a heterogeneous compartment exhibiting stem cell characteristics. Oncotarget 2014, 5, 7027–7039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Testa, U.; Petrucci, E.; Pasquini, L.; Castelli, G.; Pelosi, E. Ovarian Cancers: Genetic Abnormalities, Tumor Heterogeneity and Progression, Clonal Evolution and Cancer Stem Cells. Medicines 2018, 5, 16. [Google Scholar] [CrossRef] [Green Version]
- Gaudet, H.; Cheng, S.; Christensen, E.; Filardo, E. The G-protein coupled estrogen receptor, GPER: The inside and inside-out story. Mol. Cell. Endocrinol. 2015, 418, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Osmond, R.I.W.; Sheehan, A.; Borowicz, R.; Barnett, E.; Harvey, G.; Turner, C.; Brown, A.; Crouch, M.F.; Dyer, A.R. GPCR Screening via ERK 1/2: A Novel Platform for Screening G Protein–Coupled Receptors. J. Biomol. Screen. 2005, 10, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.-J.; Wei, W.-D.; Jiang, G.-M.; Liu, H.; Yang, X.; Wu, Y.-M.; Liu, H.; Wong, C.K.; Du, J.; Wang, H.-S. Activation of GPER suppresses epithelial mesenchymal transition of triple negative breast cancer cells via NF-κB signals. Mol. Oncol. 2016, 10, 775–788. [Google Scholar] [CrossRef] [Green Version]
- Chimento, A.; Sirianni, R.; Casaburi, I.; Zolea, F.; Rizza, P.; Avena, P.; Malivindi, R.; De Luca, A.; Campana, C.; Martire, E.; et al. GPER agonist G-1 decreases adrenocortical carcinoma (ACC) cell growth in vitro and in vivo. Oncotarget 2015, 6, 19190–19203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dennis, M.K.; Field, A.S.; Burai, R.; Ramesh, C.; Petrie, W.K.; Bologa, C.G.; Oprea, T.I.; Yamaguchi, Y.; Hayashi, S.-I.; Sklar, S.L.A.; et al. Identification of a GPER/GPR30 antagonist with improved estrogen receptor counterselectivity. J. Steroid Biochem. Mol. Biol. 2011, 127, 358–366. [Google Scholar] [CrossRef] [Green Version]
- Jenei-Lanzl, Z.; Straub, R.H.; Dienstknecht, T.; Huber, M.; Hager, M.; Grässel, S.; Kujat, R.; Angele, M.K.; Nerlich, M.; Angele, P. Estradiol inhibits chondrogenic differentiation of mesenchymal stem cells via nonclassic signaling. Arthritis Rheum. 2010, 62, 1088–1096. [Google Scholar] [CrossRef]
- Lindsey, S.H.; Carver, K.A.; Prossnitz, E.R.; Chappell, M.C. Vasodilation in Response to the GPR30 Agonist G-1 is Not Different from Estradiol in the mRen2.Lewis Female Rat. J. Cardiovasc. Pharmacol. 2011, 57, 598–603. [Google Scholar] [CrossRef] [Green Version]
- Bologa, C.G.; Revankar, C.M.; Young, S.M.; Edwards, B.S.; Arterburn, J.B.; Kiselyov, A.S.; Parker, M.A.; Tkachenko, S.E.; Savchuck, N.P.; Sklar, L.A.; et al. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat. Chem. Biol. 2006, 2, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Liu, M.; Luo, H.; Wu, C.; Tang, X.; Tang, S.; Hu, P.; Yan, Y.; Wang, Z.; Tu, G. GPER mediates enhanced cell viability and motility via non-genomic signaling induced by 17β-estradiol in triple-negative breast cancer cells. J. Steroid Biochem. Mol. Biol. 2014, 143, 392–403. [Google Scholar] [CrossRef]
- Meloche, S.; Pouysségur, J. The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene 2007, 26, 3227–3239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mebratu, Y.; Tesfaigzi, Y. How ERK1/2 activation controls cell proliferation and cell death: Is subcellular localization the answer? Cell Cycle 2009, 8, 1168–1175. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-R.; Plotkin, L.I.; Aguirre, J.I.; Han, L.; Jilka, R.L.; Kousteni, S.; Bellido, T.; Manolagas, S.C. Transient Versus Sustained Phosphorylation and Nuclear Accumulation of ERKs Underlie Anti-Versus Pro-apoptotic Effects of Estrogens. J. Biol. Chem. 2005, 280, 4632–4638. [Google Scholar] [CrossRef] [Green Version]
- Marshall, C. Specificity of receptor tyrosine kinase signaling: Transient versus sustained extracellular signal-regulated kinase activation. Cell 1995, 80, 179–185. [Google Scholar] [CrossRef] [Green Version]
- Adachi, T.; Kar, S.; Wang, M.; Carr, B.I. Transient and sustained ERK phosphorylation and nuclear translocation in growth control. J. Cell. Physiol. 2002, 192, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Goeman, F.; De Nicola, F.; De Meo, P.D.; Pallocca, M.; Elmi, B.; Castrignanò, T.; Pesole, G.; Strano, S.; Blandino, G.; Fanciulli, M.; et al. VDR primary targets by genome-wide transcriptional profiling. J. Steroid Biochem. Mol. Biol. 2014, 143, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Dreijerink, K.M.; Mulder, K.W.; Winkler, G.S.; Höppener, J.W.; Lips, C.J.; Timmers, H.M. Menin Links Estrogen Receptor Activation to Histone H3K4 Trimethylation. Cancer Res. 2006, 66, 4929–4935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, W.; Chen, Z.-J.; Zhang, K.-S.; Yang, X.-L.; Wu, Y.-M.; Chen, X.-H.; Huang, H.-B.; Liu, H.-L.; Cai, S.-H.; Du, J.; et al. The activation of G protein-coupled receptor 30 (GPR30) inhibits proliferation of estrogen receptor-negative breast cancer cells in vitro and in vivo. Cell Death Dis. 2014, 5, e1428. [Google Scholar] [CrossRef] [PubMed]
- Mertens-Walker, I.; Bolitho, C.; Baxter, R.C.; Marsh, D.J. Gonadotropin-induced ovarian cancer cell migration and proliferation require extracellular signal-regulated kinase 1/2 activation regulated by calcium and protein kinase Cδ. Endocr. Relat. Cancer 2010, 17, 335–349. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.-F.; Wu, J.; Luo, J.-H.; Li, K.-S.; Wang, F.; Huang, W.; Wu, Y.; Gao, S.-P.; Zhang, X.-M. SNHG22 overexpression indicates poor prognosis and induces chemotherapy resistance via the miR-2467/Gal-1 signaling pathway in epithelial ovarian carcinoma. Aging 2019, 11, 8204–8216. [Google Scholar] [CrossRef]
- Imesch, P.; Samartzis, E.P.; Dedes, K.J.; Fink, D.; Fedier, A. Histone deacetylase inhibitors down-regulate G-protein-coupled estrogen receptor and the GPER-antagonist G-15 inhibits proliferation in endometriotic cells. Fertil. Steril. 2013, 100, 770–776. [Google Scholar] [CrossRef]













Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, N.; Heublein, S.; Jeschke, U.; Kuhn, C.; Hester, A.; Czogalla, B.; Mahner, S.; Rottmann, M.; Mayr, D.; Schmoeckel, E.; et al. The G-Protein-Coupled Estrogen Receptor (GPER) Regulates Trimethylation of Histone H3 at Lysine 4 and Represses Migration and Proliferation of Ovarian Cancer Cells In Vitro. Cells 2021, 10, 619. https://doi.org/10.3390/cells10030619
Han N, Heublein S, Jeschke U, Kuhn C, Hester A, Czogalla B, Mahner S, Rottmann M, Mayr D, Schmoeckel E, et al. The G-Protein-Coupled Estrogen Receptor (GPER) Regulates Trimethylation of Histone H3 at Lysine 4 and Represses Migration and Proliferation of Ovarian Cancer Cells In Vitro. Cells. 2021; 10(3):619. https://doi.org/10.3390/cells10030619
Chicago/Turabian StyleHan, Nan, Sabine Heublein, Udo Jeschke, Christina Kuhn, Anna Hester, Bastian Czogalla, Sven Mahner, Miriam Rottmann, Doris Mayr, Elisa Schmoeckel, and et al. 2021. "The G-Protein-Coupled Estrogen Receptor (GPER) Regulates Trimethylation of Histone H3 at Lysine 4 and Represses Migration and Proliferation of Ovarian Cancer Cells In Vitro" Cells 10, no. 3: 619. https://doi.org/10.3390/cells10030619
APA StyleHan, N., Heublein, S., Jeschke, U., Kuhn, C., Hester, A., Czogalla, B., Mahner, S., Rottmann, M., Mayr, D., Schmoeckel, E., & Trillsch, F. (2021). The G-Protein-Coupled Estrogen Receptor (GPER) Regulates Trimethylation of Histone H3 at Lysine 4 and Represses Migration and Proliferation of Ovarian Cancer Cells In Vitro. Cells, 10(3), 619. https://doi.org/10.3390/cells10030619