
Progression of Metastasis through Lymphatic System


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Lymphatic Vessels
3. Lymph Nodes
4. Lymph Node Metastasis
4.1. Tumor Cell Migration to Draining Lymph Node
4.2. Tumor Cell Survival in Draining Lymph Nodes
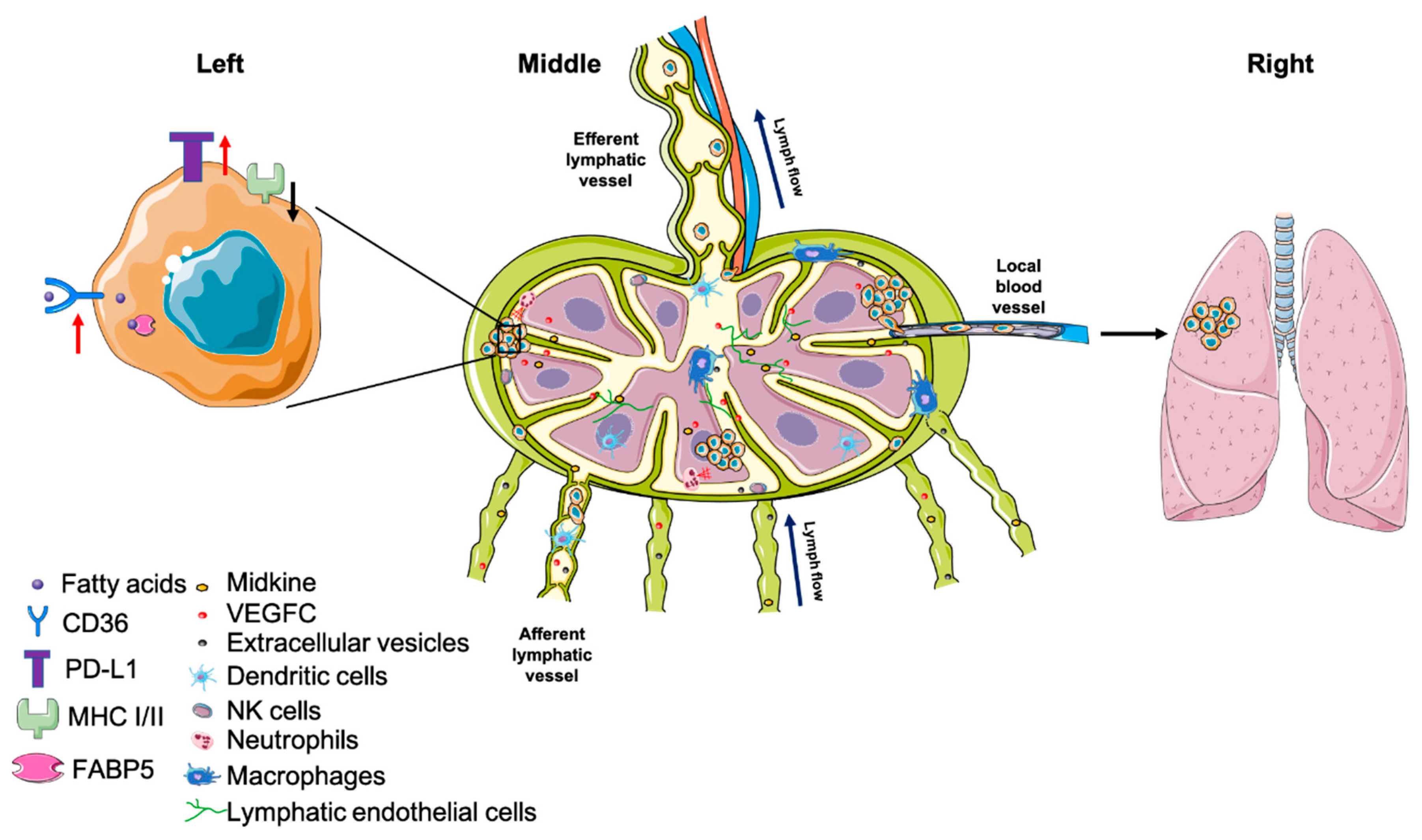
4.2.1. Premetastatic Niche
4.2.2. Major Histocompatibility Complex (MHC)
4.2.3. Tumor-Immune Cell Interaction
4.2.4. Tumor–Stromal Cell Interaction
4.2.5. Metabolic Adaptation
4.3. Lymph Node Metastases Spread Further to Other Organs
5. Targeting Lymph Node Metastasis
5.1. Controversy over Extensive Resection of Metastatic LNs
5.2. Strategies to Treat LN Metastases
5.2.1. Targeted LN Surgical Removal
5.2.2. Targeted LN Agent Delivery
5.3. Potential Targets in LN Metastases
5.3.1. Targeting Lymphangiogenesis
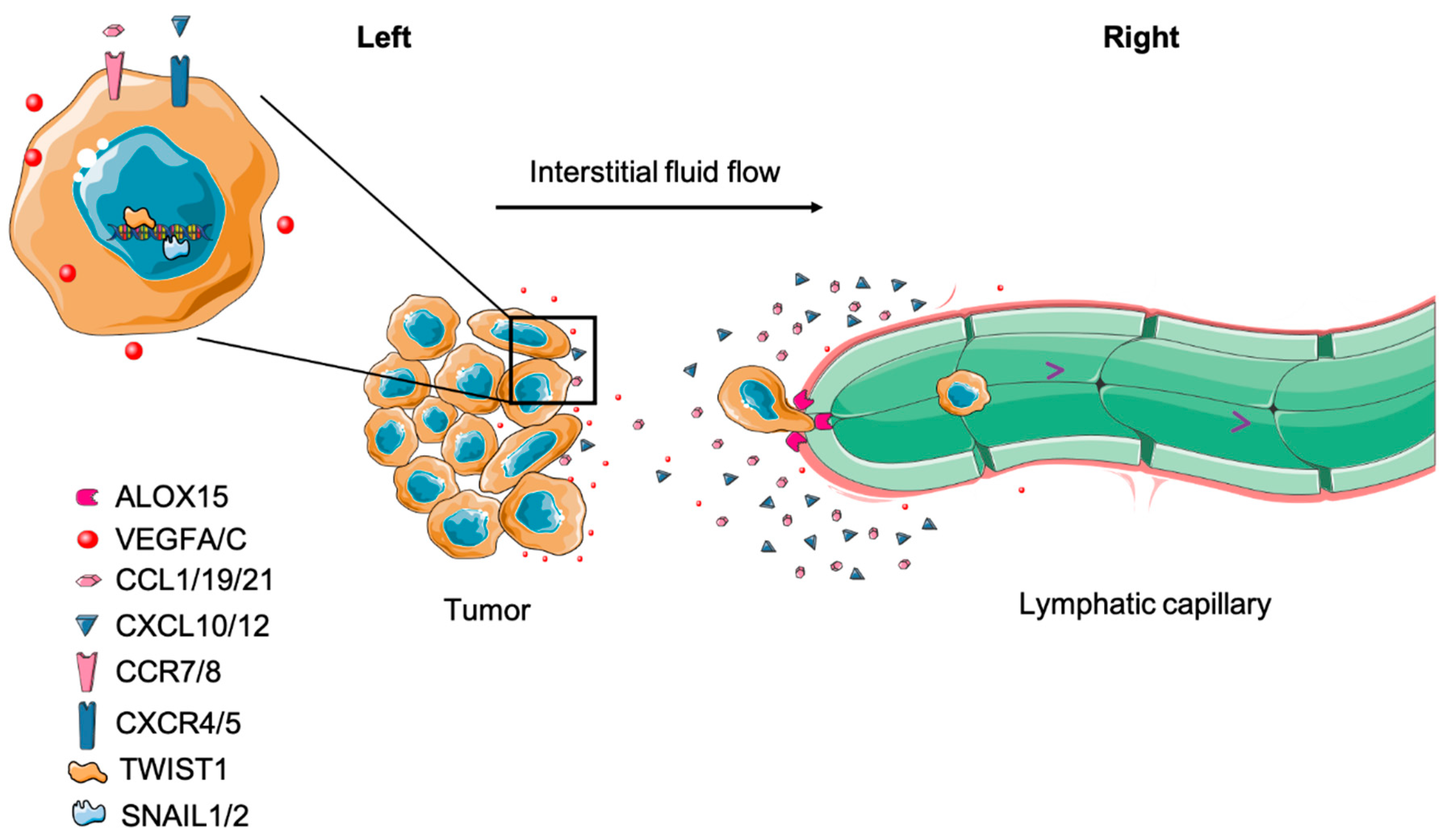
5.3.2. Targeting Chemotaxis
5.3.3. Targeting Lipid Metabolism
6. Concluding Remarks
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Padera, T.P.; Meijer, E.F.; Munn, L.L. The Lymphatic System in Disease Processes and Cancer Progression. Annu. Rev. Biomed. Eng. 2016, 18, 125–158. [Google Scholar] [CrossRef] [Green Version]
- Randolph, G.J.; Ivanov, S.; Zinselmeyer, B.H.; Scallan, J.P. The Lymphatic System: Integral Roles in Immunity. Annu. Rev. Immunol. 2017, 35, 31–52. [Google Scholar] [CrossRef] [Green Version]
- Liao, S.; Bouta, E.M.; Morris, L.M.; Jones, D.; Jain, R.K.; Padera, T.P. Inducible Nitric Oxide Synthase and CD11b(+)Gr1(+) Cells Impair Lymphatic Contraction of Tumor-Draining Lymphatic Vessels. Lymphat. Res. Biol. 2019, 17, 294–300. [Google Scholar] [CrossRef]
- Liao, S.; Cheng, G.; Conner, D.A.; Huang, Y.; Kucherlapati, R.S.; Munn, L.L.; Ruddle, N.H.; Jain, R.K.; Fukumura, D.; Padera, T.P. Impaired lymphatic contraction associated with immunosuppression. Proc. Natl. Acad. Sci. USA 2011, 108, 18784–18789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padera, T.P.; Kadambi, A.; di Tomaso, E.; Mouta Carreira, C.; Brown, E.B.; Boucher, Y.; Choi, N.C.; Mathisen, D.; Wain, J.; Mark, E.J.; et al. Lymphatic metastasis in the absence of functional intratumor lymphatics. Science 2002, 296, 1883–1886. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.; Pereira, E.R.; Padera, T.P. Growth and Immune Evasion of Lymph Node Metastasis. Front. Oncol. 2018, 8, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baluk, P.; Fuxe, J.; Hashizume, H.; Romano, T.; Lashnits, E.; Butz, S.; Vestweber, D.; Corada, M.; Molendini, C.; Dejana, E.; et al. Functionally specialized junctions between endothelial cells of lymphatic vessels. J. Exp. Med. 2007, 204, 2349–2362. [Google Scholar] [CrossRef] [PubMed]
- Drayton, D.L.; Liao, S.; Mounzer, R.H.; Ruddle, N.H. Lymphoid organ development: From ontogeny to neogenesis. Nat. Immunol. 2006, 7, 344–353. [Google Scholar] [CrossRef]
- Oliver, G.; Alitalo, K. The lymphatic vasculature: Recent progress and paradigms. Annu. Rev. Cell Dev. Biol. 2005, 21, 457–483. [Google Scholar] [CrossRef]
- Schmid-Schonbein, G.W. Microlymphatics and lymph flow. Physiol. Rev. 1990, 70, 987–1028. [Google Scholar] [CrossRef]
- Miteva, D.O.; Rutkowski, J.M.; Dixon, J.B.; Kilarski, W.; Shields, J.D.; Swartz, M.A. Transmural flow modulates cell and fluid transport functions of lymphatic endothelium. Circ. Res. 2010, 106, 920–931. [Google Scholar] [CrossRef] [PubMed]
- Schumann, K.; Lämmermann, T.; Bruckner, M.; Legler, D.F.; Polleux, J.; Spatz, J.P.; Schuler, G.; Förster, R.; Lutz, M.B.; Sorokin, L.; et al. Immobilized chemokine fields and soluble chemokine gradients cooperatively shape migration patterns of dendritic cells. Immunity 2010, 32, 703–713. [Google Scholar] [CrossRef] [Green Version]
- Weber, M.; Hauschild, R.; Schwarz, J.; Moussion, C.; de Vries, I.; Legler, D.F.; Luther, S.A.; Bollenbach, T.; Sixt, M. Interstitial dendritic cell guidance by haptotactic chemokine gradients. Science 2013, 339, 328–332. [Google Scholar] [CrossRef] [Green Version]
- Russo, E.; Teijeira, A.; Vaahtomeri, K.; Willrodt, A.-H.; Bloch, J.S.; Nitschké, M.; Santambrogio, L.; Kerjaschki, D.; Sixt, M.; Halin, C. Intralymphatic CCL21 Promotes Tissue Egress of Dendritic Cells through Afferent Lymphatic Vessels. Cell Rep. 2016, 14, 1723–1734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, E.; Nitschke, M.; Halin, C. Dendritic cell interactions with lymphatic endothelium. Lymphat. Res. Biol. 2013, 11, 172–182. [Google Scholar] [CrossRef]
- Teijeira, A.; Russo, E.; Halin, C. Taking the lymphatic route: Dendritic cell migration to draining lymph nodes. Semin. Immunopathol. 2014, 36, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Randolph, G.J.; Angeli, V.; Swartz, M.A. Dendritic-cell trafficking to lymph nodes through lymphatic vessels. Nat. Rev. Immunol. 2005, 5, 617–628. [Google Scholar] [CrossRef]
- Skalak, T.C.; Schmid-Schonbein, G.W.; Zweifach, B.W. New morphological evidence for a mechanism of lymph formation in skeletal muscle. Microvasc. Res. 1984, 28, 95–112. [Google Scholar] [CrossRef]
- Bohlen, H.G.; Gasheva, O.Y.; Zawieja, D.C. Nitric oxide formation by lymphatic bulb and valves is a major regulatory component of lymphatic pumping. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1897–H1906. [Google Scholar] [CrossRef] [Green Version]
- Kaji, D.M.; Thakkar, U.; Kahn, T. Glucocorticoid-induced alterations in the sodium potassium pump of the human erythrocyte. J. Clin. Investig. 1981, 68, 422–430. [Google Scholar] [CrossRef] [Green Version]
- To, K.H.T.; Gui, P.; Li, M.; Zawieja, S.D.; Castorena-Gonzalez, J.A.; Davis, M.J. T-type, but not L-type, voltage-gated calcium channels are dispensable for lymphatic pacemaking and spontaneous contractions. Sci. Rep. 2020, 10, 70. [Google Scholar] [CrossRef] [PubMed]
- Schmid-Schonbein, G.W. Nitric oxide (NO) side of lymphatic flow and immune surveillance. Proc. Natl. Acad. Sci. USA 2012, 109, 3–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucas, E.D.; Tamburini, B.A.J. Lymph Node Lymphatic Endothelial Cell Expansion and Contraction and the Programming of the Immune Response. Front. Immunol. 2019, 10, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santambrogio, L.; Berendam, S.J.; Engelhard, V.H. The Antigen Processing and Presentation Machinery in Lymphatic Endothelial Cells. Front. Immunol. 2019, 10, 1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girard, J.P.; Moussion, C.; Forster, R. HEVs, lymphatics and homeostatic immune cell trafficking in lymph nodes. Nat. Rev. Immunol. 2012, 12, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Jafarnejad, M.; Woodruff, M.C.; Zawieja, D.C.; Carroll, M.C.; Moore, J.E., Jr. Modeling Lymph Flow and Fluid Exchange with Blood Vessels in Lymph Nodes. Lymphat. Res. Biol. 2015, 13, 234–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivetic, A.; Green, H.L.H.; Hart, S.J. L-selectin: A Major Regulator of Leukocyte Adhesion, Migration and Signaling. Front. Immunol. 2019, 10, 1068. [Google Scholar] [CrossRef] [Green Version]
- Miyasaka, M.; Tanaka, T. Lymphocyte trafficking across high endothelial venules: Dogmas and enigmas. Nat. Rev. Immunol. 2004, 4, 360–370. [Google Scholar] [CrossRef]
- von Andrian, U.H.; Mempel, T.R. Homing and cellular traffic in lymph nodes. Nat. Rev. Immunol. 2003, 3, 867–878. [Google Scholar] [CrossRef]
- Carlsen, H.S.; Haraldsen, G.; Brandtzaeg, P.; Baekkevold, E.S. Disparate lymphoid chemokine expression in mice and men: No evidence of CCL21 synthesis by human high endothelial venules. Blood 2005, 106, 444–446. [Google Scholar] [CrossRef] [Green Version]
- Shamri, R.; Grabovsky, V.; Gauguet, J.-M.; Feigelson, S.; Manevich, E.; Kolanus, W.; Robinson, M.K.; Staunton, D.E.; von Andrian, U.H.; Alon, R. Lymphocyte arrest requires instantaneous induction of an extended LFA-1 conformation mediated by endothelium-bound chemokines. Nat. Immunol. 2005, 6, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Bajenoff, M.; Egen, J.G.; Koo, L.Y.; Laugier, J.P.; Brau, F.; Glaichenhaus, N.; Germain, R.N. Stromal cell networks regulate lymphocyte entry, migration, and territoriality in lymph nodes. Immunity 2006, 25, 989–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mionnet, C.; Sanos, S.L.; Mondor, I.; Jorquera, A.; Laugier, J.-P.; Germain, R.N.; Bajénoff, M. High endothelial venules as traffic control points maintaining lymphocyte population homeostasis in lymph nodes. Blood 2011, 118, 6115–6122. [Google Scholar] [CrossRef]
- Qu, C.; Edwards, E.W.; Tacke, F.; Angeli, V.; Llodrá, J.; Sanchez-Schmitz, G.; Garin, A.; Haque, N.S.; Peters, W.; van Rooijen, N.; et al. Role of CCR8 and other chemokine pathways in the migration of monocyte-derived dendritic cells to lymph nodes. J. Exp. Med. 2004, 200, 1231–1241. [Google Scholar] [CrossRef]
- Bousso, P. T-cell activation by dendritic cells in the lymph node: Lessons from the movies. Nat. Rev. Immunol. 2008, 8, 675–684. [Google Scholar] [CrossRef]
- Tai, Y.; Wang, Q.; Korner, H.; Zhang, L.; Wei, W. Molecular Mechanisms of T Cells Activation by Dendritic Cells in Autoimmune Diseases. Front. Pharmacol. 2018, 9, 642. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.V.; Connors, T.J.; Farber, D.L. Human T Cell Development, Localization, and Function throughout Life. Immunity 2018, 48, 202–213. [Google Scholar] [CrossRef] [Green Version]
- Cyster, J.G.; Schwab, S.R. Sphingosine-1-phosphate and lymphocyte egress from lymphoid organs. Annu. Rev. Immunol. 2012, 30, 69–94. [Google Scholar] [CrossRef]
- Liu, Y.N.; Zhang, H.; Zhang, L.; Cai, T.-T.; Huang, D.-J.; He, J.; Ni, H.-H.; Zhou, F.-J.; Zhang, X.-S.; Li, J. Sphingosine 1 phosphate receptor-1 (S1P1) promotes tumor-associated regulatory T cell expansion: Leading to poor survival in bladder cancer. Cell Death Dis. 2019, 10, 50. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Yuan, Y.; Lin, W.; Zhong, H.; Xu, K.; Qi, X. Roles of sphingosine-1-phosphate signaling in cancer. Cancer Cell Int. 2019, 19, 295. [Google Scholar] [CrossRef]
- Fletcher, A.L.; Acton, S.E.; Knoblich, K. Lymph node fibroblastic reticular cells in health and disease. Nat. Rev. Immunol. 2015, 15, 350–361. [Google Scholar] [CrossRef] [Green Version]
- Dasoveanu, D.C.; Park, H.J.; Ly, C.L.; Shipman, W.D.; Chyou, S.; Kumar, V.; Tarlinton, D.; Ludewig, B.; Mehrara, B.J.; Lum, T.T. Lymph node stromal CCL2 limits antibody responses. Sci. Immunol. 2020, 5, eaaw0693. [Google Scholar] [CrossRef] [PubMed]
- Nadafi, R.; Gago de Graça, C.; Keuning, F.D.; Koning, J.J.; de Kivit, S.; Konijn, T.; Henri, S.; Borst, J.; Reijmers, R.M.; van Baarsen, L.G.M.; et al. Lymph Node Stromal Cells Generate Antigen-Specific Regulatory T Cells and Control Autoreactive T and B Cell Responses. Cell Rep. 2020, 30, 4110–4123. [Google Scholar] [CrossRef]
- Carter, C.L.; Allen, C.; Henson, D.E. Relation of tumor size, lymph node status, and survival in 24,740 breast cancer cases. Cancer 1989, 63, 181–187. [Google Scholar] [CrossRef]
- Hoshida, T.; Isaka, N.; Hagendoorn, J.; di Tomaso, E.; Chen, Y.-L.; Pytowski, B.; Fukumura, D.; Padera, T.P.; Jain, R.K. Imaging steps of lymphatic metastasis reveals that vascular endothelial growth factor-C increases metastasis by increasing delivery of cancer cells to lymph nodes: Therapeutic implications. Cancer Res. 2006, 66, 8065–8075. [Google Scholar] [CrossRef] [Green Version]
- Boucher, Y.; Baxter, L.T.; Jain, R.K. Interstitial pressure gradients in tissue-isolated and subcutaneous tumors: Implications for therapy. Cancer Res. 1990, 50, 4478–4484. [Google Scholar]
- Rofstad, E.K.; Tunheim, S.H.; Mathiesen, B.; Graff, B.A.; Halsør, E.F.; Nilsen, K.; Galappathi, K. Pulmonary and lymph node metastasis is associated with primary tumor interstitial fluid pressure in human melanoma xenografts. Cancer Res. 2002, 62, 661–664. [Google Scholar]
- Hompland, T.; Ellingsen, C.; Øvrebø, K.M.; Rofstad, E.K. Interstitial fluid pressure and associated lymph node metastasis revealed in tumors by dynamic contrast-enhanced MRI. Cancer Res. 2012, 72, 4899–4908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, R.K.; Tong, R.T.; Munn, L.L. Effect of vascular normalization by antiangiogenic therapy on interstitial hypertension, peritumor edema, and lymphatic metastasis: Insights from a mathematical model. Cancer Res. 2007, 67, 2729–2735. [Google Scholar] [CrossRef] [Green Version]
- Rofstad, E.K.; Galappathi, K.; Mathiesen, B.S. Tumor interstitial fluid pressure-a link between tumor hypoxia, microvascular density, and lymph node metastasis. Neoplasia 2014, 16, 586–594. [Google Scholar] [CrossRef] [Green Version]
- Shields, J.D.; Fleury, M.E.; Yong, C.; Tomei, A.A.; Randolph, G.J.; Swartz, M.A. Autologous chemotaxis as a mechanism of tumor cell homing to lymphatics via interstitial flow and autocrine CCR7 signaling. Cancer Cell 2007, 11, 526–538. [Google Scholar] [CrossRef] [Green Version]
- Polacheck, W.J.; Charest, J.L.; Kamm, R.D. Interstitial flow influences direction of tumor cell migration through competing mechanisms. Proc. Natl. Acad. Sci. USA 2011, 108, 11115–11120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evje, S.; Waldeland, J.O. How Tumor Cells Can Make Use of Interstitial Fluid Flow in a Strategy for Metastasis. Cell Mol. Bioeng. 2019, 12, 227–254. [Google Scholar] [CrossRef]
- Shieh, A.C.; Rozansky, H.A.; Hinz, B.; Swartz, M.A. Tumor cell invasion is promoted by interstitial flow-induced matrix priming by stromal fibroblasts. Cancer Res. 2011, 71, 790–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingber, D.E. Mechanical signaling and the cellular response to extracellular matrix in angiogenesis and cardiovascular physiology. Circ. Res. 2002, 91, 877–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janmey, P.A.; Miller, R.T. Mechanisms of mechanical signaling in development and disease. J. Cell Sci. 2011, 124, 9–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martino, F.; Perestrelo, A.R.; Vinarský, V.; Pagliari, S.; Forte, G. Cellular Mechanotransduction: From Tension to Function. Front. Physiol. 2018, 9, 824. [Google Scholar] [CrossRef]
- Borsig, L.; Wolf, M.J.; Roblek, M.; Lorentzen, A.; Heikenwalder, H. Inflammatory chemokines and metastasis--tracing the accessory. Oncogene 2014, 33, 3217–3224. [Google Scholar] [CrossRef] [Green Version]
- Farnsworth, R.H.; Karnezis, T.; Maciburko, S.J.; Mueller, S.N.; Stacker, S.A. The Interplay Between Lymphatic Vessels and Chemokines. Front. Immunol. 2019, 10, 518. [Google Scholar] [CrossRef] [Green Version]
- Stacker, S.A.; Williams, S.P.; Karnezis, T.; Shayan, R.; Fox, S.B.; Achen, M.G. Lymphangiogenesis and lymphatic vessel remodelling in cancer. Nat. Rev. Cancer 2014, 14, 159–172. [Google Scholar] [CrossRef]
- Fankhauser, M.; Broggi, M.A.S.; Potin, L.; Bordry, N.; Jeanbart, L.; Lund, A.W.; Da Costa, E.; Hauert, S.; Rincon-Restrepo, M.; Tremblay, C.; et al. Tumor lymphangiogenesis promotes T cell infiltration and potentiates immunotherapy in melanoma. Sci. Transl. Med. 2017, 9, eaal4712. [Google Scholar] [CrossRef] [Green Version]
- Alitalo, A.; Detmar, M. Interaction of tumor cells and lymphatic vessels in cancer progression. Oncogene 2012, 31, 4499–4508. [Google Scholar] [CrossRef] [Green Version]
- Dadiani, M.; Kalchenko, V.; Yosepovich, A.; Margalit, R.; Hassid, Y.; Degani, H.; Seger, D. Real-time imaging of lymphogenic metastasis in orthotopic human breast cancer. Cancer Res. 2006, 66, 8037–8041. [Google Scholar] [CrossRef] [Green Version]
- Kerjaschki, D.; Bago-Horvath, Z.; Rudas, M.; Sexl, V.; Schneckenleithner, C.; Wolbank, S.; Bartel, G.; Krieger, S.; Kalt, R.; Hantusch, B.; et al. Lipoxygenase mediates invasion of intrametastatic lymphatic vessels and propagates lymph node metastasis of human mammary carcinoma xenografts in mouse. J. Clin. Investig. 2011, 121, 2000–2012. [Google Scholar] [CrossRef]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, J.H.; Yang, J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev. 2013, 27, 2192–2206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olmeda, D.; Montes, A.; Moreno-Bueno, G.; Flores, J.M.; Portillo, F.; Cano, A. Snai1 and Snai2 collaborate on tumor growth and metastasis properties of mouse skin carcinoma cell lines. Oncogene 2008, 27, 4690–4701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olmeda, D.; Moreno-Bueno, G.; Flores, J.M.; Fabra, A.; Portillo, F.; Cano, A. SNAI1 is required for tumor growth and lymph node metastasis of human breast carcinoma MDA-MB-231 cells. Cancer Res. 2007, 67, 11721–11731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miro, C.; Di Cicco, E.; Ambrosio, R.; Mancino, G.; Di Girolamo, D.; Gaetana Cicatiello, A.; Sagliocchi, S.; Nappi, A.; De Stefano, M.A.; Luongo, C.; et al. Thyroid hormone induces progression and invasiveness of squamous cell carcinomas by promoting a ZEB-1/E-cadherin switch. Nat. Commun. 2019, 10, 5410. [Google Scholar] [CrossRef] [PubMed]
- Ou, D.L.; Chien, H.-F.; Chen, C.-L.; Lin, T.-C.; Lin, L.I. Role of Twist in head and neck carcinoma with lymph node metastasis. Anticancer Res. 2008, 28, 1355–1359. [Google Scholar] [PubMed]
- Paz, H.; Pathak, N.; Yang, J. Invading one step at a time: The role of invadopodia in tumor metastasis. Oncogene 2014, 33, 4193–4202. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, M.C.; Gonzalez, S.F.; Welin, J.; Fuxe, J. Epithelial-mesenchymal transition in cancer metastasis through the lymphatic system. Mol. Oncol. 2017, 11, 781–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Q.; Hu, X.; He, W.; Zhao, Y.; Hao, S.; Wu, Q.; Li, S.; Zhang, S.; Shi, M. Fluid shear stress and tumor metastasis. Am. J. Cancer Res. 2018, 8, 763–777. [Google Scholar] [PubMed]
- Dixon, J.B.; Greiner, S.T.; Gashev, A.A.; Cote, G.L.; Moore, J.E.; Zawieja, D.C. Lymph flow, shear stress, and lymphocyte velocity in rat mesenteric prenodal lymphatics. Microcirculation 2006, 13, 597–610. [Google Scholar] [CrossRef]
- Cooper, J.; Giancotti, F.G. Integrin Signaling in Cancer: Mechanotransduction, Stemness, Epithelial Plasticity, and Therapeutic Resistance. Cancer Cell 2019, 35, 347–367. [Google Scholar] [CrossRef]
- Pereira, E.R.; Jones, D.; Jung, K.; Padera, T.P. The lymph node microenvironment and its role in the progression of metastatic cancer. Semin. Cell Dev. Biol. 2015, 38, 98–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, F.; Amano, H.; Eshima, K.; Ito, Y.; Matsui, Y.; Hosono, K.; Kitasato, H.; Iyoda, A.; Iwabuchi, K.; Kumagai, Y.; et al. Prostanoid induces premetastatic niche in regional lymph nodes. J. Clin. Investig. 2014, 124, 4882–4894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Torisu-Itakara, H.; Cochran, A.J.; Kadison, A.; Huynh, Y.; Morton, D.L.; Essner, R. Quantitative analysis of melanoma-induced cytokine-mediated immunosuppression in melanoma sentinel nodes. Clin. Cancer Res. 2005, 11, 107–112. [Google Scholar]
- Mansfield, A.S.; Holtan, S.G.; Grotz, T.E.; Allred, J.B.; Jakub, J.W.; Erickson, L.A.; Markovic, S.N. Regional Immunity in melanoma: Immunosuppressive changes precede nodal metastasis. Mod. Pathol. 2011, 24, 487–494. [Google Scholar] [CrossRef] [Green Version]
- Sleeman, J.P. The lymph node pre-metastatic niche. J. Mol. Med. (Berl.) 2015, 93, 1173–1184. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, S.; Brown, L.F.; Kodama, S.; Paavonen, K.; Alitalo, K.; Detmar, M. VEGF-C-induced lymphangiogenesis in sentinel lymph nodes promotes tumor metastasis to distant sites. Blood 2007, 109, 1010–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirakawa, S.; Kodama, S.; Kunstfeld, R.; Kajiya, K.; Brown, L.F.; Detmar, M. VEGF-A induces tumor and sentinel lymph node lymphangiogenesis and promotes lymphatic metastasis. J. Exp. Med. 2005, 201, 1089–1099. [Google Scholar] [CrossRef] [Green Version]
- Commerford, C.D.; Dieterich, L.C.; He, Y.; Hell, T.; Montoya-Zegarra, J.A.; Noerrelykke, S.F.; Russo, E.; Röcken, M.; Detmar, M. Mechanisms of Tumor-Induced Lymphovascular Niche Formation in Draining Lymph Nodes. Cell Rep. 2018, 25, 3554.e4–3563.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olmeda, D.; Cerezo-Wallis, D.; Riveiro-Falkenbach, E.; Pennacchi, P.C.; Contreras-Alcalde, M.; Ibarz, N.; Cifdaloz, M.; Catena, X.; Calvo, T.G.; Cañón, E.; et al. Whole-body imaging of lymphovascular niches identifies pre-metastatic roles of midkine. Nature 2017, 546, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Hood, J.L.; San, R.S.; Wickline, S.A. Exosomes released by melanoma cells prepare sentinel lymph nodes for tumor metastasis. Cancer Res. 2011, 71, 3792–3801. [Google Scholar] [CrossRef] [Green Version]
- Peinado, H.; Alečković, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; García-Santos, G.; Ghajar, C.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pucci, F.; Garris, C.; Lai, C.P.; Newton, A.; Pfirschke, C.; Engblom, C.; Alvarez, D.; Sprachman, M.; Evavold, C.; Magnuson, A.; et al. SCS macrophages suppress melanoma by restricting tumor-derived vesicle-B cell interactions. Science 2016, 352, 242–246. [Google Scholar] [CrossRef] [Green Version]
- Broggi, M.A.S.; Maillat, L.; Clement, C.C.; Bordry, N.; Corthésy, P.; Auger, A.; Matter, M.; Hamelin, R.; Potin, L.; Demurtas, D.; et al. Tumor-associated factors are enriched in lymphatic exudate compared to plasma in metastatic melanoma patients. J. Exp. Med. 2019, 216, 1091–1107. [Google Scholar] [CrossRef] [Green Version]
- Garrido, F.; Aptsiauri, N.; Doorduijn, E.M.; Garcia Lora, A.M.; van Hall, T. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr. Opin. Immunol. 2016, 39, 44–51. [Google Scholar] [CrossRef] [Green Version]
- de Charette, M.; Marabelle, A.; Houot, R. Turning tumour cells into antigen presenting cells: The next step to improve cancer immunotherapy? Eur. J. Cancer 2016, 68, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Pantel, K.; Schlimok, G.; Kutter, D.; Schaller, G.; Genz, T.; Wiebecke, B.; Backmann, R.; Funke, I.; Riethmüller, G. Frequent down-regulation of major histocompatibility class I antigen expression on individual micrometastatic carcinoma cells. Cancer Res. 1991, 51, 4712–4715. [Google Scholar] [PubMed]
- Erdogdu, I.H. MHC Class 1 and PDL-1 Status of Primary Tumor and Lymph Node Metastatic Tumor Tissue in Gastric Cancers. Gastroenterol. Res. Pract. 2019, 2019, 4785098. [Google Scholar] [CrossRef] [PubMed]
- Axelrod, M.L.; Cook, R.S.; Johnson, D.B.; Balko, J.M. Biological Consequences of MHC-II Expression by Tumor Cells in Cancer. Clin. Cancer Res. 2019, 25, 2392–2402. [Google Scholar] [CrossRef]
- Cromme, F.V.; van Bommel, P.F.; Walboomers, J.M.; Gallee, M.P.; Stern, P.L.; Kenemans, P.; Helmerhorst, T.J.; Stukart, M.J.; Meijer, C.J. Differences in MHC and TAP-1 expression in cervical cancer lymph node metastases as compared with the primary tumours. Br. J. Cancer 1994, 69, 1176–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warabi, M.; Kitagawa, M.; Hirokawa, K. Loss of MHC class II expression is associated with a decrease of tumor-infiltrating T cells and an increase of metastatic potential of colorectal cancer: Immunohistological and histopathological analyses as compared with normal colonic mucosa and adenomas. Pathol. Res. Pract. 2000, 196, 807–815. [Google Scholar] [CrossRef]
- Lin, Y.; Xu, J.; Lan, H. Tumor-associated macrophages in tumor metastasis: Biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019, 12, 76. [Google Scholar] [CrossRef]
- Guo, Q.; Jin, Z.; Yuan, Y.; Liu, R.; Xu, T.; Wei, H.; Xu, X.; He, S.; Chen, S.; Shi, Z.; et al. New Mechanisms of Tumor-Associated Macrophages on Promoting Tumor Progression: Recent Research Advances and Potential Targets for Tumor Immunotherapy. J. Immunol. Res. 2016, 2016, 9720912. [Google Scholar] [CrossRef] [Green Version]
- Ries, C.H.; Cannarile, M.A.; Hoves, S.; Benz, J.; Wartha, K.; Runza, V.; Rey-Giraud, F.; Pradel, L.P.; Feuerhake, F.; Klaman, I.; et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell 2014, 25, 846–859. [Google Scholar] [CrossRef] [Green Version]
- Hollmen, M.; Karaman, S.; Schwager, S.; Lisibach, A.; Christiansen, A.J.; Maksimow, M.; Varga, Z.; Jalkanen, S.; Detmar, M. G-CSF regulates macrophage phenotype and associates with poor overall survival in human triple-negative breast cancer. Oncoimmunology 2016, 5, e1115177. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Xu, J.; Li, M.; Zhang, Y.; Yi, H.; Chen, J.; Dong, L.; Zhang, J.; Huang, Z. Targeting Lymph Node Sinus Macrophages to Inhibit Lymph Node Metastasis. Mol. Ther. Nucleic Acids 2019, 16, 650–662. [Google Scholar] [CrossRef] [Green Version]
- Domogalla, M.P.; Rostan, P.V.; Raker, V.K.; Steinbrink, K. Tolerance through Education: How Tolerogenic Dendritic Cells Shape Immunity. Front. Immunol. 2017, 8, 1764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeVito, N.C.; Plebanek, M.P.; Theivanthiran, B.; Hanks, B.A. Role of Tumor-Mediated Dendritic Cell Tolerization in Immune Evasion. Front. Immunol. 2019, 10, 2876. [Google Scholar] [CrossRef] [PubMed]
- van Pul, K.M.; Vuylsteke, R.J.C.L.M.; van de Ven, R.; Te Velde, E.A.; Rutgers, E.J.T.; van den Tol, P.M.; Stockmann, H.B.A.C.; de Gruijl, T.D. Selectively hampered activation of lymph node-resident dendritic cells precedes profound T cell suppression and metastatic spread in the breast cancer sentinel lymph node. J. Immunother. Cancer 2019, 7, 133. [Google Scholar] [CrossRef] [Green Version]
- Masucci, M.T.; Minopoli, M.; Carriero, M.V. Tumor Associated Neutrophils. Their Role in Tumorigenesis, Metastasis, Prognosis and Therapy. Front. Oncol. 2019, 9, 1146. [Google Scholar] [CrossRef] [Green Version]
- Hiramatsu, S.; Tanaka, H.; Nishimura, J.; Sakimura, C.; Tamura, T.; Toyokawa, T.; Muguruma, K.; Yashiro, M.; Hirakawa, K.; Ohira, M. Neutrophils in primary gastric tumors are correlated with neutrophil infiltration in tumor-draining lymph nodes and the systemic inflammatory response. BMC Immunol. 2018, 19, 13. [Google Scholar] [CrossRef] [Green Version]
- Coffelt, S.B.; Kersten, K.; Doornebal, C.W.; Weiden, J.; Vrijland, K.; Hau, C.-S.; Verstegen, N.J.M.; Ciampricotti, M.; Hawinkels, L.J.A.C.; Jonkers, J.; et al. IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis. Nature 2015, 522, 345–348. [Google Scholar] [CrossRef]
- Wculek, S.K.; Malanchi, I. Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature 2015, 528, 413–417. [Google Scholar] [CrossRef] [Green Version]
- Kessler, D.J.; Mickel, R.A.; Lichtenstein, A. Depressed natural killer cell activity in cervical lymph nodes containing focal metastatic squamous cell carcinoma. Arch. Otolaryngol. Head Neck Surg. 1988, 114, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Schantz, S.P.; Poisson, L. Natural killer cell response to regional lymph node metastases. Arch. Otolaryngol. Head Neck Surg. 1986, 112, 545–551. [Google Scholar] [CrossRef]
- Frazao, A.; Messaoudene, M.; Nunez, N.; Dulphy, N.; Roussin, F.; Sedlik, C.; Zitvogel, L.; Piaggio, E.; Toubert, A.; Caignard, A. CD16(+)NKG2A(high) Natural Killer Cells Infiltrate Breast Cancer-Draining Lymph Nodes. Cancer Immunol. Res. 2019, 7, 208–218. [Google Scholar] [CrossRef]
- Messaoudene, M.; Fregni, G.; Fourmentraux-Neves, E.; Chanal, J.; Maubec, E.; Mazouz-Dorval, S.; Couturaud, B.; Girod, A.; Sastre-Garau, X.; Albert, S.; et al. Mature cytotoxic CD56(bright)/CD16(+) natural killer cells can infiltrate lymph nodes adjacent to metastatic melanoma. Cancer Res. 2014, 74, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Sarvaria, A.; Madrigal, J.A.; Saudemont, A. B cell regulation in cancer and anti-tumor immunity. Cell. Mol. Immunol. 2017, 14, 662–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosser, E.C.; Mauri, C. Regulatory B cells: Origin, phenotype, and function. Immunity 2015, 42, 607–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Zhu, Y.; Du, R.; Pang, N.; Zhang, F.; Dong, D.; Ding, J.; Ding, Y. Role of Regulatory B Cells in the Progression of Cervical Cancer. Mediat. Inflamm. 2019, 2019, 6519427. [Google Scholar] [CrossRef] [PubMed]
- Ganti, S.N.; Albershardt, T.C.; Iritani, B.M.; Ruddell, A. Regulatory B cells preferentially accumulate in tumor-draining lymph nodes and promote tumor growth. Sci. Rep. 2015, 5, 12255. [Google Scholar] [CrossRef] [Green Version]
- Murakami, Y.; Saito, H.; Shimizu, S.; Kono, Y.; Shishido, Y.; Miyatani, K.; Matsunaga, T.; Fukumoto, Y.; Ashida, K.; Sakabe, T.; et al. Increased regulatory B cells are involved in immune evasion in patients with gastric cancer. Sci. Rep. 2019, 9, 13083. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.; Zhang, Y.; Rosenblatt, J.D. B cell regulation of the anti-tumor response and role in carcinogenesis. J. Immunother. Cancer 2016, 4, 40. [Google Scholar] [CrossRef] [Green Version]
- Ho, W.J.; Yarchoan, M.; Charmsaz, S.; Munday, R.M.; Danilova, L.; Sztein, M.B.; Fertig, E.J.; Jaffee, E.M. Multipanel mass cytometry reveals anti-PD-1 therapy-mediated B and T cell compartment remodeling in tumor-draining lymph nodes. JCI Insight 2020, 5, e132286. [Google Scholar] [CrossRef]
- Bruni, D.; Angell, H.K.; Galon, J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat. Rev. Cancer 2020, 20, 662–680. [Google Scholar] [CrossRef]
- Xia, A.; Zhang, Y.; Xu, J.; Yin, T.; Lu, X.-J. T Cell Dysfunction in Cancer Immunity and Immunotherapy. Front. Immunol. 2019, 10, 1719. [Google Scholar] [CrossRef] [Green Version]
- Preynat-Seauve, O.; Contassot, E.; Schuler, P.; Piguet, V.; French, L.E.; Huard, B. Extralymphatic tumors prepare draining lymph nodes to invasion via a T-cell cross-tolerance process. Cancer Res. 2007, 67, 5009–5016. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, S.; Deguchi, K.; Zheng, R.; Tamai, H.; Wang, L.-X.; Cohen, P.A.; Shu, S. Tumor-induced CD11b+Gr-1+ myeloid cells suppress T cell sensitization in tumor-draining lymph nodes. J. Immunol. 2008, 181, 3291–3300. [Google Scholar] [CrossRef] [Green Version]
- Alonso, R.; Flament, H.; Lemoine, S.; Sedlik, C.; Bottasso, E.; Péguillet, I.; Prémel, V.; Denizeau, J.; Salou, M.; Darbois, A.; et al. Induction of anergic or regulatory tumor-specific CD4(+) T cells in the tumor-draining lymph node. Nat. Commun. 2018, 9, 2113. [Google Scholar] [CrossRef] [PubMed]
- Nunez, N.G.; Tosello Boari, J.; Nalio Ramos, R.; Richer, W.; Cagnard, N.; Anderfuhren, C.D.; Niborski, L.L.; Bigot, J.; Meseure, D.; De La Rochere, P.; et al. Tumor invasion in draining lymph nodes is associated with Treg accumulation in breast cancer patients. Nat. Commun. 2020, 11, 3272. [Google Scholar] [CrossRef] [PubMed]
- Hargadon, K.M.; Brinkman, C.C.; Sheasley-O’neill, S.L.; Nichols, L.A.; Bullock, T.N.J.; Engelhard, V.H. Incomplete differentiation of antigen-specific CD8 T cells in tumor-draining lymph nodes. J. Immunol. 2006, 177, 6081–6090. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.D.; Baban, B.; Chandler, P.; Hou, D.Y.; Singh, N.; Yagita, H.; Azuma, M.; Blazar, B.R.; Mellor, A.L.; Munn, D.H. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. J. Clin. Investig. 2007, 117, 2570–2582. [Google Scholar] [CrossRef] [Green Version]
- Krishnamurty, A.T.; Turley, S.J. Lymph node stromal cells: Cartographers of the immune system. Nat. Immunol. 2020, 21, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Roozendaal, R.; Mempel, T.R.; Pitcher, L.A.; Gonzalez, S.F.; Verschoor, A.; Mebius, R.E.; von Andrian, U.H.; Carroll, M.C. Conduits mediate transport of low-molecular-weight antigen to lymph node follicles. Immunity 2009, 30, 264–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soudja, S.M.; Henri, S.; Mello, M.; Chasson, L.; Mas, A.; Wehbe, M.; Auphan-Anezin, N.; Leserman, L.; Van den Eynde, B.; Schmitt-Verhulst, A.-M. Disrupted lymph node and splenic stroma in mice with induced inflammatory melanomas is associated with impaired recruitment of T and dendritic cells. PLoS ONE 2011, 6, e22639. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Zhao, L.; Liu, L.; Yang, Y.; Guo, B.; Zhu, B. Disrupted fibroblastic reticular cells and interleukin-7 expression in tumor draining lymph nodes. Oncol. Lett. 2017, 14, 2954–2960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riedel, A.; Shorthouse, D.; Haas, L.; Hall, B.A.; Shields, J. Tumor-induced stromal reprogramming drives lymph node transformation. Nat. Immunol. 2016, 17, 1118–1127. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, C.; He, Y.; Wu, H.; Wang, Z.; Song, W.; Li, W.; He, W.; Cai, S.; Zhan, W. Lymphatic endothelial cell-secreted CXCL1 stimulates lymphangiogenesis and metastasis of gastric cancer. Int. J. Cancer 2012, 130, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Sarrou, E.; Podgrabinska, S.; Cassella, M.; Kumar Mungamuri, S.; Feirt, N.; Gordon, R.; Nagi, C.S.; Wang, Y.; Entenberg, D.; et al. Tumor cell entry into the lymph node is controlled by CCL1 chemokine expressed by lymph node lymphatic sinuses. J. Exp. Med. 2013, 210, 1509–1528. [Google Scholar] [CrossRef] [Green Version]
- Tokumoto, M.; Tanaka, H.; Tauchi, Y.; Tamura, T.; Toyokawa, T.; Kimura, K.; Muguruma, K.; Yashiro, M.; Maeda, K.; Hirakawa, K.; et al. Immunoregulatory Function of Lymphatic Endothelial Cells in Tumor-draining Lymph Nodes of Human Gastric Cancer. Anticancer Res. 2017, 37, 2875–2883. [Google Scholar] [PubMed] [Green Version]
- Lund, A.W.; Duraes, F.V.; Hirosue, S.; Raghavan, V.R.; Nembrini, C.; Thomas, S.N.; Issa, A.; Hugues, S.; Swartz, M.A. VEGF-C promotes immune tolerance in B16 melanomas and cross-presentation of tumor antigen by lymph node lymphatics. Cell Rep. 2012, 1, 191–199. [Google Scholar] [CrossRef]
- Jeong, H.S.; Jones, D.; Liao, S.; Wattson, D.A.; Cui, C.H.; Duda, D.G.; Willett, C.G.; Jain, R.K.; Padera, T.P. Investigation of the Lack of Angiogenesis in the Formation of Lymph Node Metastases. J. Natl. Cancer Inst. 2015, 107, djv155. [Google Scholar] [CrossRef] [Green Version]
- Elia, I.; Doglioni, G.; Fendt, S.M. Metabolic Hallmarks of Metastasis Formation. Trends Cell Biol. 2018, 28, 673–684. [Google Scholar] [CrossRef]
- Pascual, G.; Avgustinova, A.; Mejetta, S.; Martín, M.; Castellanos, A.; Attolini, C.S.-O.; Berenguer, A.; Prats, N.; Toll, A.; Hueto, J.A.; et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 2017, 541, 41–45. [Google Scholar] [CrossRef]
- Lee, C.K.; Jeong, S.-H.; Jang, C.; Bae, H.; Kim, Y.H.; Park, I.; Kim, S.K.; Koh, G.Y. Tumor metastasis to lymph nodes requires YAP-dependent metabolic adaptation. Science 2019, 363, 644–649. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Liao, Y.; Liu, P.; Du, Q.; Liang, Y.; Ooi, S.; Qin, S.; He, S.; Yao, S.; Wang, W. FABP5 promotes lymph node metastasis in cervical cancer by reprogramming fatty acid metabolism. Theranostics 2020, 10, 6561–6580. [Google Scholar] [CrossRef]
- Maulucci, G.; Cohen, O.; Daniel, B.; Sansone, A.; Petropoulou, P.I.; Filou, S.; Spyridonidis, A.; Pani, G.; De Spirito, M.; Chatgilialoglu, C.; et al. Fatty acid-related modulations of membrane fluidity in cells: Detection and implications. Free Radic. Res. 2016, 50, S40–S50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taraboletti, G.; Perin, L.; Bottazzi, B.; Mantovani, A.; Giavazzi, R.; Salmona, M. Membrane fluidity affects tumor-cell motility, invasion and lung-colonizing potential. Int. J. Cancer 1989, 44, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Ubellacker, J.M.; Tasdogan, A.; Ramesh, V.; Shen, B.; Mitchell, E.C.; Martin-Sandoval, M.S.; Gu, Z.; McCormick, M.L.; Durham, A.B.; Spitz, D.R.; et al. Lymph protects metastasizing melanoma cells from ferroptosis. Nature 2020, 585, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Naxerova, K.; Reiter, J.G.; Brachtel, E.; Lennerz, J.K.; van de Wetering, M.; Rowan, A.; Cai, T.; Clevers, H.; Swanton, C.; Nowak, M.A.; et al. Origins of lymphatic and distant metastases in human colorectal cancer. Science 2017, 357, 55–60. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Zhang, L.; Xu, T.; Xue, R.; Yu, L.; Zhu, Y.; Wu, Y.; Zhang, Q.; Li, D.; Shen, S.; et al. Mapping the spreading routes of lymphatic metastases in human colorectal cancer. Nat. Commun. 2020, 11, 1993. [Google Scholar] [CrossRef]
- Siegel, M.B.; He, X.; Hoadley, K.A.; Hoyle, A.; Pearce, J.B.; Garrett, A.L.; Kumar, S.; Moylan, V.J.; Brady, C.M.; Van Swearingen, A.E.; et al. Integrated RNA and DNA sequencing reveals early drivers of metastatic breast cancer. J. Clin. Investig. 2018, 128, 1371–1383. [Google Scholar] [CrossRef]
- Ullah, I.; Karthik, G.-M.; Alkodsi, A.; Kjällquist, U.; Stålhammar, G.; Lövrot, J.; Martinez, N.-F.; Lagergren, J.; Hautaniemi, S.; Hartman, J.; et al. Evolutionary history of metastatic breast cancer reveals minimal seeding from axillary lymph nodes. J. Clin. Investig. 2018, 128, 1355–1370. [Google Scholar] [CrossRef] [Green Version]
- Quinn, J.J.; Jones, M.G.; Okimoto, R.A.; Nanjo, S.; Chan, M.M.; Yosef, N.; Bivona, T.G.; Weissman, J.S. Single-cell lineages reveal the rates, routes, and drivers of metastasis in cancer xenografts. Science 2021, 371, eabc1944. [Google Scholar] [CrossRef]
- Giuliano, A.E.; Hunt, K.K.; Ballman, K.V.; Beitsch, P.D.; Whitworth, P.W.; Blumencranz, P.W.; Marilyn Leitch, A.; Saha, S.; McCall, L.M.; Morrow, M. Axillary dissection vs no axillary dissection in women with invasive breast cancer and sentinel node metastasis: A randomized clinical trial. JAMA 2011, 305, 569–575. [Google Scholar] [CrossRef] [Green Version]
- Faries, M.B.; Thompson, J.F.; Cochran, A.J.; Andtbacka, R.H.; Mozzillo, N.; Zager, J.S.; Jahkola, T.; Bowles, T.L.; Testori, A.; Beitsch, P.D.; et al. Completion Dissection or Observation for Sentinel-Node Metastasis in Melanoma. N. Engl. J. Med. 2017, 376, 2211–2222. [Google Scholar] [CrossRef]
- Galimberti, V.; Cole, B.F.; Zurrida, S.; Viale, G.; Luini, A.; Veronesi, P.; Baratella, P.; Chifu, C.; Sargenti, M.; Intra, M.; et al. Axillary dissection versus no axillary dissection in patients with sentinel-node micrometastases (IBCSG 23-01): A phase 3 randomised controlled trial. Lancet Oncol. 2013, 14, 297–305. [Google Scholar] [CrossRef] [Green Version]
- Pereira, E.R.; Kedrin, D.; Seano, G.; Gautier, O.; Meijer, E.F.J.; Jones, D.; Chin, S.-M.; Kitahara, S.; Bouta, E.M.; Chang, J.; et al. Lymph node metastases can invade local Blood vessels, exit the node, and colonize distant organs in mice. Science 2018, 359, 1403–1407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, M.; Assen, F.P.; Leithner, A.; Abe, J.; Schachner, H.; Asfour, G.; Bago-Horvath, Z.; Stein, J.V.; Uhrin, P.; Sixt, M.; et al. Lymph node blood vessels provide exit routes for metastatic tumor cell dissemination in mice. Science 2018, 359, 1408–1411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, A.; Lang, J. Survival Analysis of Lymph Node Resection in Ovarian Cancer: A Population-Based Study. Front. Oncol. 2020, 10, 355. [Google Scholar] [CrossRef] [PubMed]
- Jagsi, R.; Chadha, M.; Moni, J.; Ballman, K.; Laurie, F.; Buchholz, T.A.; Giuliano, A.; Haffty, B.G. Radiation field design in the ACOSOG Z0011 (Alliance) Trial. J. Clin. Oncol. 2014, 32, 3600–3606. [Google Scholar] [CrossRef] [Green Version]
- Whelan, T.J.; Olivoto, I.A.; Parulekar, W.R.; Ackerman, I.; Chua, B.H.; Nabid, A.; Vallis, K.A.; White, J.R.; Rousseau, P.; Fortin, A.; et al. Regional Nodal Irradiation in Early-Stage Breast Cancer. N. Engl. J. Med. 2015, 373, 307–316. [Google Scholar] [CrossRef] [Green Version]
- Poortmans, P.M.; Collette, S.; Kirkove, C.; Van Limbergen, E.; Budach, V.; Stuikmans, H.; Collete, L.; Fourquet, A.; Maingon, P.; Valli, M.; et al. Internal Mammary and Medial Supraclavicular Irradiation in Breast Cancer. N. Engl. J. Med. 2015, 373, 317–327. [Google Scholar] [CrossRef] [Green Version]
- Hiller, J.G.; Perry, N.J.; Poulogiannis, G.; Riedel, B.; Sloan, E.K. Perioperative events influence cancer recurrence risk after surgery. Nat. Rev. Clin. Oncol. 2018, 15, 205–218. [Google Scholar] [CrossRef]
- Tohme, S.; Simmons, R.L.; Tsung, A. Surgery for Cancer: A Trigger for Metastases. Cancer Res. 2017, 77, 1548–1552. [Google Scholar] [CrossRef] [Green Version]
- Alieva, M.; van Rheenen, J.; Broekman, M.L.D. Potential impact of invasive surgical procedures on primary tumor growth and metastasis. Clin. Exp. Metastasis 2018, 35, 319–331. [Google Scholar] [CrossRef] [Green Version]
- Hsiao, P.C.; Liu, J.-T.; Lin, C.-L.; Chou, W.; Lu, S.-R. Risk of breast cancer recurrence in patients receiving manual lymphatic drainage: A hospital-based cohort study. Ther. Clin. Risk Manag. 2015, 11, 349–358. [Google Scholar] [CrossRef] [Green Version]
- Fransen, M.F.; Schoonderwoerd, M.; Knopf, P.; Camps, M.G.; Hawinkels, L.J.; Kneilling, M.; van Hall, T.; Ossendorp, F. Tumor-draining lymph nodes are pivotal in PD-1/PD-L1 checkpoint therapy. JCI Insight 2018, 3, e124507. [Google Scholar] [CrossRef] [Green Version]
- Caudle, A.S.; Yang, W.T.; Mittendorf, E.A.; Black, D.M.; Hwang, R.; Hobbs, B.; Hunt, K.K.; Krishnamurthy, S.; Kuerer, H.M. Selective surgical localization of axillary lymph nodes containing metastases in patients with breast cancer: A prospective feasibility trial. JAMA Surg. 2015, 150, 137–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boughey, J.C.; Ballman, K.V.; Le-Petross, H.T.; McCall, L.M.; Mittendorf, E.A.; Ahrendt, G.M.; Wilke, L.G.; Taback, B.; Feliberti, E.C.; Hunt, K.K. Identification and Resection of Clipped Node Decreases the False-negative Rate of Sentinel Lymph Node Surgery in Patients Presenting with Node-positive Breast Cancer (T0-T4, N1-N2) Who Receive Neoadjuvant Chemotherapy: Results From ACOSOG Z1071 (Alliance). Ann. Surg. 2016, 263, 802–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donker, M.; Straver, M.E.; Wesseling, J.; Loo, C.E.; Schot, M.; Drukker, C.A.; van Tinteren, H.; Sonke, G.S.; Rutgers, E.J.T.; Vrancken Peeters, M.J.T.F.D. Marking axillary lymph nodes with radioactive iodine seeds for axillary staging after neoadjuvant systemic treatment in breast cancer patients: The MARI procedure. Ann. Surg. 2015, 261, 378–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Moreno, J.L.; Benjumeda-Gonzalez, A.M.; Amerigo-Góngora, M.; Landra-Dulanto, P.J.; Gonzalez-Corena, Y.; Gomez-Menchero, J. Targeted axillary dissection in breast cancer by marking lymph node metastasis with a magnetic seed before starting neoadjuvant treatment. J. Surg. Case Rep. 2019, 2019, rjz344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Wang, T.; Yang, L.; Wang, Y.; Li, H.; Zhou, X.; Zhao, W.; Ren, J.; Li, X.; Tian, J.; et al. Preoperative Prediction of Axillary Lymph Node Metastasis in Breast Cancer Using Mammography-Based Radiomics Method. Sci. Rep. 2019, 9, 4429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fletcher, C.V.; Staskus, K.; Wietgrefe, S.W.; Rothenberger, M.; Reilly, C.; Chipman, J.G.; Beilman, G.J.; Khoruts, A.; Thorkelson, A.; Schmidt, T.E.; et al. Persistent HIV-1 replication is associated with lower antiretroviral drug concentrations in lymphatic tissues. Proc. Natl. Acad. Sci. USA 2014, 111, 2307–2312. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Bagby, T.R.; Cohen, M.S.; Forrest, M.L. Drug delivery to the lymphatic system: Importance in future cancer diagnosis and therapies. Expert Opin. Drug Deliv. 2009, 6, 785–792. [Google Scholar] [CrossRef]
- Schudel, A.; Francis, D.M.; Thomas, S.N. Material design for lymph node drug delivery. Nat. Rev. Mater. 2019, 4, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Y.; Lu, W.Y. Recent advances in lymphatic targeted drug delivery system for tumor metastasis. Cancer Biol. Med. 2014, 11, 247–254. [Google Scholar]
- Akamo, Y.; Mizuno, I.; Yotsuyanagi, T.; Ichino, T.; Tanimoto, N.; Yamamoto, T.; Nagata, M.; Takeyama, H.; Shinagawa, N.; Yura, J.; et al. Chemotherapy targeting regional lymph nodes by gastric submucosal injection of liposomal adriamycin in patients with gastric carcinoma. Jpn. J. Cancer Res. 1994, 85, 652–658. [Google Scholar] [CrossRef]
- Cai, S.; Yang, Q.; Bagby, T.R.; Forrest, M.L. Lymphatic drug delivery using engineered liposomes and solid lipid nanoparticles. Adv. Drug Deliv. Rev. 2011, 63, 901–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabral, H.; Makino, J.; Matsumoto, Y.; Mi, P.; Wu, H.; Nomoto, T.; Toh, K.; Yamada, N.; Higuchi, Y.; Konishi, S.; et al. Systemic Targeting of Lymph Node Metastasis through the Blood Vascular System by Using Size-Controlled Nanocarriers. ACS Nano 2015, 9, 4957–4967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Li, H.-J.; Luo, Y.-L.; Xu, C.-F.; Du, X.-J.; Du, J.-Z.; Wang, J. Enhanced Primary Tumor Penetration Facilitates Nanoparticle Draining into Lymph Nodes after Systemic Injection for Tumor Metastasis Inhibition. ACS Nano 2019, 13, 8648–8658. [Google Scholar] [CrossRef] [PubMed]
- Fromen, C.A.; Robbins, G.R.; Shen, T.W.; Kai, M.P.; Ting, J.P.Y.; DeSimone, J.M. Controlled analysis of nanoparticle charge on mucosal and systemic antibody responses following pulmonary immunization. Proc. Natl. Acad. Sci. USA 2015, 112, 488–493. [Google Scholar] [CrossRef] [Green Version]
- Kaminskas, L.M.; Porter, C.J. Targeting the lymphatics using dendritic polymers (dendrimers). Adv. Drug Deliv. Rev. 2011, 63, 890–900. [Google Scholar] [CrossRef]
- Mehta, D.; Leong, N.; McLeod, V.M.; Kelly, B.D.; Pathak, R.; Owen, D.J.; Porter, C.J.H.; Kaminskas, L.M. Reducing Dendrimer Generation and PEG Chain Length Increases Drug Release and Promotes Anticancer Activity of PEGylated Polylysine Dendrimers Conjugated with Doxorubicin via a Cathepsin-Cleavable Peptide Linker. Mol. Pharm. 2018, 15, 4568–4576. [Google Scholar] [CrossRef]
- Ryan, G.M.; McLeod, V.M.; Mehta, D.; Kelly, B.D.; Stanislawski, P.C.; Owen, D.J.; Kaminskas, L.M.; Porter, C.J.H. Lymphatic transport and lymph node targeting of methotrexate-conjugated PEGylated dendrimers are enhanced by reducing the length of the drug linker or masking interactions with the injection site. Nanomedicine 2017, 13, 2485–2494. [Google Scholar] [CrossRef]
- Reddy, S.T.; Rehor, A.; Schmoekel, H.G.; Hubbell, J.A.; Swartz, M.A. In vivo targeting of dendritic cells in lymph nodes with poly(propylene sulfide) nanoparticles. J. Control. Release 2006, 112, 26–34. [Google Scholar] [CrossRef]
- Hirosue, S.; Kourtis, I.C.; van der Vlies, A.J.; Hubbell, J.A.; Swartz, M.A. Antigen delivery to dendritic cells by poly(propylene sulfide) nanoparticles with disulfide conjugated peptides: Cross-presentation and T cell activation. Vaccine 2010, 28, 7897–7906. [Google Scholar] [CrossRef]
- Jeanbart, L.; Ballester, M.; de Titta, A.; Corthésy, P.; Romero, P.; Hubbell, J.A.; Swartz, M.A. Enhancing efficacy of anticancer vaccines by targeted delivery to tumor-draining lymph nodes. Cancer Immunol. Res. 2014, 2, 436–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, S.N.; Vokali, E.; Lund, A.W.; Hubbell, J.A.; Swartz, M.A. Targeting the tumor-draining lymph node with adjuvanted nanoparticles reshapes the anti-tumor immune response. Biomaterials 2014, 35, 814–824. [Google Scholar] [CrossRef] [Green Version]
- Reddy, S.T.; van der Vlies, A.J.; Simeoni, E.; Angeli, V.; Randolph, G.J.; O’Neil, C.P.; Lee, L.K.; Swartz, M.A.; Hubbell, J.A. Exploiting lymphatic transport and complement activation in nanoparticle vaccines. Nat. Biotechnol. 2007, 25, 1159–1164. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.N.; van der Vlies, A.J.; O’Neil, C.P.; Reddy, S.T.; Yu, S.T.; Giorgio, T.D.; Swartz, M.A.; Hubbell, J.A. Engineering complement activation on polypropylene sulfide vaccine nanoparticles. Biomaterials 2011, 32, 2194–2203. [Google Scholar] [CrossRef] [PubMed]
- Schudel, A.; Poole Chapman, A.; Yau, M.-K.; Higginson, C.J.; Francis, D.M.; Manspeaker, M.P.; Chua Avecilla, A.R.; Rohner, N.A.; Finn, M.G.; Napier, S. Thomas Programmable multistage drug delivery to lymph nodes. Nat. Nanotechnol. 2020, 15, 491–499. [Google Scholar] [CrossRef]
- Yan, Z.; Zhan, C.; Wen, Z.; Feng, L.; Wang, F.; Liu, Y.; Yang, X.; Dong, Q.; Liu, M.; Lu, W. LyP-1-conjugated doxorubicin-loaded liposomes suppress lymphatic metastasis by inhibiting lymph node metastases and destroying tumor lymphatics. Nanotechnology 2011, 22, 415103. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Stephan, M.T.; Gai, S.A.; Abraham, W.; Shearer, A.; Irvin, D.J. In vivo targeting of adoptively transferred T-cells with antibody- and cytokine-conjugated liposomes. J. Control. Release 2013, 172, 426–435. [Google Scholar] [CrossRef] [Green Version]
- Cruz, L.J.; Rosalia, R.A.; Kleinovink, J.W.; Rueda, F.; Löwik, C.W.G.M.; Ossendorp, F. Targeting nanoparticles to CD40, DEC-205 or CD11c molecules on dendritic cells for efficient CD8(+) T cell response: A comparative study. J. Control. Release 2014, 192, 209–218. [Google Scholar] [CrossRef]
- Hayashi, K.; Zhao, M.; Yamauchi, K.; Yamamoto, N.; Tsuchiya, H.; Tomita, K.; Hoffman, R.M. Cancer metastasis directly eradicated by targeted therapy with a modified Salmonella typhimurium. J. Cell Biochem. 2009, 106, 992–998. [Google Scholar] [CrossRef] [Green Version]
- Goel, H.L.; Mercurio, A.M. VEGF targets the tumour cell. Nat. Rev. Cancer 2013, 13, 871–882. [Google Scholar] [CrossRef]
- Karaman, S.; Detmar, M. Mechanisms of lymphatic metastasis. J. Clin. Investig. 2014, 124, 922–928. [Google Scholar] [CrossRef] [Green Version]
- Meadows, K.L.; Hurwitz, H.I. Anti-VEGF therapies in the clinic. Cold Spring Harb. Perspect. Med. 2012, 2, a006577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, T.A.; Gore, M.E. Sunitinib in the treatment of metastatic renal cell carcinoma. Ther. Adv. Urol. 2016, 8, 348–371. [Google Scholar] [CrossRef] [Green Version]
- Padera, T.P.; Kuo, A.H.; Hoshida, T.; Liao, S.; Lobo, J.; Kozak, K.R.; Fukumura, D.; Jain, R.K. Differential response of primary tumor versus lymphatic metastasis to VEGFR-2 and VEGFR-3 kinase inhibitors cediranib and vandetanib. Mol. Cancer Ther. 2008, 7, 2272–2279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gengenbacher, N.; Singhal, M.; Mogler, C.; Hai, L.; Milde, L.; Ahmed, A.; Pari, A.; Besemfelder, E.; Fricke, C.; Baumann, D.; et al. Timed Ang2-Targeted Therapy Identifies the Angiopoietin-Tie Pathway as Key Regulator of Fatal Lymphogenous Metastasis. Cancer Discov. 2021, 11, 424–445. [Google Scholar] [CrossRef]
- Muller, A.; Homey, B.; Soto, H.; Ge, N.; Catron, D.; Buchanan, M.E.; McClanahan, T.; Murphy, E.; Yuan, W.; Wagner, S.N.; et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001, 410, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Mollica Poeta, V.; Massara, M.; Capucetti, A.; Bonecchi, R. Chemokines and Chemokine Receptors: New Targets for Cancer Immunotherapy. Front. Immunol. 2019, 10, 379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walenkamp, A.M.E.; Lapa, C.; Herrmann, K.; Wester, H.-J. CXCR4 Ligands: The Next Big Hit? J. Nucl. Med. 2017, 58, 77S–82S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pernas, S.; Martin, M.; Kaufman, P.A.; Gil-Martin, M.; Gomez Pardo, P.; Lopez-Tarruella, S.; Manso, L.; Ciruelos, E.; Perez-Fidalgo, J.A.; Hernando, C.; et al. Balixafortide plus eribulin in HER2-negative metastatic breast cancer: A phase 1, single-arm, dose-escalation trial. Lancet Oncol. 2018, 19, 812–824. [Google Scholar] [CrossRef]
- Bockorny, B.; Semenisty, V.; Macarulla, T.; Borazanci, E.; Wolpin, B.M.; Stemmer, S.M.; Golan, T.; Geva, R.; Borad, M.J.; Pedersen, K.S.; et al. BL-8040, a CXCR4 antagonist, in combination with pembrolizumab and chemotherapy for pancreatic cancer: The COMBAT trial. Nat. Med. 2020, 26, 878–885. [Google Scholar] [CrossRef] [PubMed]
- Fatehi Hassanabad, A. Current perspectives on statins as potential anti-cancer therapeutics: Clinical outcomes and underlying molecular mechanisms. Transl. Lung Cancer Res. 2019, 8, 692–699. [Google Scholar] [CrossRef] [PubMed]
- Toloczko-Iwaniuk, N.; Dziemiańczyk-Pakieła, D.; Nowaszewska, B.K.; Celińska-Janowicz, K.; Miltyk, W. Celecoxib in Cancer Therapy and Prevention-Review. Curr. Drug Targets 2019, 20, 302–315. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Huang, J. The expanded role of fatty acid metabolism in cancer: New aspects and targets. Precis. Clin. Med. 2019, 2, 183–191. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, H.; Lei, P.-j.; Padera, T.P. Progression of Metastasis through Lymphatic System. Cells 2021, 10, 627. https://doi.org/10.3390/cells10030627
Zhou H, Lei P-j, Padera TP. Progression of Metastasis through Lymphatic System. Cells. 2021; 10(3):627. https://doi.org/10.3390/cells10030627
Chicago/Turabian StyleZhou, Hengbo, Pin-ji Lei, and Timothy P. Padera. 2021. "Progression of Metastasis through Lymphatic System" Cells 10, no. 3: 627. https://doi.org/10.3390/cells10030627
APA StyleZhou, H., Lei, P. -j., & Padera, T. P. (2021). Progression of Metastasis through Lymphatic System. Cells, 10(3), 627. https://doi.org/10.3390/cells10030627