
Cell–Matrix Interactions in the Eye: From Cornea to Choroid

 , , and
, , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
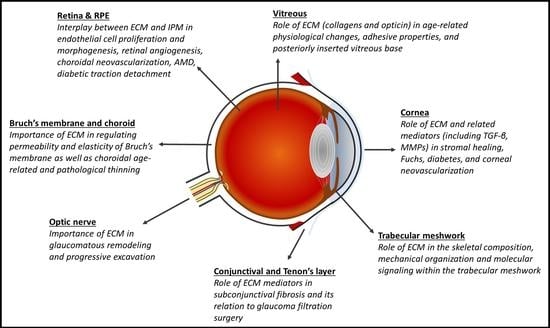
1. Introduction
2. Cornea
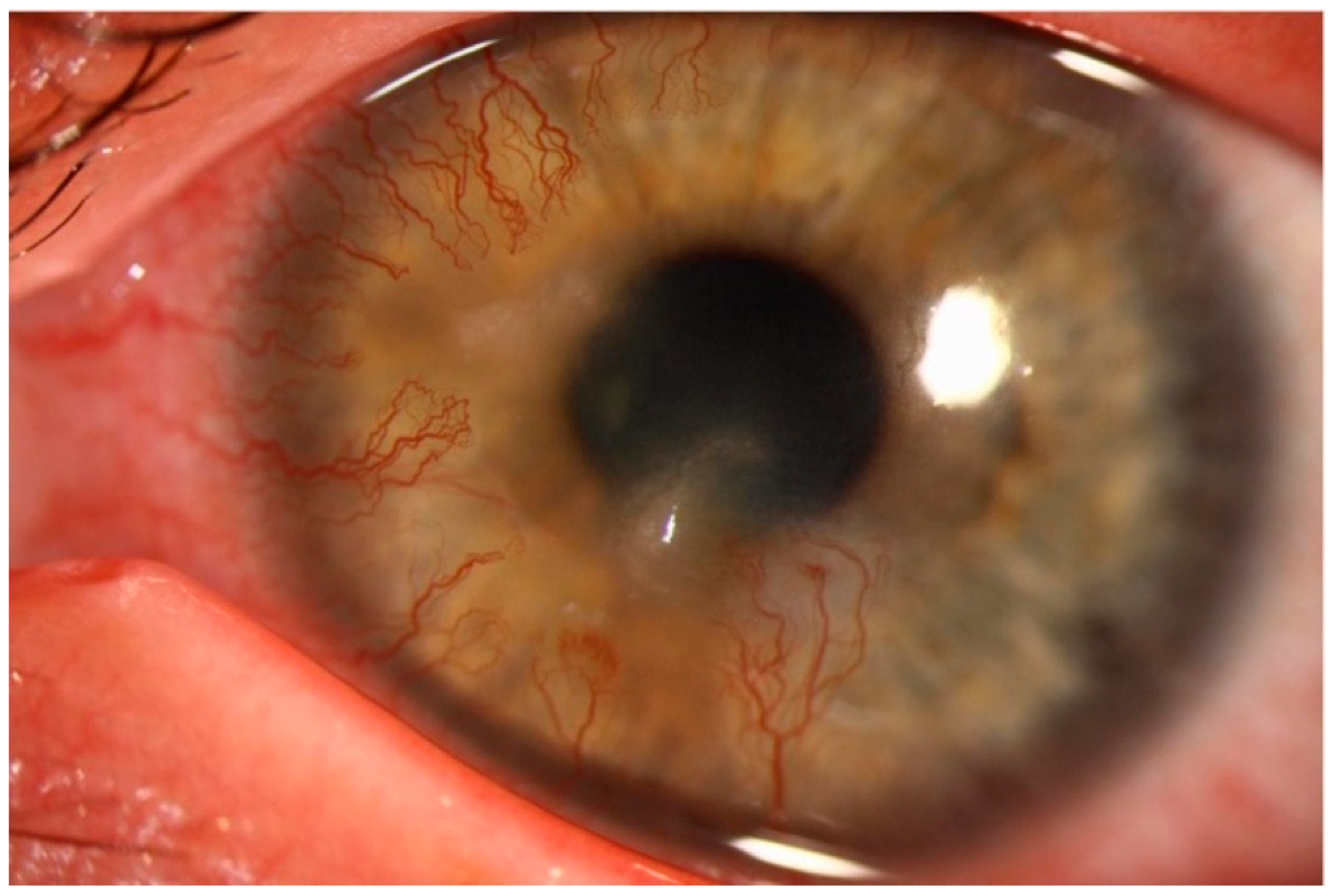
2.1. Stromal Extracellular Matrix Alterations and Corneal Neovascularization
2.2. Fuchs Endothelial Corneal Dystrophy (FECD) and EDM Pathology
2.3. Diabetes and EDM Pathology
3. Glaucoma
3.1. Trabecular Meshwork
3.2. Optic Nerve
3.3. Conjunctival and Tenon’s Layer
4. Vitreous
4.1. Age-Induced ECM Changes in the Vitreous Causing Retinal Detachment
4.2. Posteriorly Inserted Vitreous Base
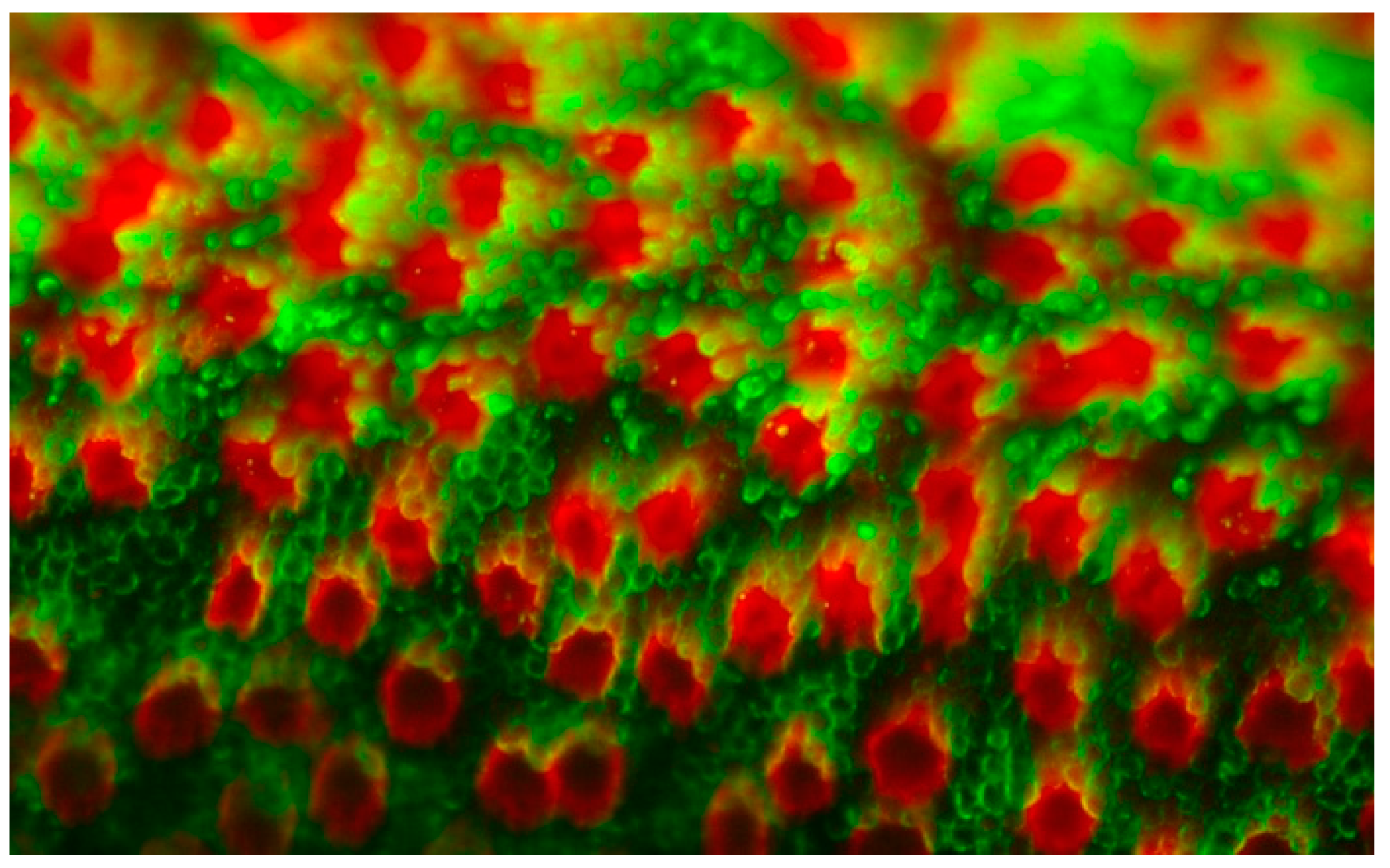
5. Retina and Retinal Pigment Epithelium (RPE)
5.1. Retinal Endothelial Cells and Angiogenesis
5.2. Angiogenesis and Fibrosis in Proliferative Diabetic Retinopathy
5.3. Choroidal Neovascularization, Autosomal Dominant Radial Drusen and AMD
6. Bruch’s Membrane and Choroid
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Reinhard, J.; Joachim, S.C.; Faissner, A. Extracellular matrix remodeling during retinal development. Exp. Eye Res. 2015, 133, 132–140. [Google Scholar] [CrossRef]
- Yue, B. Biology of the extracellular matrix: An overview. J. Glaucoma 2014, 23, S20–S23. [Google Scholar] [CrossRef]
- Hubmacher, D.; Apte, S.S. The biology of the extracellular matrix: Novel insights. Curr. Opin. Rheumatol. 2013, 25, 65–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Ubaidi, M.R.; Naash, M.I.; Conley, S.M. A perspective on the role of the extracellular matrix in progressive retinal degenerative disorders. Investig. Ophthalmol. Vis. Sci. 2013, 54, 8119–8124. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.H.; Huang, Y.H.; Cunningham, C.M.; Han, K.Y.; Chang, M.; Seiki, M.; Zhou, Z.; Azar, D.T. Matrix metalloproteinase 14 modulates signal transduction and angiogenesis in the cornea. Surv. Ophthalmol. 2016, 61, 478–497. [Google Scholar] [CrossRef]
- Tshionyi, M.; Shay, E.; Lunde, E.; Lin, A.; Han, K.Y.; Jain, S.; Chang, J.H.; Azar, D.T. Hemangiogenesis and lymphangiogenesis in corneal pathology. Cornea 2012, 31, 74–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourne, W.M.; Nelson, L.R.; Hodge, D.O. Central corneal endothelial cell changes over a ten-year period. Investig. Ophthalmol. Vis. Sci. 1997, 38, 779–782. [Google Scholar]
- Greiner, M.A.; Rixen, J.J.; Wagoner, M.D.; Schmidt, G.A.; Stoeger, C.G.; Straiko, M.D.; Zimmerman, M.B.; Kitzmann, A.S.; Goins, K.M. Diabetes mellitus increases risk of unsuccessful graft preparation in Descemet membrane endothelial keratoplasty: A multicenter study. Cornea 2014, 33, 1129–1133. [Google Scholar] [CrossRef]
- Lass, J.H.; Benetz, B.A.; Patel, S.V.; Szczotka-Flynn, L.B.; O’Brien, R.; Ayala, A.R.; Maguire, M.G.; Daoud, Y.J.; Greiner, M.A.; Hannush, S.B.; et al. Donor, Recipient, and Operative Factors Associated With Increased Endothelial Cell Loss in the Cornea Preservation Time Study. JAMA Ophthalmol. 2019, 137, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Terry, M.A.; Aldave, A.J.; Szczotka-Flynn, L.B.; Liang, W.; Ayala, A.R.; Maguire, M.G.; Croasdale, C.; Daoud, Y.J.; Dunn, S.P.; Hoover, C.K.; et al. Donor, Recipient, and Operative Factors Associated with Graft Success in the Cornea Preservation Time Study. Ophthalmology 2018, 125, 1700–1709. [Google Scholar] [CrossRef]
- Cursiefen, C.; Küchle, M.; Naumann, G.O. Angiogenesis in corneal diseases: Histopathologic evaluation of 254 human corneal buttons with neovascularization. Cornea 1998, 17, 611–613. [Google Scholar] [CrossRef]
- Netto, M.V.; Mohan, R.R.; Ambrosio, R., Jr.; Hutcheon, A.E.; Zieske, J.D.; Wilson, S.E. Wound healing in the cornea: A review of refractive surgery complications and new prospects for therapy. Cornea 2005, 24, 509–522. [Google Scholar] [CrossRef] [PubMed]
- Anitua, E.; de la Fuente, M.; Muruzabal, F.; Riestra, A.; Merayo-Lloves, J.; Orive, G. Plasma rich in growth factors (PRGF) eye drops stimulates scarless regeneration compared to autologous serum in the ocular surface stromal fibroblasts. Exp. Eye Res. 2015, 135, 118–126. [Google Scholar] [CrossRef]
- Warejcka, D.J.; Vaughan, K.A.; Bernstein, A.M.; Twining, S.S. Differential conversion of plasminogen to angiostatin by human corneal cell populations. Mol. Vis. 2005, 11, 859–868. [Google Scholar] [PubMed]
- Onguchi, T.; Han, K.Y.; Chang, J.H.; Azar, D.T. Membrane type-1 matrix metalloproteinase potentiates basic fibroblast growth factor-induced corneal neovascularization. Am. J. Pathol. 2009, 174, 1564–1571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kvanta, A.; Sarman, S.; Fagerholm, P.; Seregard, S.; Steen, B. Expression of matrix metalloproteinase-2 (MMP-2) and vascular endothelial growth factor (VEGF) in inflammation-associated corneal neovascularization. Exp. Eye Res. 2000, 70, 419–428. [Google Scholar] [CrossRef]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [Green Version]
- Page-McCaw, A.; Ewald, A.J.; Werb, Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 2007, 8, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Sounni, N.E.; Paye, A.; Host, L.; Noel, A. MT-MMPS as Regulators of Vessel Stability Associated with Angiogenesis. Front. Pharm. 2011, 2, 111. [Google Scholar] [CrossRef] [Green Version]
- Sarnicola, C.; Farooq, A.V.; Colby, K. Fuchs Endothelial Corneal Dystrophy: Update on Pathogenesis and Future Directions. Eye Contact Lens 2019, 45, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Vedana, G.; Villarreal, G., Jr.; Jun, A.S. Fuchs endothelial corneal dystrophy: Current perspectives. Clin. Ophthalmol. 2016, 10, 321–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanda, G.G.; Alone, D.P. REVIEW: Current understanding of the pathogenesis of Fuchs’ endothelial corneal dystrophy. Mol. Vis. 2019, 25, 295–310. [Google Scholar]
- Okumura, N.; Minamiyama, R.; Ho, L.T.; Kay, E.P.; Kawasaki, S.; Tourtas, T.; Schlotzer-Schrehardt, U.; Kruse, F.E.; Young, R.D.; Quantock, A.J.; et al. Involvement of ZEB1 and Snail1 in excessive production of extracellular matrix in Fuchs endothelial corneal dystrophy. Lab. Investig. 2015, 95, 1291–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kocaba, V.; Katikireddy, K.R.; Gipson, I.; Price, M.O.; Price, F.W.; Jurkunas, U.V. Association of the Gutta-Induced Microenvironment With Corneal Endothelial Cell Behavior and Demise in Fuchs Endothelial Corneal Dystrophy. JAMA Ophthalmol. 2018, 136, 886–892. [Google Scholar] [CrossRef] [Green Version]
- Liaboe, C.A.; Aldrich, B.T.; Carter, P.C.; Skeie, J.M.; Burckart, K.A.; Schmidt, G.A.; Reed, C.R.; Zimmerman, M.B.; Greiner, M.A. Assessing the Impact of Diabetes Mellitus on Donor Corneal Endothelial Cell Density. Cornea 2017, 36, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Aldrich, B.T.; Schlötzer-Schrehardt, U.; Skeie, J.M.; Burckart, K.A.; Schmidt, G.A.; Reed, C.R.; Zimmerman, M.B.; Kruse, F.E.; Greiner, M.A. Mitochondrial and Morphologic Alterations in Native Human Corneal Endothelial Cells Associated With Diabetes Mellitus. Investig. Ophthalmol. Vis. Sci. 2017, 58, 2130–2138. [Google Scholar] [CrossRef]
- Schwarz, C.; Aldrich, B.T.; Burckart, K.A.; Schmidt, G.A.; Zimmerman, M.B.; Reed, C.R.; Greiner, M.A.; Sander, E.A. Descemet membrane adhesion strength is greater in diabetics with advanced disease compared to healthy donor corneas. Exp. Eye Res. 2016, 153, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Rehany, U.; Ishii, Y.; Lahav, M.; Rumelt, S. Ultrastructural changes in corneas of diabetic patients: An electron-microscopy study. Cornea 2000, 19, 534–538. [Google Scholar] [CrossRef]
- Acott, T.S.; Kelley, M.J. Extracellular matrix in the trabecular meshwork. Exp. Eye Res. 2008, 86, 543–561. [Google Scholar] [CrossRef] [Green Version]
- Vranka, J.A.; Kelley, M.J.; Acott, T.S.; Keller, K.E. Extracellular matrix in the trabecular meshwork: Intraocular pressure regulation and dysregulation in glaucoma. Exp. Eye Res. 2015, 133, 112–125. [Google Scholar] [CrossRef] [Green Version]
- Pasquale, L.R. Vascular and autonomic dysregulation in primary open-angle glaucoma. Curr. Opin. Ophthalmol. 2016, 27, 94–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buys, E.S.; Ko, Y.C.; Alt, C.; Hayton, S.R.; Jones, A.; Tainsh, L.T.; Ren, R.; Giani, A.; Clerté, M.; Abernathy, E.; et al. Soluble guanylate cyclase α1-deficient mice: A novel murine model for primary open angle glaucoma. PLoS ONE 2013, 8, e60156. [Google Scholar] [CrossRef] [PubMed]
- Cavet, M.E.; Vittitow, J.L.; Impagnatiello, F.; Ongini, E.; Bastia, E. Nitric oxide (NO): An emerging target for the treatment of glaucoma. Investig. Ophthalmol. Vis. Sci. 2014, 55, 5005–5015. [Google Scholar] [CrossRef] [Green Version]
- Weinreb, R.N.; Ong, T.; Scassellati Sforzolini, B.; Vittitow, J.L.; Singh, K.; Kaufman, P.L. A randomised, controlled comparison of latanoprostene bunod and latanoprost 0.005% in the treatment of ocular hypertension and open angle glaucoma: The VOYAGER study. Br. J. Ophthalmol. 2015, 99, 738–745. [Google Scholar] [CrossRef] [Green Version]
- Tamm, E.R. The trabecular meshwork outflow pathways: Structural and functional aspects. Exp. Eye Res. 2009, 88, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Bradley, J.M.; Vranka, J.; Colvis, C.M.; Conger, D.M.; Alexander, J.P.; Fisk, A.S.; Samples, J.R.; Acott, T.S. Effect of matrix metalloproteinases activity on outflow in perfused human organ culture. Investig. Ophthalmol. Vis. Sci. 1998, 39, 2649–2658. [Google Scholar]
- Hopkins, A.A.; Murphy, R.; Irnaten, M.; Wallace, D.M.; Quill, B.; O’Brien, C. The role of lamina cribrosa tissue stiffness and fibrosis as fundamental biomechanical drivers of pathological glaucoma cupping. Am. J. Physiol. Cell Physiol. 2020, 319, C611–C623. [Google Scholar] [CrossRef] [PubMed]
- Bradley, J.M.; Kelley, M.J.; Zhu, X.; Anderssohn, A.M.; Alexander, J.P.; Acott, T.S. Effects of mechanical stretching on trabecular matrix metalloproteinases. Investig. Ophthalmol. Vis. Sci. 2001, 42, 1505–1513. [Google Scholar]
- Chudgar, S.M.; Deng, P.; Maddala, R.; Epstein, D.L.; Rao, P.V. Regulation of connective tissue growth factor expression in the aqueous humor outflow pathway. Mol. Vis. 2006, 12, 1117–1126. [Google Scholar]
- Bradley, J.M.; Kelley, M.J.; Rose, A.; Acott, T.S. Signaling pathways used in trabecular matrix metalloproteinase response to mechanical stretch. Investig. Ophthalmol. Vis. Sci. 2003, 44, 5174–5181. [Google Scholar] [CrossRef] [Green Version]
- Vittal, V.; Rose, A.; Gregory, K.E.; Kelley, M.J.; Acott, T.S. Changes in Gene Expression by Trabecular Meshwork Cells in Response to Mechanical Stretching. Investig. Opthalmol. Vis. Sci. 2005, 46, 2857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirwan, R.P.; Fenerty, C.H.; Crean, J.; Wordinger, R.J.; Clark, A.F.; O’Brien, C.J. Influence of cyclical mechanical strain on extracellular matrix gene expression in human lamina cribrosa cells in vitro. Mol. Vis. 2005, 11, 798–810. [Google Scholar]
- Parshley, D.E.; Bradley, J.M.; Samples, J.R.; Van Buskirk, E.M.; Acott, T.S. Early changes in matrix metalloproteinases and inhibitors after in vitro laser treatment to the trabecular meshwork. Curr. Eye Res. 1995, 14, 537–544. [Google Scholar] [CrossRef]
- Alexander, J.P.; Samples, J.R.; Van Buskirk, E.M.; Acott, T.S. Expression of matrix metalloproteinases and inhibitor by human trabecular meshwork. Investig. Ophthalmol. Vis. Sci. 1991, 32, 172–180. [Google Scholar]
- Alexander, J.P.; Samples, J.R.; Acott, T.S. Growth factor and cytokine modulation of trabecular meshwork matrix metalloproteinase and TIMP expression. Curr. Eye Res. 1998, 17, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Keller, K.E.; Vranka, J.A.; Haddadin, R.I.; Kang, M.H.; Oh, D.J.; Rhee, D.J.; Yang, Y.F.; Sun, Y.Y.; Kelley, M.J.; Acott, T.S. The effects of tenascin C knockdown on trabecular meshwork outflow resistance. Investig. Ophthalmol. Vis. Sci. 2013, 54, 5613–5623. [Google Scholar] [CrossRef] [PubMed]
- Haddadin, R.I.; Oh, D.J.; Kang, M.H.; Filippopoulos, T.; Gupta, M.; Hart, L.; Sage, E.H.; Rhee, D.J. SPARC-null mice exhibit lower intraocular pressures. Investig. Ophthalmol. Vis. Sci. 2009, 50, 3771–3777. [Google Scholar] [CrossRef] [Green Version]
- Haddadin, R.I.; Oh, D.-J.; Kang, M.H.; Villarreal, G.; Kang, J.-H.; Jin, R.; Gong, H.; Rhee, D.J. Thrombospondin-1 (TSP1)–Null and TSP2-Null Mice Exhibit Lower Intraocular Pressures. Investig. Opthalmol. Vis. Sci. 2012, 53, 6708. [Google Scholar] [CrossRef]
- Yan, Q.; Clark, J.I.; Sage, E.H. Expression and characterization of SPARC in human lens and in the aqueous and vitreous humors. Exp. Eye Res. 2000, 71, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Rhee, D.J.; Fariss, R.N.; Brekken, R.; Helene Sage, E.; Russell, P. The matricellular protein SPARC is expressed in human trabecular meshwork. Exp. Eye Res. 2003, 77, 601–607. [Google Scholar] [CrossRef]
- Kang, M.H.; Oh, D.-J.; Kang, J.-H.; Rhee, D.J. Regulation of SPARC by Transforming Growth Factor β2 in Human Trabecular Meshwork. Investig. Opthalmol. Vis. Sci. 2013, 54, 2523. [Google Scholar] [CrossRef] [Green Version]
- Bollinger, K.E.; Crabb, J.S.; Yuan, X.; Putliwala, T.; Clark, A.F.; Crabb, J.W. Quantitative Proteomics: TGFβ2Signaling in Trabecular Meshwork Cells. Investig. Opthalmol. Vis. Sci. 2011, 52, 8287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomarev, S.I.; Wistow, G.; Raymond, V.; Dubois, S.P.; Malyukova, I. Gene Expression Profile of the Human Trabecular Meshwork: NEIBank Sequence Tag Analysis. Investig. Opthalmol. Vis. Sci. 2003, 44, 2588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junglas, B.; Yu, A.H.; Welge-Lüssen, U.; Tamm, E.R.; Fuchshofer, R. Connective tissue growth factor induces extracellular matrix deposition in human trabecular meshwork cells. Exp. Eye Res. 2009, 88, 1065–1075. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.M.; Clark, A.F.; Lipson, K.E.; Andrews, D.; Crean, J.K.; O’Brien, C.J. Anti-Connective Tissue Growth Factor Antibody Treatment Reduces Extracellular Matrix Production in Trabecular Meshwork and Lamina Cribrosa Cells. Investig. Opthalmol. Vis. Sci. 2013, 54, 7836. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, B.J.; Tripathi, R.C.; Yang, C.; Millard, C.B.; Dixit, V.M. Synthesis of a thrombospondin-like cytoadhesion molecule by cells of the trabecular meshwork. Investig. Ophthalmol. Vis. Sci. 1991, 32, 181–188. [Google Scholar]
- Hiscott, P.; Schlötzer-Schrehardt, U.; Naumann, G.O.H. Unexpected expression of thrombospondin 1 by corneal and iris fibroblasts in the pseudoexfoliation syndrome. Hum. Pathol. 1996, 27, 1255–1258. [Google Scholar] [CrossRef]
- Faralli, J.A.; Filla, M.S.; Peters, D.M. Role of Fibronectin in Primary Open Angle Glaucoma. Cells 2019, 8, 1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faralli, J.A.; Schwinn, M.K.; Gonzalez, J.M., Jr.; Filla, M.S.; Peters, D.M. Functional properties of fibronectin in the trabecular meshwork. Exp. Eye Res. 2009, 88, 689–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heimark, R.L.; Kaochar, S.; Stamer, W.D. Human Schlemm’s canal cells express the endothelial adherens proteins, VE-cadherin and PECAM-1. Curr. Eye Res. 2002, 25, 299–308. [Google Scholar] [CrossRef]
- Filla, M.S.; Faralli, J.A.; Peotter, J.L.; Peters, D.M. The role of integrins in glaucoma. Exp. Eye Res. 2017, 158, 124–136. [Google Scholar] [CrossRef] [Green Version]
- Murphy-Ullrich, J.E.; Downs, J.C. The Thrombospondin1-TGF-beta Pathway and Glaucoma. J. Ocul Pharm. Ther. 2015, 31, 371–375. [Google Scholar] [CrossRef]
- Nikhalashree, S.; Karthikkeyan, G.; George, R.; Shantha, B.; Vijaya, L.; Ratra, V.; Sulochana, K.N.; Coral, K. Lowered Decorin With Aberrant Extracellular Matrix Remodeling in Aqueous Humor and Tenon’s Tissue From Primary Glaucoma Patients. Investig. Opthalmol. Vis. Sci. 2019, 60, 4661. [Google Scholar] [CrossRef] [Green Version]
- Overby, D.R.; Bertrand, J.; Tektas, O.Y.; Boussommier-Calleja, A.; Schicht, M.; Ethier, C.R.; Woodward, D.F.; Stamer, W.D.; Lütjen-Drecoll, E. Ultrastructural changes associated with dexamethasone-induced ocular hypertension in mice. Investig. Ophthalmol. Vis. Sci. 2014, 55, 4922–4933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Lee, C.; Agrahari, V.; Wang, K.; Navarro, I.; Sherwood, J.M.; Crews, K.; Farsiu, S.; Gonzalez, P.; Lin, C.W.; et al. In vivo measurement of trabecular meshwork stiffness in a corticosteroid-induced ocular hypertensive mouse model. Proc. Natl. Acad. Sci. USA 2019, 116, 1714–1722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohd Nasir, N.A.; Agarwal, R.; Krasilnikova, A.; Sheikh Abdul Kadir, S.H.; Iezhitsa, I. Effect of dexamethasone on the expression of MMPs, adenosine A1 receptors and NFKB by human trabecular meshwork cells. J. Basic Clin. Physiol. Pharm. 2020. [Google Scholar] [CrossRef]
- Wallace, D.M.; Murphy-Ullrich, J.E.; Downs, J.C.; O’Brien, C.J. The role of matricellular proteins in glaucoma. Matrix Biol. 2014, 37, 174–182. [Google Scholar] [CrossRef] [Green Version]
- Buller, C.; Johnson, D.H.; Tschumper, R.C. Human trabecular meshwork phagocytosis. Observations in an organ culture system. Investig. Ophthalmol. Vis. Sci. 1990, 31, 2156–2163. [Google Scholar]
- Porter, K.M.; Epstein, D.L.; Liton, P.B. Up-Regulated Expression of Extracellular Matrix Remodeling Genes in Phagocytically Challenged Trabecular Meshwork Cells. PLoS ONE 2012, 7, e34792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, K.; Lin, Y.; Liton, P.B. Cathepsin B Is Up-Regulated and Mediates Extracellular Matrix Degradation in Trabecular Meshwork Cells Following Phagocytic Challenge. PLoS ONE 2013, 8, e68668. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Yang, X.; Fang, J.; Zhang, Y.; Zhu, W.; Yang, X. Rho-Associated Protein Kinase Inhibitor Treatment Promotes Proliferation and Phagocytosis in Trabecular Meshwork Cells. Front. Pharmacol. 2020, 11, 302. [Google Scholar] [CrossRef]
- Tian, B.; Kaufman, P.L. Effects of the Rho kinase inhibitor Y-27632 and the phosphatase inhibitor calyculin A on outflow facility in monkeys. Exp. Eye Res. 2005, 80, 215–225. [Google Scholar] [CrossRef]
- Yu, M.; Chen, X.; Wang, N.; Cai, S.; Li, N.; Qiu, J.; Brandt, C.R.; Kaufman, P.L.; Liu, X. H-1152 effects on intraocular pressure and trabecular meshwork morphology of rat eyes. J. Ocul. Pharm. Ther. 2008, 24, 373–379. [Google Scholar] [CrossRef]
- Hernandez, M.R. The optic nerve head in glaucoma: Role of astrocytes in tissue remodeling. Prog. Retin. Eye Res. 2000, 19, 297–321. [Google Scholar] [CrossRef]
- Yuan, L.; Neufeld, A.H. Activated microglia in the human glaucomatous optic nerve head. J. Neurosci. Res. 2001, 64, 523–532. [Google Scholar] [CrossRef]
- Wallace, D.M.; O’Brien, C.J. The role of lamina cribrosa cells in optic nerve head fibrosis in glaucoma. Exp. Eye Res. 2016, 142, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Albon, J. Age related compliance of the lamina cribrosa in human eyes. Br. J. Ophthalmol. 2000, 84, 318–323. [Google Scholar] [CrossRef] [Green Version]
- Vogel, H. Influence of maturation and aging on mechanical and biochemical properties of connective tissue in rats. Mech. Ageing Dev. 1980, 14, 283–292. [Google Scholar] [CrossRef]
- Burgoyne, C.F.; Downs, J.C.; Bellezza, A.J.; Suh, J.K.; Hart, R.T. The optic nerve head as a biomechanical structure: A new paradigm for understanding the role of IOP-related stress and strain in the pathophysiology of glaucomatous optic nerve head damage. Prog. Retin. Eye Res. 2005, 24, 39–73. [Google Scholar] [CrossRef]
- Qu, J.; Chen, H.; Zhu, L.; Ambalavanan, N.; Girkin, C.A.; Murphy-Ullrich, J.E.; Downs, J.C.; Zhou, Y. High-Magnitude and/or High-Frequency Mechanical Strain Promotes Peripapillary Scleral Myofibroblast Differentiation. Investig. Opthalmol. Vis. Sci. 2015, 56, 7821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, X.; Tezel, G.; Wax, M.B.; Edward, D.P. Matrix metalloproteinases and tumor necrosis factor alpha in glaucomatous optic nerve head. Arch. Ophthalmol. 2000, 118, 666–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khaw, P.T.; Occleston, N.L.; Schultz, G.; Grierson, I.; Sherwood, M.B.; Larkin, G. Activation and suppression of fibroblast function. Eye 1994, 8, 188–195. [Google Scholar] [CrossRef] [Green Version]
- Daniels, J.T.; Occleston, N.L.; Crowston, J.G.; Cordeiro, M.F.; Alexander, R.A.; Wilkins, M.; Porter, R.; Brown, R.; Khaw, P.T. Understanding and controlling the scarring response: The contribution of histology and microscopy. Microsc Res. Tech. 1998, 42, 317–333. [Google Scholar] [CrossRef]
- Skuta, G.L.; Parrish, R.K. Wound healing in glaucoma filtering surgery. Surv. Ophthalmol. 1987, 32, 149–170. [Google Scholar] [CrossRef] [Green Version]
- Wu, N.; Chen, L.; Yan, D.; Zhou, M.; Shao, C.; Lu, Y.; Yao, Q.; Sun, H.; Fu, Y. Trehalose attenuates TGF-β1-induced fibrosis of hSCFs by activating autophagy. Mol. Cell. Biochem. 2020, 470, 175–188. [Google Scholar] [CrossRef]
- Fuchshofer, R.; Tamm, E.R. The role of TGF-β in the pathogenesis of primary open-angle glaucoma. Cell Tissue Res. 2012, 347, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Seet, L.-F.; Su, R.; Barathi, V.A.; Lee, W.S.; Poh, R.; Heng, Y.M.; Manser, E.; Vithana, E.N.; Aung, T.; Weaver, M.; et al. SPARC Deficiency Results in Improved Surgical Survival in a Novel Mouse Model of Glaucoma Filtration Surgery. PLoS ONE 2010, 5, e9415. [Google Scholar] [CrossRef] [Green Version]
- Esson, D.W.; Neelakantan, A.; Iyer, S.A.; Blalock, T.D.; Balasubramanian, L.; Grotendorst, G.R.; Schultz, G.S.; Sherwood, M.B. Expression of Connective Tissue Growth Factor after Glaucoma Filtration Surgery in a Rabbit Model. Investig. Opthalmol. Vis. Sci. 2004, 45, 485. [Google Scholar] [CrossRef] [Green Version]
- Yuan, H.P.; Li, X.H.; Yang, B.B.; Shao, Z.B.; Yan, L.P. Expression of connective tissue growth factor after trabeculectomy in rabbits. Zhonghua Yan Ke Za Zhi 2009, 45, 168–174. [Google Scholar]
- Wang, J.-M.; Hui, N.; Fan, Y.-Z.; Xiong, L.; Sun, N.-X. Filtering bleb area and intraocular pressure following subconjunctival injection of CTGF antibody after glaucoma filtration surgery in rabbits. Int. J. Ophthalmol. 2011, 4, 480–483. [Google Scholar] [CrossRef]
- Qiao, H.; Hisatomi, T.; Sonoda, K.H.; Kura, S.; Sassa, Y.; Kinoshita, S.; Nakamura, T.; Sakamoto, T.; Ishibashi, T. The characterisation of hyalocytes: The origin, phenotype, and turnover. Br. J. Ophthalmol. 2005, 89, 513–517. [Google Scholar] [CrossRef]
- Bishop, P.N. Structural macromolecules and supramolecular organisation of the vitreous gel. Prog. Retin. Eye Res. 2000, 19, 323–344. [Google Scholar] [CrossRef]
- Bishop, P.N.; Crossman, M.V.; McLeod, D.; Ayad, S. Extraction and characterization of the tissue forms of collagen types II and IX from bovine vitreous. Biochem. J. 1994, 299 Pt 2, 497–505. [Google Scholar] [CrossRef] [Green Version]
- Bishop, P.N.; Reardon, A.J.; McLeod, D.; Ayad, S. Identification of alternatively spliced variants of type II procollagen in vitreous. Biochem. Biophys. Res. Commun. 1994, 203, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Bos, K.J.; Holmes, D.F.; Kadler, K.E.; McLeod, D.; Morris, N.P.; Bishop, P.N. Axial structure of the heterotypic collagen fibrils of vitreous humour and cartilage. J. Mol. Biol. 2001, 306, 1011–1022. [Google Scholar] [CrossRef]
- Kielty, C.M.; Whittaker, S.P.; Grant, M.E.; Shuttleworth, C.A. Type VI collagen microfibrils: Evidence for a structural association with hyaluronan. J. Cell Biol. 1992, 118, 979–990. [Google Scholar] [CrossRef] [Green Version]
- Foos, R.Y. Posterior vitreous detachment. Trans. Am. Acad. Ophthalmol. Otolaryngol. 1972, 76, 480–497. [Google Scholar] [PubMed]
- Matsumoto, B.; Blanks, J.C.; Ryan, S.J. Topographic variations in the rabbit and primate internal limiting membrane. Investig. Ophthalmol. Vis. Sci. 1984, 25, 71–82. [Google Scholar]
- Bishop, P.N.; Holmes, D.F.; Kadler, K.E.; McLeod, D.; Bos, K.J. Age-related changes on the surface of vitreous collagen fibrils. Investig. Ophthalmol. Vis. Sci. 2004, 45, 1041–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bos, K.J.; Holmes, D.F.; Meadows, R.S.; Kadler, K.E.; McLeod, D.; Bishop, P.N. Collagen fibril organisation in mammalian vitreous by freeze etch/rotary shadowing electron microscopy. Micron 2001, 32, 301–306. [Google Scholar] [CrossRef]
- Ramesh, S.; Bonshek, R.E.; Bishop, P.N. Immunolocalisation of opticin in the human eye. Br. J. Ophthalmol. 2004, 88, 697–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hindson, V.J.; Gallagher, J.T.; Halfter, W.; Bishop, P.N. Opticin binds to heparan and chondroitin sulfate proteoglycans. Investig. Ophthalmol. Vis. Sci. 2005, 46, 4417–4423. [Google Scholar] [CrossRef]
- Teng, C.C.; Chi, H.H. Vitreous changes and the mechanism of retinal detachment. Am. J. Ophthalmol. 1957, 44, 335–356. [Google Scholar] [CrossRef]
- Mitry, D.; Fleck, B.W.; Wright, A.F.; Campbell, H.; Charteris, D.G. Pathogenesis of rhegmatogenous retinal detachment: Predisposing anatomy and cell biology. Retina 2010, 30, 1561–1572. [Google Scholar] [CrossRef] [PubMed]
- Streeten, B.W.; Bert, M. The retinal surface in lattice degeneration of the retina. Am. J. Ophthalmol. 1972, 74, 1201–1209. [Google Scholar] [CrossRef]
- Straatsma, B.R.; Zeegen, P.D.; Foos, R.Y.; Feman, S.S.; Shabo, A.L. Lattice degeneration of the retina. XXX Edward Jackson Memorial Lecture. Am. J. Ophthalmol. 1974, 77, 619–649. [Google Scholar] [CrossRef]
- Wang, J.; McLeod, D.; Henson, D.B.; Bishop, P.N. Age-dependent changes in the basal retinovitreous adhesion. Investig. Ophthalmol. Vis. Sci. 2003, 44, 1793–1800. [Google Scholar] [CrossRef]
- Schepens, C.L. Clinical aspects of pathologic changes in the vitreous body. Am. J. Ophthalmol. 1954, 38, 8–21. [Google Scholar] [CrossRef]
- Foos, R.Y.; Allen, R.A. Retinal tears and lesser lesions of the peripheral retina in autopsy eyes. Am. J. Ophthalmol. 1967, 64, 643–655. [Google Scholar] [CrossRef]
- Hogan, M.J. The vitreous, its structure, and relation to the ciliary body and retina. proctor award lecture. Investig. Ophthalmol. 1963, 2, 418–445. [Google Scholar]
- Sohn, E.H.; Strohbehn, A.; Stryjewski, T.; Brodowska, K.; Flamme-Wiese, M.J.; Mullins, R.F.; Eliott, D. Posteriorly inserted vitreous base: Preoperative Characteristics, Intraoperative Findings, and Outcomes After Vitrectomy. Retina 2020, 40, 943–950. [Google Scholar] [CrossRef]
- Shukla, D. Correspondence. Retina 2020, 40, e68. [Google Scholar] [CrossRef]
- Sohn, E.H.; Mullins, R.F.; Eliott, D. Reply. Retina 2020, 40, e68–e69. [Google Scholar] [CrossRef]
- Quinlan, R.; Nilsson, M. Reloading the retina by modifying the glial matrix. Trends Neurosci. 2004, 27, 241–242. [Google Scholar] [CrossRef]
- Adler, A.J.; Severin, K.M. Proteins of the bovine interphotoreceptor matrix: Tissues of origin. Exp. Eye Res. 1981, 32, 755–769. [Google Scholar] [CrossRef]
- De Smet, M.D.; Gad Elkareem, A.M.; Zwinderman, A.H. The vitreous, the retinal interface in ocular health and disease. Ophthalmologica 2013, 230, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Candiello, J.; Cole, G.J.; Halfter, W. Age-dependent changes in the structure, composition and biophysical properties of a human basement membrane. Matrix Biol. 2010, 29, 402–410. [Google Scholar] [CrossRef]
- Hausman, R.E. Ocular extracellular matrices in development. Prog. Retin. Eye Res. 2007, 26, 162–188. [Google Scholar] [CrossRef]
- Oster, S.F.; Sretavan, D.W. Connecting the eye to the brain: The molecular basis of ganglion cell axon guidance. Br. J. Ophthalmol. 2003, 87, 639–645. [Google Scholar] [CrossRef]
- Pires Neto, M.A.; Braga-de-Souza, S.; Lent, R. Extracellular matrix molecules play diverse roles in the growth and guidance of central nervous system axons. Braz. J. Med. Biol Res. 1999, 32, 633–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ardi, V.C.; Kupriyanova, T.A.; Deryugina, E.I.; Quigley, J.P. Human neutrophils uniquely release TIMP-free MMP-9 to provide a potent catalytic stimulator of angiogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 20262–20267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giraudo, E.; Inoue, M.; Hanahan, D. An amino-bisphosphonate targets MMP-9-expressing macrophages and angiogenesis to impair cervical carcinogenesis. J. Clin. Investig. 2004, 114, 623–633. [Google Scholar] [CrossRef]
- Nelissen, I.; Martens, E.; Van den Steen, P.E.; Proost, P.; Ronsse, I.; Opdenakker, G. Gelatinase B/matrix metalloproteinase-9 cleaves interferon-beta and is a target for immunotherapy. Brain A J. Neurol. 2003, 126, 1371–1381. [Google Scholar] [CrossRef] [Green Version]
- Vu, T.H.; Shipley, J.M.; Bergers, G.; Berger, J.E.; Helms, J.A.; Hanahan, D.; Shapiro, S.D.; Senior, R.M.; Werb, Z. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell 1998, 93, 411–422. [Google Scholar] [CrossRef] [Green Version]
- Bishop, P.N. The role of extracellular matrix in retinal vascular development and preretinal neovascularization. Exp. Eye Res. 2015, 133, 30–36. [Google Scholar] [CrossRef]
- McLeod, D. A chronic grey matter penumbra, lateral microvascular intussusception and venous peduncular avulsion underlie diabetic vitreous haemorrhage. Br. J. Ophthalmol. 2007, 91, 677–689. [Google Scholar] [CrossRef] [PubMed]
- Davis, G.E.; Senger, D.R. Endothelial extracellular matrix: Biosynthesis, remodeling, and functions during vascular morphogenesis and neovessel stabilization. Circ. Res. 2005, 97, 1093–1107. [Google Scholar] [CrossRef] [Green Version]
- Davis, G.E.; Bayless, K.J.; Mavila, A. Molecular basis of endothelial cell morphogenesis in three-dimensional extracellular matrices. Anat. Rec. 2002, 268, 252–275. [Google Scholar] [CrossRef]
- Davis, G.E.; Bayless, K.J. An integrin and Rho GTPase-dependent pinocytic vacuole mechanism controls capillary lumen formation in collagen and fibrin matrices. Microcirculation 2003, 10, 27–44. [Google Scholar] [CrossRef]
- Heissig, B.; Hattori, K.; Friedrich, M.; Rafii, S.; Werb, Z. Angiogenesis: Vascular remodeling of the extracellular matrix involves metalloproteinases. Curr. Opin. Hematol. 2003, 10, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Pepper, M.S. Role of the matrix metalloproteinase and plasminogen activator-plasmin systems in angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1104–1117. [Google Scholar] [CrossRef] [Green Version]
- Bayless, K.J.; Davis, G.E. Sphingosine-1-phosphate markedly induces matrix metalloproteinase and integrin-dependent human endothelial cell invasion and lumen formation in three-dimensional collagen and fibrin matrices. Biochem. Biophys. Res. Commun. 2003, 312, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Fassina, G.; Ferrari, N.; Brigati, C.; Benelli, R.; Santi, L.; Noonan, D.M.; Albini, A. Tissue inhibitors of metalloproteases: Regulation and biological activities. Clin. Exp. Metastasis 2000, 18, 111–120. [Google Scholar] [CrossRef]
- Yana, I.; Sagara, H.; Takaki, S.; Takatsu, K.; Nakamura, K.; Nakao, K.; Katsuki, M.; Taniguchi, S.; Aoki, T.; Sato, H.; et al. Crosstalk between neovessels and mural cells directs the site-specific expression of MT1-MMP to endothelial tip cells. J. Cell Sci. 2007, 120, 1607–1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saunders, W.B.; Bohnsack, B.L.; Faske, J.B.; Anthis, N.J.; Bayless, K.J.; Hirschi, K.K.; Davis, G.E. Coregulation of vascular tube stabilization by endothelial cell TIMP-2 and pericyte TIMP-3. J. Cell Biol. 2006, 175, 179–191. [Google Scholar] [CrossRef] [Green Version]
- Ausprunk, D.H.; Folkman, J. Migration and proliferation of endothelial cells in preformed and newly formed blood vessels during tumor angiogenesis. Microvasc. Res. 1977, 14, 53–65. [Google Scholar] [CrossRef]
- Akiyama, S.K.; Yamada, S.S.; Chen, W.T.; Yamada, K.M. Analysis of fibronectin receptor function with monoclonal antibodies: Roles in cell adhesion, migration, matrix assembly, and cytoskeletal organization. J. Cell Biol. 1989, 109, 863–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hynes, R.O. Integrins: Versatility, modulation, and signaling in cell adhesion. Cell 1992, 69, 11–25. [Google Scholar] [CrossRef]
- Giancotti, F.G.; Ruoslahti, E. Integrin signaling. Science 1999, 285, 1028–1032. [Google Scholar] [CrossRef] [PubMed]
- Meredith, J.E., Jr.; Schwartz, M.A. Integrins, adhesion and apoptosis. Trends Cell Biol. 1997, 7, 146–150. [Google Scholar] [CrossRef]
- Roovers, K.; Assoian, R.K. Integrating the MAP kinase signal into the G1 phase cell cycle machinery. Bioessays 2000, 22, 818–826. [Google Scholar] [CrossRef]
- Short, S.M.; Talbott, G.A.; Juliano, R.L. Integrin-mediated signaling events in human endothelial cells. Mol. Biol. Cell 1998, 9, 1969–1980. [Google Scholar] [CrossRef] [Green Version]
- Vinals, F.; Pouyssegur, J. Confluence of vascular endothelial cells induces cell cycle exit by inhibiting p42/p44 mitogen-activated protein kinase activity. Mol. Cell Biol. 1999, 19, 2763–2772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoudjit, F.; Vuori, K. Matrix attachment regulates Fas-induced apoptosis in endothelial cells: A role for c-flip and implications for anoikis. J. Cell Biol. 2001, 152, 633–643. [Google Scholar] [CrossRef]
- Salvesen, G.S. Programmed cell death and the caspases. APMIS 1999, 107, 73–79. [Google Scholar] [CrossRef]
- Holderfield, M.T.; Hughes, C.C. Crosstalk between vascular endothelial growth factor, notch, and transforming growth factor-beta in vascular morphogenesis. Circ. Res. 2008, 102, 637–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fouser, L.; Iruela-Arispe, L.; Bornstein, P.; Sage, E.H. Transcriptional activity of the alpha 1(I)-collagen promoter is correlated with the formation of capillary-like structures by endothelial cells in vitro. J. Biol. Chem. 1991, 266, 18345–18351. [Google Scholar] [CrossRef]
- Montesano, R.; Orci, L.; Vassalli, P. In vitro rapid organization of endothelial cells into capillary-like networks is promoted by collagen matrices. J. Cell Biol. 1983, 97, 1648–1652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweeney, S.M.; Guy, C.A.; Fields, G.B.; San Antonio, J.D. Defining the domains of type I collagen involved in heparin- binding and endothelial tube formation. Proc. Natl. Acad. Sci. USA 1998, 95, 7275–7280. [Google Scholar] [CrossRef] [Green Version]
- Whelan, M.C.; Senger, D.R. Collagen I initiates endothelial cell morphogenesis by inducing actin polymerization through suppression of cyclic AMP and protein kinase A. J. Biol. Chem. 2003, 278, 327–334. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Senger, D.R. Matrix-specific activation of Src and Rho initiates capillary morphogenesis of endothelial cells. FASEB J. 2004, 18, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Otey, C.A.; Burridge, K. Patterning of the membrane cytoskeleton by the extracellular matrix. Semin Cell Biol 1990, 1, 391–399. [Google Scholar] [PubMed]
- Nichols, S.A.; Roberts, B.W.; Richter, D.J.; Fairclough, S.R.; King, N. Origin of metazoan cadherin diversity and the antiquity of the classical cadherin/β-catenin complex. Proc. Natl. Acad. Sci. USA 2012, 109, 13046–13051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suga, H.; Chen, Z.; de Mendoza, A.; Sebé-Pedrós, A.; Brown, M.W.; Kramer, E.; Carr, M.; Kerner, P.; Vervoort, M.; Sánchez-Pons, N.; et al. The Capsaspora genome reveals a complex unicellular prehistory of animals. Nat. Commun. 2013, 4, 2325. [Google Scholar] [CrossRef] [Green Version]
- King, N.; Westbrook, M.J.; Young, S.L.; Kuo, A.; Abedin, M.; Chapman, J.; Fairclough, S.; Hellsten, U.; Isogai, Y.; Letunic, I.; et al. The genome of the choanoflagellate Monosiga brevicollis and the origin of metazoans. Nature 2008, 451, 783–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, F.; Tew, H.A.; Paul, C.E.; Adams, J.C. The predicted secretomes of Monosiga brevicollis and Capsaspora owczarzaki, close unicellular relatives of metazoans, reveal new insights into the evolution of the metazoan extracellular matrix. Matrix Biol. 2014, 37, 60–68. [Google Scholar] [CrossRef]
- Erkenbrack, E.M.; Petsios, E. A Conserved Role for VEGF Signaling in Specification of Homologous Mesenchymal Cell Types Positioned at Spatially Distinct Developmental Addresses in Early Development of Sea Urchins. J. Exp. Zool B Mol. Dev. Evol. 2017, 328, 423–432. [Google Scholar] [CrossRef]
- Vega-Macaya, F.; Manieu, C.; Valdivia, M.; Mlodzik, M.; Olguín, P. Establishment of the Muscle-Tendon Junction During Thorax Morphogenesis in Drosophila Requires the Rho-Kinase. Genetics 2016, 204, 1139–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiozzo, S.; Voskoboynik, A.; Brown, F.D.; De Tomaso, A.W. A conserved role of the VEGF pathway in angiogenesis of an ectodermally-derived vasculature. Dev. Biol. 2008, 315, 243–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vorotnikova, E.; McIntosh, D.; Dewilde, A.; Zhang, J.; Reing, J.E.; Zhang, L.; Cordero, K.; Bedelbaeva, K.; Gourevitch, D.; Heber-Katz, E.; et al. Extracellular matrix-derived products modulate endothelial and progenitor cell migration and proliferation in vitro and stimulate regenerative healing in vivo. Matrix Biol. 2010, 29, 690–700. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.C. Matricellular Proteins: Functional Insights From Non-mammalian Animal Models. Curr. Top. Dev. Biol. 2018, 130, 39–105. [Google Scholar] [CrossRef]
- Witjas, F.M.R.; van den Berg, B.M.; van den Berg, C.W.; Engelse, M.A.; Rabelink, T.J. Concise Review: The Endothelial Cell Extracellular Matrix Regulates Tissue Homeostasis and Repair. Stem Cells Transl. Med. 2019, 8, 375–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casaroli Marano, R.P.; Preissner, K.T.; Vilaro, S. Fibronectin, laminin, vitronectin and their receptors at newly-formed capillaries in proliferative diabetic retinopathy. Exp. Eye Res. 1995, 60, 5–17. [Google Scholar] [CrossRef]
- Hosoda, Y.; Okada, M.; Matsumura, M.; Ogino, N.; Honda, Y.; Nagai, Y. Intravitreal neovascular tissue of proliferative diabetic retinopathy: An immunohistochemical study. Ophthalmic Res. 1992, 24, 260–264. [Google Scholar] [CrossRef] [PubMed]
- Maatta, M.; Heljasvaara, R.; Pihlajaniemi, T.; Uusitalo, M. Collagen XVIII/endostatin shows a ubiquitous distribution in human ocular tissues and endostatin-containing fragments accumulate in ocular fluid samples. Graefe’s Arch. Clin. Exp. Ophthalmol. Albrecht Graefes Arch. Klin. Exp. Ophthalmol. 2007, 245, 74–81. [Google Scholar] [CrossRef]
- Sun, J.; Hopkins, B.D.; Tsujikawa, K.; Perruzzi, C.; Adini, I.; Swerlick, R.; Bornstein, P.; Lawler, J.; Benjamin, L.E. Thrombospondin-1 modulates VEGF-A-mediated Akt signaling and capillary survival in the developing retina. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1344–H1351. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Gottlieb, J.L.; Sorenson, C.M.; Sheibani, N. Modulation of thrombospondin 1 and pigment epithelium-derived factor levels in vitreous fluid of patients with diabetes. Arch. Ophthalmol. 2009, 127, 507–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Goff, M.M.; Hindson, V.J.; Jowitt, T.A.; Scott, P.G.; Bishop, P.N. Characterization of opticin and evidence of stable dimerization in solution. J. Biol. Chem. 2003, 278, 45280–45287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Goff, M.M.; Sutton, M.J.; Slevin, M.; Latif, A.; Humphries, M.J.; Bishop, P.N. Opticin exerts its anti-angiogenic activity by regulating extracellular matrix adhesiveness. J. Biol. Chem. 2012, 287, 28027–28036. [Google Scholar] [CrossRef] [Green Version]
- Tio, L.; Martel-Pelletier, J.; Pelletier, J.P.; Bishop, P.N.; Roughley, P.; Farran, A.; Benito, P.; Monfort, J. Characterization of opticin digestion by proteases involved in osteoarthritis development. Jt. Bone Spine 2014, 81, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Kuiper, E.J.; Van Nieuwenhoven, F.A.; de Smet, M.D.; van Meurs, J.C.; Tanck, M.W.; Oliver, N.; Klaassen, I.; Van Noorden, C.J.; Goldschmeding, R.; Schlingemann, R.O. The angio-fibrotic switch of VEGF and CTGF in proliferative diabetic retinopathy. PLoS ONE 2008, 3, e2675. [Google Scholar] [CrossRef]
- Van Geest, R.J.; Lesnik-Oberstein, S.Y.; Tan, H.S.; Mura, M.; Goldschmeding, R.; Van Noorden, C.J.; Klaassen, I.; Schlingemann, R.O. A shift in the balance of vascular endothelial growth factor and connective tissue growth factor by bevacizumab causes the angiofibrotic switch in proliferative diabetic retinopathy. Br. J. Ophthalmol. 2012, 96, 587–590. [Google Scholar] [CrossRef]
- Sohn, E.H.; He, S.; Kim, L.A.; Salehi-Had, H.; Javaheri, M.; Spee, C.; Dustin, L.; Hinton, D.R.; Eliott, D. Angiofibrotic response to vascular endothelial growth factor inhibition in diabetic retinal detachment: Report no. 1. Arch. Ophthalmol. 2012, 130, 1127–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, C.; Eliott, D.; Spee, C.; He, S.; Wang, K.; Mullins, R.F.; Hinton, D.R.; Sohn, E.H. Apoptosis and angiofibrosis in diabetic tractional membranes after vascular endothelial growth factor inhibition: Results of a Prospective Trial. Report No. 2. Retina 2019, 39, 265–273. [Google Scholar] [CrossRef]
- Kubota, T.; Morita, H.; Tou, N.; Nitta, N.; Tawara, A.; Satoh, H.; Shimajiri, S. Histology of fibrovascular membranes of proliferative diabetic retinopathy after intravitreal injection of bevacizumab. Retina 2010, 30, 468–472. [Google Scholar] [CrossRef]
- Nakao, S.; Ishikawa, K.; Yoshida, S.; Kohno, R.; Miyazaki, M.; Enaida, H.; Kono, T.; Ishibashi, T. Altered vascular microenvironment by bevacizumab in diabetic fibrovascular membrane. Retina 2013, 33, 957–963. [Google Scholar] [CrossRef] [PubMed]
- Pattwell, D.M.; Stappler, T.; Sheridan, C.; Heimann, H.; Gibran, S.K.; Wong, D.; Hiscott, P. Fibrous membranes in diabetic retinopathy and bevacizumab. Retina 2010, 30, 1012–1016. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.; Lajko, M.; Fawzi, A.A. Endothelin-1 is associated with fibrosis in proliferative diabetic retinopathy membranes. PLoS ONE 2018, 13, e0191285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2, e106–e116. [Google Scholar] [CrossRef] [Green Version]
- Lengyel, I.; Tufail, A.; Hosaini, H.A.; Luthert, P.; Bird, A.C.; Jeffery, G. Association of drusen deposition with choroidal intercapillary pillars in the aging human eye. Investig. Ophthalmol. Vis. Sci. 2004, 45, 2886–2892. [Google Scholar] [CrossRef] [Green Version]
- Mullins, R.F.; Johnson, M.N.; Faidley, E.A.; Skeie, J.M.; Huang, J. Choriocapillaris vascular dropout related to density of drusen in human eyes with early age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1606–1612. [Google Scholar] [CrossRef] [Green Version]
- Zeng, S.; Whitmore, S.S.; Sohn, E.H.; Riker, M.J.; Wiley, L.A.; Scheetz, T.E.; Stone, E.M.; Tucker, B.A.; Mullins, R.F. Molecular response of chorioretinal endothelial cells to complement injury: Implications for macular degeneration. J. Pathol. 2016, 238, 446–456. [Google Scholar] [CrossRef] [Green Version]
- Sohn, E.H.; Wang, K.; Thompson, S.; Riker, M.J.; Hoffmann, J.M.; Stone, E.M.; Mullins, R.F. Comparison of drusen and modifying genes in autosomal dominant radial drusen and age-related macular degeneration. Retina 2015, 35, 48–57. [Google Scholar] [CrossRef] [Green Version]
- Mullins, R.F.; Khanna, A.; Schoo, D.P.; Tucker, B.A.; Sohn, E.H.; Drack, A.V.; Stone, E.M. Is age-related macular degeneration a microvascular disease? Adv. Exp. Med. Biol. 2014, 801, 283–289. [Google Scholar] [CrossRef]
- Whitmore, S.S.; Sohn, E.H.; Chirco, K.R.; Drack, A.V.; Stone, E.M.; Tucker, B.A.; Mullins, R.F. Complement activation and choriocapillaris loss in early AMD: Implications for pathophysiology and therapy. Prog. Retin. Eye Res. 2015, 45, 1–29. [Google Scholar] [CrossRef] [Green Version]
- Anderson, D.H.; Radeke, M.J.; Gallo, N.B.; Chapin, E.A.; Johnson, P.T.; Curletti, C.R.; Hancox, L.S.; Hu, J.; Ebright, J.N.; Malek, G.; et al. The pivotal role of the complement system in aging and age-related macular degeneration: Hypothesis re-visited. Prog. Retin. Eye Res. 2010, 29, 95–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hageman, G.S.; Anderson, D.H.; Johnson, L.V.; Hancox, L.S.; Taiber, A.J.; Hardisty, L.I.; Hageman, J.L.; Stockman, H.A.; Borchardt, J.D.; Gehrs, K.M.; et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2005, 102, 7227–7232. [Google Scholar] [CrossRef] [Green Version]
- Mullins, R.F.; Schoo, D.P.; Sohn, E.H.; Flamme-Wiese, M.J.; Workamelahu, G.; Johnston, R.M.; Wang, K.; Tucker, B.A.; Stone, E.M. The membrane attack complex in aging human choriocapillaris: Relationship to macular degeneration and choroidal thinning. Am. J. Pathol. 2014, 184, 3142–3153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seth, A.; Cui, J.; To, E.; Kwee, M.; Matsubara, J. Complement-associated deposits in the human retina. Investig. Ophthalmol. Vis. Sci. 2008, 49, 743–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohn, E.H.; Flamme-Wiese, M.J.; Whitmore, S.S.; Workalemahu, G.; Marneros, A.G.; Boese, E.A.; Kwon, Y.H.; Wang, K.; Abramoff, M.D.; Tucker, B.A.; et al. Choriocapillaris Degeneration in Geographic Atrophy. Am. J. Pathol. 2019, 189, 1473–1480. [Google Scholar] [CrossRef] [PubMed]
- Zarbin, M.A. Current concepts in the pathogenesis of age-related macular degeneration. Arch. Ophthalmol. 2004, 122, 598–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambati, J.; Ambati, B.K.; Yoo, S.H.; Ianchulev, S.; Adamis, A.P. Age-related macular degeneration: Etiology, pathogenesis, and therapeutic strategies. Surv. Ophthalmol. 2003, 48, 257–293. [Google Scholar] [CrossRef]
- Sohn, E.H.; Patel, P.J.; MacLaren, R.E.; Adatia, F.A.; Pal, B.; Webster, A.R.; Tufail, A. Responsiveness of choroidal neovascular membranes in patients with R345W mutation in fibulin 3 (Doyne honeycomb retinal dystrophy) to anti-vascular endothelial growth factor therapy. Arch. Ophthalmol. 2011, 129, 1626–1628. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.F.; Maguire, M.G.; Ying, G.S.; Grunwald, J.E.; Fine, S.L.; Jaffe, G.J. Ranibizumab and bevacizumab for neovascular age-related macular degeneration. N. Engl. J. Med. 2011, 364, 1897–1908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakall, B.; Folk, J.C.; Boldt, H.C.; Sohn, E.H.; Stone, E.M.; Russell, S.R.; Mahajan, V.B. Aflibercept therapy for exudative age-related macular degeneration resistant to bevacizumab and ranibizumab. Am. J. Ophthalmol. 2013, 156, 15–22.e11. [Google Scholar] [CrossRef]
- Davis, A.S.; Folk, J.C.; Russell, S.R.; Sohn, E.H.; Boldt, H.C.; Stone, E.M.; Mahajan, V.B. Intravitreal bevacizumab for peripapillary choroidal neovascular membranes. Arch. Ophthalmol. 2012, 130, 1073–1075. [Google Scholar] [CrossRef] [Green Version]
- Krebs, I.; Glittenberg, C.; Ansari-Shahrezaei, S.; Hagen, S.; Steiner, I.; Binder, S. Non-responders to treatment with antagonists of vascular endothelial growth factor in age-related macular degeneration. Br. J. Ophthalmol. 2013, 97, 1443–1446. [Google Scholar] [CrossRef]
- Barthelmes, D.; Walton, R.; Campain, A.E.; Simpson, J.M.; Arnold, J.J.; McAllister, I.L.; Guymer, R.H.; Hunyor, A.P.; Essex, R.W.; Morlet, N.; et al. Outcomes of persistently active neovascular age-related macular degeneration treated with VEGF inhibitors: Observational study data. Br. J. Ophthalmol. 2015, 99, 359–364. [Google Scholar] [CrossRef]
- Tozer, K.; Roller, A.B.; Chong, L.P.; Sadda, S.; Folk, J.C.; Mahajan, V.B.; Russell, S.R.; Boldt, H.C.; Sohn, E.H. Combination therapy for neovascular age-related macular degeneration refractory to anti-vascular endothelial growth factor agents. Ophthalmology 2013, 120, 2029–2034. [Google Scholar] [CrossRef]
- Keane, P.A.; Liakopoulos, S.; Ongchin, S.C.; Heussen, F.M.; Msutta, S.; Chang, K.T.; Walsh, A.C.; Sadda, S.R. Quantitative subanalysis of optical coherence tomography after treatment with ranibizumab for neovascular age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2008, 49, 3115–3120. [Google Scholar] [CrossRef]
- Forooghian, F.; Cukras, C.; Meyerle, C.B.; Chew, E.Y.; Wong, W.T. Tachyphylaxis after intravitreal bevacizumab for exudative age-related macular degeneration. Retina 2009, 29, 723–731. [Google Scholar] [CrossRef] [Green Version]
- Brown, D.M.; Tuomi, L.; Shapiro, H.; Pier Study, G. Anatomical measures as predictors of visual outcomes in ranibizumab-treated eyes with neovascular age-related macular degeneration. Retina 2013, 33, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Schaal, S.; Kaplan, H.J.; Tezel, T.H. Is there tachyphylaxis to intravitreal anti-vascular endothelial growth factor pharmacotherapy in age-related macular degeneration? Ophthalmology 2008, 115, 2199–2205. [Google Scholar] [CrossRef] [PubMed]
- Binder, S. Loss of reactivity in intravitreal anti-VEGF therapy: Tachyphylaxis or tolerance? Br. J. Ophthalmol. 2012, 96, 1–2. [Google Scholar] [CrossRef] [Green Version]
- Gasperini, J.L.; Fawzi, A.A.; Khondkaryan, A.; Lam, L.; Chong, L.P.; Eliott, D.; Walsh, A.C.; Hwang, J.; Sadda, S.R. Bevacizumab and ranibizumab tachyphylaxis in the treatment of choroidal neovascularisation. Br. J. Ophthalmol. 2012, 96, 14–20. [Google Scholar] [CrossRef]
- Zeng, R.; Zhang, X.; Wu, K.; Su, Y.; Wen, F. MMP9 gene polymorphism is not associated with polypoidal choroidal vasculopathy and neovascular age-related macular degeneration in a Chinese Han population. Ophthalmic Genet. 2014, 35, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Steen, B.; Sejersen, S.; Berglin, L.; Seregard, S.; Kvanta, A. Matrix metalloproteinases and metalloproteinase inhibitors in choroidal neovascular membranes. Investig. Ophthalmol. Vis. Sci. 1998, 39, 2194–2200. [Google Scholar]
- Tatar, O.; Adam, A.; Shinoda, K.; Eckert, T.; Scharioth, G.B.; Klein, M.; Yoeruek, E.; Bartz-Schmidt, K.U.; Grisanti, S. Matrix metalloproteinases in human choroidal neovascular membranes excised following verteporfin photodynamic therapy. Br. J. Ophthalmol. 2007, 91, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- Nussenblatt, R.B.; Ferris, F., 3rd. Age-related macular degeneration and the immune response: Implications for therapy. Am. J. Ophthalmol. 2007, 144, 618–626. [Google Scholar] [CrossRef] [Green Version]
- Egeblad, M.; Werb, Z. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2002, 2, 161–174. [Google Scholar] [CrossRef]
- Overall, C.M.; López-Otín, C. Strategies for MMP inhibition in cancer: Innovations for the post-trial era. Nat. Rev. Cancer 2002, 2, 657–672. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, S.D. Matrix metalloproteinase degradation of extracellular matrix: Biological consequences. Curr. Opin. Cell Biol. 1998, 10, 602–608. [Google Scholar] [CrossRef]
- Lund, L.R.; Romer, J.; Bugge, T.H.; Nielsen, B.S.; Frandsen, T.L.; Degen, J.L.; Stephens, R.W.; Dano, K. Functional overlap between two classes of matrix-degrading proteases in wound healing. EMBO J. 1999, 18, 4645–4656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burg-Roderfeld, M.; Roderfeld, M.; Wagner, S.; Henkel, C.; Grotzinger, J.; Roeb, E. MMP-9-hemopexin domain hampers adhesion and migration of colorectal cancer cells. Int. J. Oncol. 2007, 30, 985–992. [Google Scholar] [CrossRef] [PubMed]
- Chong, N.H.; Keonin, J.; Luthert, P.J.; Frennesson, C.I.; Weingeist, D.M.; Wolf, R.L.; Mullins, R.F.; Hageman, G.S. Decreased thickness and integrity of the macular elastic layer of Bruch’s membrane correspond to the distribution of lesions associated with age-related macular degeneration. Am. J. Pathol. 2005, 166, 241–251. [Google Scholar] [CrossRef]
- Curcio, C.A.; Johnson, M. Structure, function, and pathology of Bruch’s membrane. Retina 2013, 1, 466–481. [Google Scholar]
- Johnson, L.V.; Anderson, D.H. Age-related macular degeneration and the extracellular matrix. N. Engl. J. Med. 2004, 351, 320–322. [Google Scholar] [CrossRef] [PubMed]
- Ng, E.W.; Adamis, A.P. Targeting angiogenesis, the underlying disorder in neovascular age-related macular degeneration. Can. J. Ophthalmol. 2005, 40, 352–368. [Google Scholar] [CrossRef]
- Spraul, C.W.; Lang, G.E.; Grossniklaus, H.E.; Lang, G.K. Histologic and morphometric analysis of the choroid, Bruch’s membrane, and retinal pigment epithelium in postmortem eyes with age-related macular degeneration and histologic examination of surgically excised choroidal neovascular membranes. Surv. Ophthalmol. 1999, 44 (Suppl. 1), S10–S32. [Google Scholar] [CrossRef]
- Fiotti, N.; Pedio, M.; Parodi, M.B.; Altamura, N.; Uxa, L.; Guarnieri, G.; Giansante, C.; Ravalico, G. MMP-9 microsatellite polymorphism and susceptibility to exudative form of age-related macular degeneration. Genet. Med. 2005, 7, 272–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, L.; Hussain, A.A.; Limb, G.A.; Marshall, J. Age-dependent variation in metalloproteinase activity of isolated human Bruch’s membrane and choroid. Investig. Ophthalmol. Vis. Sci. 1999, 40, 2676–2682. [Google Scholar]
- Kamei, M.; Hollyfield, J.G. TIMP-3 in Bruch’s membrane: Changes during aging and in age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 1999, 40, 2367–2375. [Google Scholar]
- Wissink, S.; van Heerde, E.C.; Schmitz, M.L.; Kalkhoven, E.; van der Burg, B.; Baeuerle, P.A.; van der Saag, P.T. Distinct domains of the RelA NF-kappaB subunit are required for negative cross-talk and direct interaction with the glucocorticoid receptor. J. Biol. Chem. 1997, 272, 22278–22284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kvanta, A.; Shen, W.Y.; Sarman, S.; Seregard, S.; Steen, B.; Rakoczy, E. Matrix metalloproteinase (MMP) expression in experimental choroidal neovascularization. Curr. Eye Res. 2000, 21, 684–690. [Google Scholar] [CrossRef]
- Voigt, A.P.; Mulfaul, K.; Mullin, N.K.; Flamme-Wiese, M.J.; Giacalone, J.C.; Stone, E.M.; Tucker, B.A.; Scheetz, T.E.; Mullins, R.F. Single-cell transcriptomics of the human retinal pigment epithelium and choroid in health and macular degeneration. Proc. Natl. Acad. Sci. USA 2019, 116, 24100–24107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oshima, Y.; Oshima, S.; Nambu, H.; Kachi, S.; Hackett, S.F.; Melia, M.; Kaleko, M.; Connelly, S.; Esumi, N.; Zack, D.J.; et al. Increased expression of VEGF in retinal pigmented epithelial cells is not sufficient to cause choroidal neovascularization. J. Cell Physiol 2004, 201, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Samtani, S.; Amaral, J.; Campos, M.M.; Fariss, R.N.; Becerra, S.P. Doxycycline-mediated inhibition of choroidal neovascularization. Investig. Ophthalmol. Vis. Sci. 2009, 50, 5098–5106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roychoudhury, J.; Herndon, J.M.; Yin, J.; Apte, R.S.; Ferguson, T.A. Targeting immune privilege to prevent pathogenic neovascularization. Investig. Ophthalmol. Vis. Sci. 2010, 51, 3560–3566. [Google Scholar] [CrossRef] [PubMed]
- Schwesinger, C.; Yee, C.; Rohan, R.M.; Joussen, A.M.; Fernandez, A.; Meyer, T.N.; Poulaki, V.; Ma, J.J.; Redmond, T.M.; Liu, S. Intrachoroidal neovascularization in transgenic mice overexpressing vascular endothelial growth factor in the retinal pigment epithelium. Am. J. Pathol. 2001, 158, 1161–1172. [Google Scholar] [CrossRef] [Green Version]
- Dawson, D.W.; Volpert, O.V.; Gillis, P.; Crawford, S.E.; Xu, H.; Benedict, W.; Bouck, N.P. Pigment epithelium-derived factor: A potent inhibitor of angiogenesis. Science 1999, 285, 245–248. [Google Scholar] [CrossRef]
- Ogata, N.; Wada, M.; Otsuji, T.; Jo, N.; Tombran-Tink, J.; Matsumura, M. Expression of pigment epithelium-derived factor in normal adult rat eye and experimental choroidal neovascularization. Investig. Ophthalmol. Vis. Sci. 2002, 43, 1168–1175. [Google Scholar]
- Becerra, S.P. Focus on Molecules: Pigment epithelium-derived factor (PEDF). Exp. Eye Res. 2006, 82, 739–740. [Google Scholar] [CrossRef] [PubMed]
- Gehlbach, P.; Demetriades, A.M.; Yamamoto, S.; Deering, T.; Duh, E.J.; Yang, H.S.; Cingolani, C.; Lai, H.; Wei, L.; Campochiaro, P.A. Periocular injection of an adenoviral vector encoding pigment epithelium-derived factor inhibits choroidal neovascularization. Gene Ther. 2003, 10, 637–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, V.; Munaut, C.; Jost, M.; Noel, A.; Werb, Z.; Foidart, J.M.; Rakic, J.M. Matrix metalloproteinase-9 contributes to choroidal neovascularization. Am. J. Pathol. 2002, 161, 1247–1253. [Google Scholar] [CrossRef] [Green Version]
- Lambert, V.; Wielockx, B.; Munaut, C.; Galopin, C.; Jost, M.; Itoh, T.; Werb, Z.; Baker, A.; Libert, C.; Krell, H.W.; et al. MMP-2 and MMP-9 synergize in promoting choroidal neovascularization. FASEB J. 2003, 17, 2290–2292. [Google Scholar] [CrossRef]
- Bergers, G.; Brekken, R.; McMahon, G.; Vu, T.H.; Itoh, T.; Tamaki, K.; Tanzawa, K.; Thorpe, P.; Itohara, S.; Werb, Z.; et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat. Cell Biol. 2000, 2, 737–744. [Google Scholar] [CrossRef]
- Hollborn, M.; Stathopoulos, C.; Steffen, A.; Wiedemann, P.; Kohen, L.; Bringmann, A. Positive feedback regulation between MMP-9 and VEGF in human RPE cells. Investig. Ophthalmol. Vis. Sci. 2007, 48, 4360–4367. [Google Scholar] [CrossRef] [Green Version]
- Liutkeviciene, R.; Lesauskaite, V.; Sinkunaite-Marsalkiene, G.; Zaliuniene, D.; Zaliaduonyte-Peksiene, D.; Mizariene, V.; Gustiene, O.; Jasinskas, V.; Jariene, G.; Tamosiunas, A. The Role of Matrix Metalloproteinases Polymorphisms in Age-Related Macular Degeneration. Ophthalmic Genet. 2015, 36, 149–155. [Google Scholar] [CrossRef]
- Chau, K.Y.; Sivaprasad, S.; Patel, N.; Donaldson, T.A.; Luthert, P.J.; Chong, N.V. Plasma levels of matrix metalloproteinase-2 and -9 (MMP-2 and MMP-9) in age-related macular degeneration. Eye 2008, 22, 855–859. [Google Scholar] [CrossRef]
- Jonas, J.B.; Tao, Y.; Neumaier, M.; Findeisen, P. Cytokine concentration in aqueous humour of eyes with exudative age-related macular degeneration. Acta Ophthalmol. 2012, 90, e381–e388. [Google Scholar] [CrossRef]
- Sivaprasad, S.; Webster, A.R.; Egan, C.A.; Bird, A.C.; Tufail, A. Clinical course and treatment outcomes of Sorsby fundus dystrophy. Am. J. Ophthalmol. 2008, 146, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Rahman, N.; Georgiou, M.; Khan, K.N.; Michaelides, M. Macular dystrophies: Clinical and imaging features, molecular genetics and therapeutic options. Br. J. Ophthalmol. 2020, 104, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Butler, G.S.; Apte, S.S.; Willenbrock, F.; Murphy, G. Human tissue inhibitor of metalloproteinases 3 interacts with both the N-and C-terminal domains of gelatinases A and B: Regulation by polyanions. J. Biol. Chem. 1999, 274, 10846–10851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohn, E.H.; Khanna, A.; Tucker, B.A.; Abramoff, M.D.; Stone, E.M.; Mullins, R.F. Structural and biochemical analyses of choroidal thickness in human donor eyes. Investig. Ophthalmol. Vis. Sci. 2014, 55, 1352–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritsche, L.G.; Igl, W.; Bailey, J.N.; Grassmann, F.; Sengupta, S.; Bragg-Gresham, J.L.; Burdon, K.P.; Hebbring, S.J.; Wen, C.; Gorski, M.; et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat. Genet. 2016, 48, 134–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohn, E.H.; Han, I.C.; Roos, B.R.; Faga, B.; Luse, M.A.; Binkley, E.M.; Boldt, H.C.; Folk, J.C.; Russell, S.R.; Mullins, R.F.; et al. Genetic association between MMP9 and choroidal neovascularization in age-related macular degeneration. Ophthalmol. Sci. 2020, 1, 100002. [Google Scholar] [CrossRef]
- Sura, A.A.; Chen, L.; Messinger, J.D.; Swain, T.A.; McGwin, G., Jr.; Freund, K.B.; Curcio, C.A. Measuring the Contributions of Basal Laminar Deposit and Bruch’s Membrane in Age-Related Macular Degeneration. Investig. Ophthalmol. Vis. Sci. 2020, 61, 19. [Google Scholar] [CrossRef]
- Mullins, R.F.; McGwin, G., Jr.; Searcey, K.; Clark, M.E.; Kennedy, E.L.; Curcio, C.A.; Stone, E.M.; Owsley, C. The ARMS2 A69S Polymorphism Is Associated with Delayed Rod-Mediated Dark Adaptation in Eyes at Risk for Incident Age-Related Macular Degeneration. Ophthalmology 2019, 126, 591–600. [Google Scholar] [CrossRef]
- Karwatowski, W.S.; Jeffries, T.E.; Duance, V.C.; Albon, J.; Bailey, A.J.; Easty, D.L. Preparation of Bruch’s membrane and analysis of the age-related changes in the structural collagens. Br. J. Ophthalmol. 1995, 79, 944–952. [Google Scholar] [CrossRef] [Green Version]
- Hussain, A.A.; Lee, Y.; Zhang, J.J.; Marshall, J. Disturbed matrix metalloproteinase activity of Bruch’s membrane in age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2011, 52, 4459–4466. [Google Scholar] [CrossRef]
- Fariss, R.N.; Apte, S.S.; Olsen, B.R.; Iwata, K.; Milam, A.H. Tissue inhibitor of metalloproteinases-3 is a component of Bruch’s membrane of the eye. Am. J. Pathol. 1997, 150, 323–328. [Google Scholar] [PubMed]
- Knupp, C.; Chong, N.H.; Munro, P.M.; Luthert, P.J.; Squire, J.M. Analysis of the collagen VI assemblies associated with Sorsby’s fundus dystrophy. J. Struct. Biol. 2002, 137, 31–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Miyamura, N.; Ninomiya, Y.; Handa, J.T. Distribution of the collagen IV isoforms in human Bruch’s membrane. Br. J. Ophthalmol. 2003, 87, 212–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhutto, I.A.; Uno, K.; Merges, C.; Zhang, L.; McLeod, D.S.; Lutty, G.A. Reduction of endogenous angiogenesis inhibitors in Bruch’s membrane of the submacular region in eyes with age-related macular degeneration. Arch. Ophthalmol. 2008, 126, 670–678. [Google Scholar] [CrossRef] [Green Version]
- Haimovici, R.; Gantz, D.L.; Rumelt, S.; Freddo, T.F.; Small, D.M. The lipid composition of drusen, Bruch’s membrane, and sclera by hot stage polarizing light microscopy. Investig. Ophthalmol. Vis. Sci. 2001, 42, 1592–1599. [Google Scholar]
- Rudolf, M.; Curcio, C.A. Esterified cholesterol is highly localized to Bruch’s membrane, as revealed by lipid histochemistry in wholemounts of human choroid. J. Histochem. Cytochem. Off. J. Histochem. Soc. 2009, 57, 731–739. [Google Scholar] [CrossRef] [Green Version]
- Uno, K.; Bhutto, I.A.; McLeod, D.S.; Merges, C.; Lutty, G.A. Impaired expression of thrombospondin-1 in eyes with age related macular degeneration. Br. J. Ophthalmol. 2006, 90, 48–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussain, A.A.; Starita, C.; Hodgetts, A.; Marshall, J. Macromolecular diffusion characteristics of ageing human Bruch’s membrane: Implications for age-related macular degeneration (AMD). Exp. Eye Res. 2010, 90, 703–710. [Google Scholar] [CrossRef]
- Chang, J.H.; Spraul, C.W.; Lynn, M.L.; Drack, A.; Grossniklaus, H.E. The two-stage mutation model in retinal hemangioblastoma. Ophthalmic Genet. 1998, 19, 123–130. [Google Scholar] [CrossRef]
- Heng, L.Z.; Comyn, O.; Peto, T.; Tadros, C.; Ng, E.; Sivaprasad, S.; Hykin, P.G. Diabetic retinopathy: Pathogenesis, clinical grading, management and future developments. Diabet Med. 2013, 30, 640–650. [Google Scholar] [CrossRef]
- Skeie, J.M.; Hernandez, J.; Hinek, A.; Mullins, R.F. Molecular responses of choroidal endothelial cells to elastin derived peptides through the elastin-binding protein (GLB1). Matrix Biol. 2012, 31, 113–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Booij, J.C.; Baas, D.C.; Beisekeeva, J.; Gorgels, T.G.; Bergen, A.A. The dynamic nature of Bruch’s membrane. Prog. Retin. Eye Res. 2010, 29, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Curcio, C.A.; Johnson, M.; Rudolf, M.; Huang, J.D. The oil spill in ageing Bruch membrane. Br. J. Ophthalmol. 2011, 95, 1638–1645. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.J.; Keenan, T.D.; Fielder, H.L.; Collinson, L.J.; Holley, R.J.; Merry, C.L.; van Kuppevelt, T.H.; Day, A.J.; Bishop, P.N. Mapping the differential distribution of glycosaminoglycans in the adult human retina, choroid, and sclera. Investig. Ophthalmol. Vis. Sci. 2011, 52, 6511–6521. [Google Scholar] [CrossRef] [PubMed]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pouw, A.E.; Greiner, M.A.; Coussa, R.G.; Jiao, C.; Han, I.C.; Skeie, J.M.; Fingert, J.H.; Mullins, R.F.; Sohn, E.H. Cell–Matrix Interactions in the Eye: From Cornea to Choroid. Cells 2021, 10, 687. https://doi.org/10.3390/cells10030687
Pouw AE, Greiner MA, Coussa RG, Jiao C, Han IC, Skeie JM, Fingert JH, Mullins RF, Sohn EH. Cell–Matrix Interactions in the Eye: From Cornea to Choroid. Cells. 2021; 10(3):687. https://doi.org/10.3390/cells10030687
Chicago/Turabian StylePouw, Andrew E., Mark A. Greiner, Razek G. Coussa, Chunhua Jiao, Ian C. Han, Jessica M. Skeie, John H. Fingert, Robert F. Mullins, and Elliott H. Sohn. 2021. "Cell–Matrix Interactions in the Eye: From Cornea to Choroid" Cells 10, no. 3: 687. https://doi.org/10.3390/cells10030687
APA StylePouw, A. E., Greiner, M. A., Coussa, R. G., Jiao, C., Han, I. C., Skeie, J. M., Fingert, J. H., Mullins, R. F., & Sohn, E. H. (2021). Cell–Matrix Interactions in the Eye: From Cornea to Choroid. Cells, 10(3), 687. https://doi.org/10.3390/cells10030687