
Fenamates as Potential Therapeutics for Neurodegenerative Disorders


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
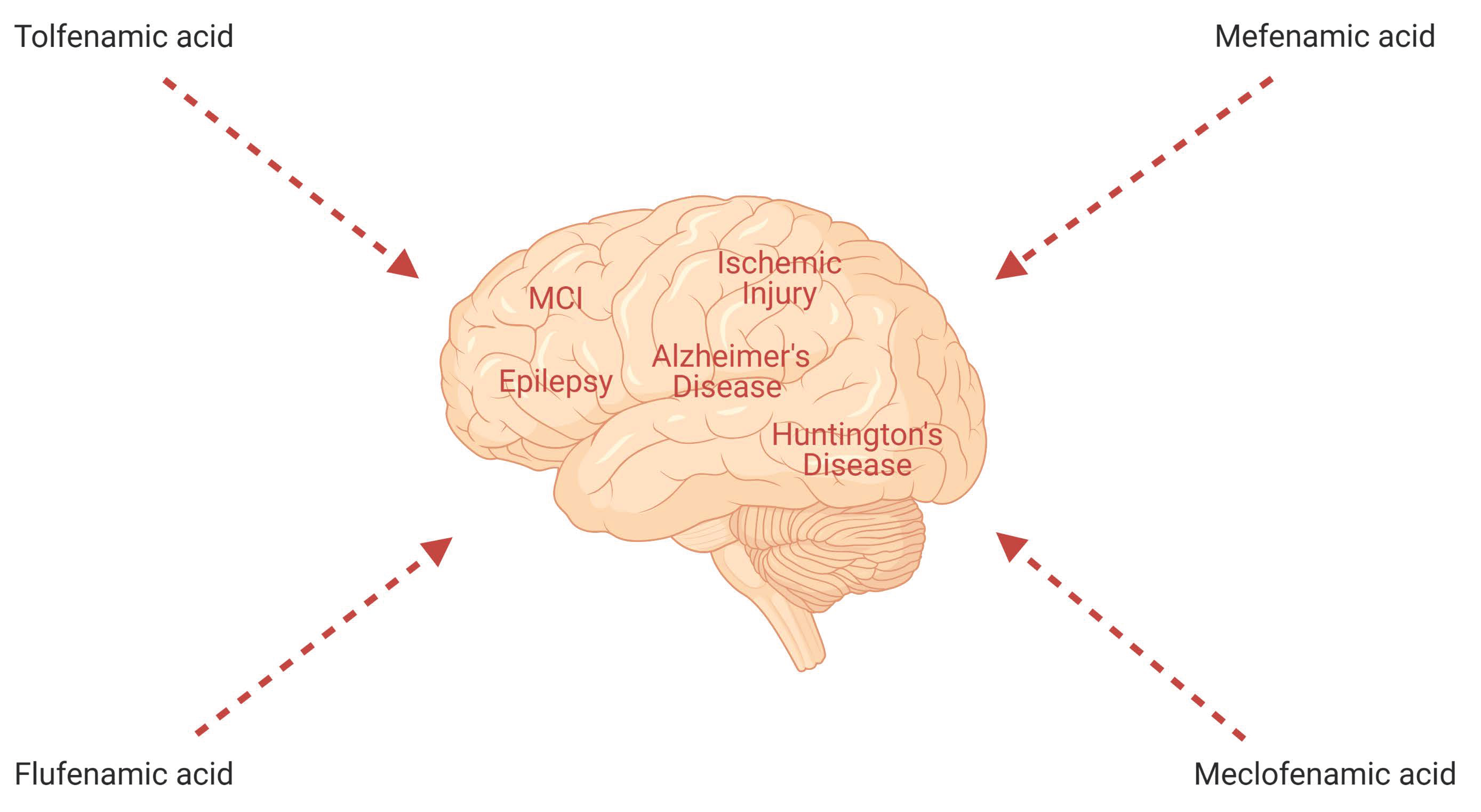
1. Introduction
2. Fenamate Pharmacokinetics and Pharmacodynamics
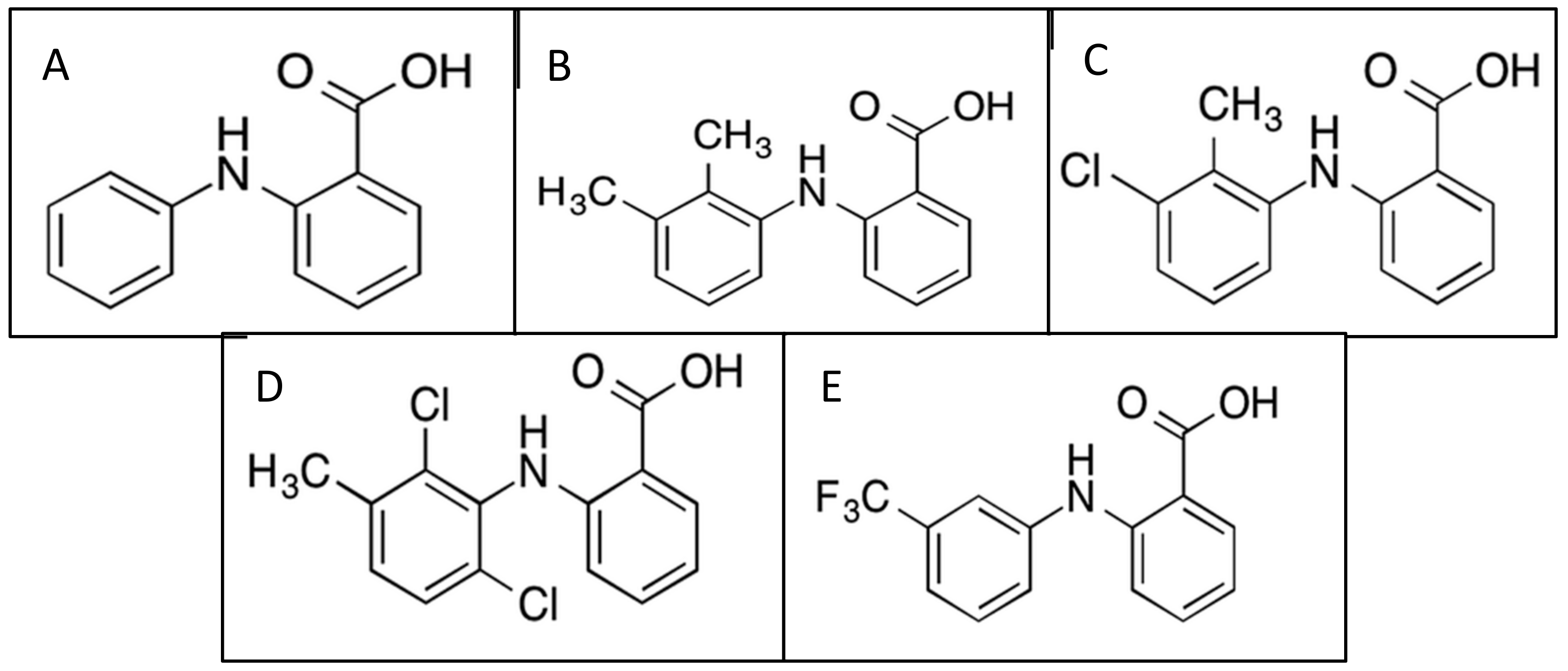
2.1. Tolfenamic Acid
2.2. Mefenamic Acid
2.3. Meclofenamic Acid
2.4. Flufenamic Acid
2.5. Adverse Drug Reactions
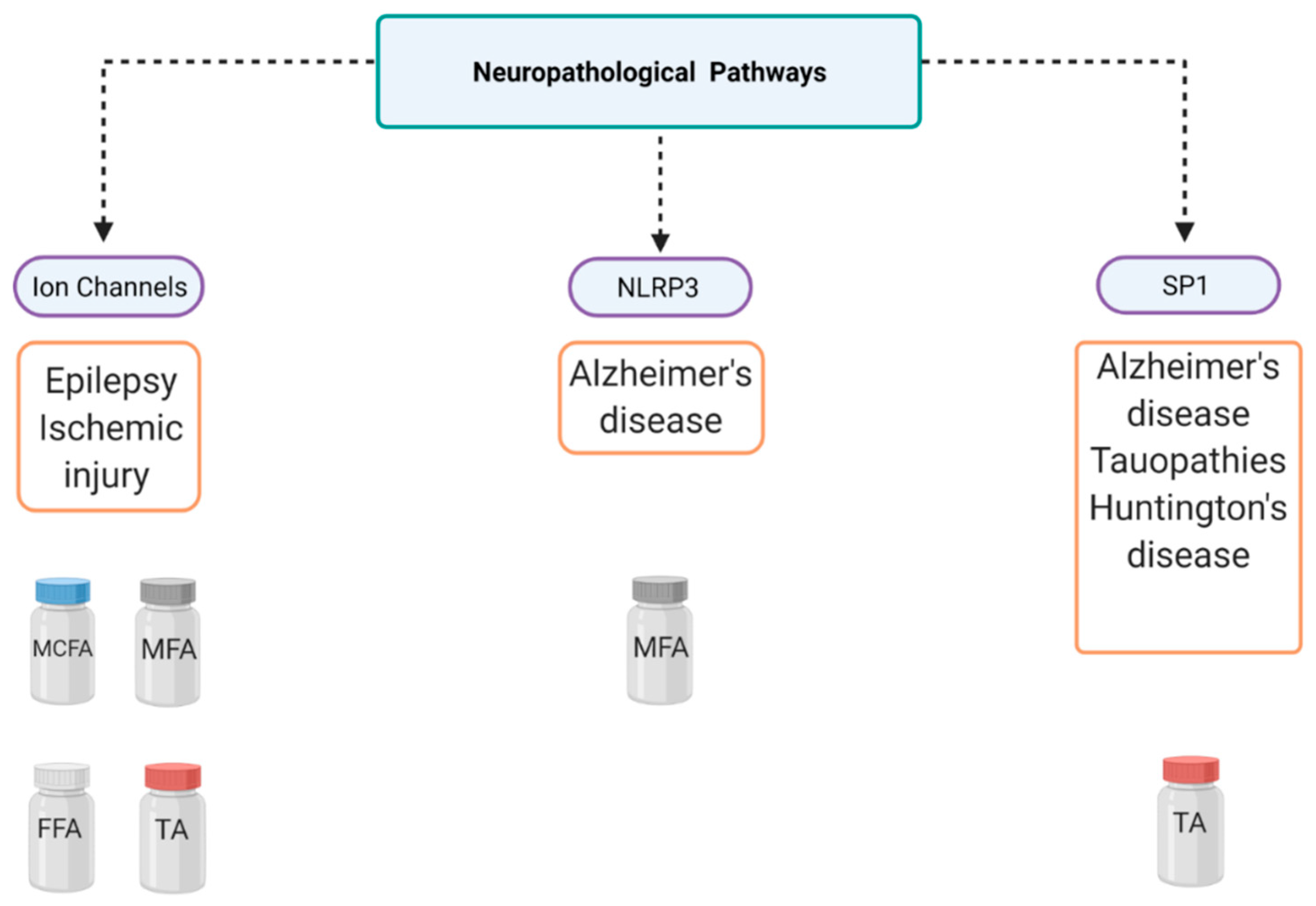
3. Fenamates and Alzheimer’s Disease (AD)
4. Fenamates and Cognitive Impairment
4.1. Fenamates and Tauopathies
4.2. Fenamates and Chronic Alcohol Exposure
4.3. Fenamates and Ischemic Injury
5. Fenamates and Huntington’s Disease
6. Fenamates and Epilepsy
7. Overview of Alternative Drugs
8. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| NSAIDS | Nonsteroidal anti-inflammatory drugs |
| COX | Cyclooxygenases |
| PG | Prostaglandin |
| IC50 | Inhibitory concentration |
| MFA | Mefenamic acid |
| TA | Tolfenamic acid |
| MCFA | Meclofenamic acid |
| FFA | Flufenamic acid |
| PHGS | Prostaglandin H synthase |
| AD | Alzheimer’s disease |
| MRI | Magnetic resonance imaging |
| CSF | Cerebral spinal fluid |
| LOAD | Late-onset Alzheimer’s disease |
| EOAD | Early-onset Alzheimer’s disease |
| FDA | Federal Drug Administration |
| NMDA | N-Methyl-D-aspartic acid |
| APP | Amyloid precursor protein |
| BACE-1 | β-Secretase-1 |
| SP1 | Specificity protein 1 |
| MWM | Morris water maze |
| MAPT | Microtubule-associated protein tau |
| CDK5 | Cyclin-dependent kinase-5 |
| siRNA | small interfering RNA |
| GSK3β | Glycogen synthase kinase-3 beta |
| ptau | Phosphotau |
| hTau | Human tau |
| NLRP3 | NLR family pyrin domain-containing 3 |
| NF-κB | Nuclear factor kappa B |
| MCI | Mild cognitive impairment |
| NFT | Neurofibrillary tau tangles |
| FTD-17 | Frontotemporal dementia with parkinsonism-17 |
| PSP | Progressive supranuclear palsy |
| PD | Parkinson’s disease |
| EMA | European Medicines Agency |
| TLRs | Toll-like receptors |
| AChE | Acetylcholine esterase |
| ROS | Reactive oxygen species |
| ICV | Intracerebroventricular |
| HD | Huntington’s disease |
| CAG | Cytosine–adenine–guanine |
| mHtt | Mutant huntingtin |
| Htt | Huntingtin protein |
| MSNs | Medium spiny neurons |
| ASC | Associated speck-like protein |
| VRAC | Voltage-gated anion channels |
| NOR | Novel object recognition |
| VGSC | Voltage-gated sodium channels |
| MTM | Mithramycin |
| ASD | Anti-seizure drugs |
| GAT | GABA transporter 1 |
References
- Joo, Y.; Kim, H.S.; Woo, R.S.; Cheol, H.P.; Shin, K.Y.; Lee, J.P.; Chang, K.A.; Kim, S.; Suh, Y.H. Mefenamic Acid Shows Neuroprotective Effects and Improves Cognitive Impairment in in Vitro and in Vivo Alzheimer’s Disease Models. Mol. Pharmacol. 2006, 69, 76–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniels, M.J.D.; Rivers-Auty, J.; Schilling, T.; Spencer, N.G.; Watremez, W.; Fasolino, V.; Booth, S.J.; White, C.S.; Baldwin, A.G.; Freeman, S.; et al. Fenamate NSAIDs Inhibit the NLRP3 Inflammasome and Protect against Alzheimer’s Disease in Rodent Models. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- DuBois, R.N.; Abramson, S.B.; Crofford, L.; Gupta, R.A.; Simon, L.S.; Putte, L.B.A.; Lipsky, P.E. Cyclooxygenase in Biology and Disease. FASEB J. 1998, 12, 1063–1073. [Google Scholar] [CrossRef] [Green Version]
- Fitzpatrick, F.A. Cyclooxygenase Enzymes: Regulation and Function. Curr. Pharm. Des. 2004, 10, 577–588. [Google Scholar] [CrossRef]
- Bombardier, C.; Laine, L.; Reicin, A.; Shapiro, D.; Burgos-Vargas, R.; Davis, B.; Day, R.; Ferraz, M.B.; Hawkey, C.J.; Hochberg, M.C.; et al. Comparison of Upper Gastrointestinal Toxicity of Rofecoxib and Naproxen in Patients with Rheumatoid Arthritis. N. Engl. J. Med. 2000, 343, 1520–1528. [Google Scholar] [CrossRef]
- Patrono, C. Cardiovascular Effects of Cyclooxygenase-2 Inhibitors: A Mechanistic and Clinical Perspective. Br. J. Pharmacol. 2016, 82, 957–964. [Google Scholar] [CrossRef]
- Hawkey, C.J. COX-1 and COX-2 Inhibitors. Best Pract. Res. Clin. Gastroenterol. 2001, 15, 801–820. [Google Scholar] [CrossRef]
- Drini, M. Peptic Ulcer Disease and Non-Steroidal Anti-Inflammatory Drugs. Aust. Prescr. 2017, 40, 91–93. [Google Scholar] [CrossRef]
- Krumholz, H.; Ross, J.S.; Presler, A.H.; Egilman, D.S. What Have We Learnt from Vioxx? BMJ 2007, 334, 120–123. [Google Scholar] [CrossRef] [Green Version]
- Graham, G.G. Fenamates. In Compendium of Inflammatory Diseases; Parnham, M.J., Ed.; Springer, Birkhauser: Basel, Switzerland, 2016; pp. 1–6. [Google Scholar] [CrossRef]
- Lees, P.; Giraudel, J.; Landoni, M.F.; Toutain, P.L. PK-PD Integration and PK-PD Modelling of Nonsteroidal Anti-Inflammatory Drugs: Principles and Applications in Veterinary Pharmacology. J. Veter Pharmacol. Ther. 2004, 27, 491–502. [Google Scholar] [CrossRef]
- Bindu, S.; Mazumder, S.; Bandyopadhyay, U. Non-Steroidal Anti-Inflammatory Drugs (NSAIDs) and Organ Damage: A Current Perspective. Biochem. Pharmacol. 2020, 180, 114147. [Google Scholar] [CrossRef]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A Comprehensive Resource for in Silico Drug Discovery and Exploration. Nucleic Acids Res. 2006, 34, D668. [Google Scholar] [CrossRef]
- Subaiea, G.M.; Alansi, B.H.; Serra, D.A.; Alwan, M.; Zawia, N.H. The ability of tolfenamic acid to penetrate the brain: A model for testing the brain disposition of candidate Alzheimer’s drugs using multiple platforms. Curr. Alzheimer Res. 2011, 8, 860–867. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. PONSTEL® (Mefenamic Acid Capsules, USP); U.S. Food and Drug Administration: Washington, DC, USA, 2008. [Google Scholar]
- Cimolai, N. The Potential and Promise of Mefenamic Acid. Expert Rev. Clin. Pharmacol. 2013, 6, 289–305. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 Update: Improved Access to Chemical Data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef] [Green Version]
- Sostres, C.; Gargallo, C.J.; Arroyo, M.T.; Lanas, A. Adverse Effects of Non-Steroidal Anti-Inflammatory Drugs (NSAIDs, Aspirin and Coxibs) on Upper Gastrointestinal Tract. Best Pract. Res. Clin. Gastroenterol. 2010, 24, 121–132. [Google Scholar] [CrossRef]
- Grosser, T.; Fries, S.; FitzGerald, G.A. Biological Basis for the Cardiovascular Consequences of COX-2 Inhibition: Therapeutic Challenges and Opportunities. J. Clin. Investig. 2005, 116, 4–15. [Google Scholar] [CrossRef]
- Varga, Z.; Sabzwari, S.; Rafay, A.; Vargova, V. Cardiovascular Risk of Nonsteroidal Anti-Inflammatory Drugs: An Under-Recognized Public Health Issue. Cureus 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Ricciotti, E.; Scalia, R.; Tang, S.Y.; Grant, G.; Yu, Z.; Landesberg, G.; Crichton, I.; Wu, W.; Puré, E.; et al. Vascular COX-2 Modulates Blood Pressure and Thrombosis in Mice. Sci. Transl. Med. 2012. [Google Scholar] [CrossRef] [Green Version]
- Muraoka, S.; Miura, T. Inactivation of Cholinesterase Induced by Non-Steroidal Anti-Inflammatory Drugs with Horseradish Peroxidase: Implication for Alzheimer’s Disease. Life Sci. 2009, 84, 272–277. [Google Scholar] [CrossRef]
- Etminan, M.; Gill, S.; Samii, A. Effect of Non-Steroidal Anti-Inflammatory Drugs on Risk of Alzheimer’s Disease: Systematic Review and Meta-Analysis of Observational Studies. BMJ 2003, 327, 128–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlad, S.C.; Miller, D.R.; Kowall, N.W.; Felson, D.T. Protective Effects of NSAIDs on the Development of Alzheimer Disease. Neurology 2008, 70, 1672–1677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, P.F.; Tremblay-Mercier, J.; Leoutsakos, J.; Madjar, C.; Lafaille-Maignan, M.É.; Savard, M.; Rosa-Neto, P.; Poirier, J.; Etienne, P.; Breitner, J. A Randomized Trial of Naproxen to Slow Progress of Presymptomatic Alzheimer Disease. Neurology 2019, 92, E2070–E2080. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Wang, Y.; Wang, D.; Zhang, J.; Zhang, F. NSAID Exposure and Risk of Alzheimer’s Disease: An Updated Meta-Analysis from Cohort Studies. Front. Aging Neurosci. 2018, 10, 83. [Google Scholar] [CrossRef]
- 2020 Alzheimer’s Disease Facts and Figures. Alzheimer’s Dement. 2020, 16, 391–460. [CrossRef]
- Yassine, H.N.; Finch, C.E. APOE Alleles and Diet in Brain Aging and Alzheimer’s Disease. Front. Aging Neurosci. 2020, 12, 150. [Google Scholar] [CrossRef]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- Venkataraman, A.; Kalk, N.; Sewell, G.; Ritchie, C.W.; Lingford-Hughes, A. Alcohol and Alzheimer’s Disease-Does Alcohol Dependence Contribute to Beta-Amyloid Deposition, Neuroinflammation and Neurodegeneration in Alzheimer’s Disease? Alcohol Alcohol. 2017, 52, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ma, Q.; Zhang, Y.; Xu, H. Proteolytic Processing of Alzheimer’s β-Amyloid Precursor Protein. J. Neurochem. 2012, 120 (Suppl. 1), 9–21. [Google Scholar] [CrossRef] [Green Version]
- Tang, B.L. Amyloid Precursor Protein (APP) and GABAergic Neurotransmission. Cells 2019, 8, 550. [Google Scholar] [CrossRef] [Green Version]
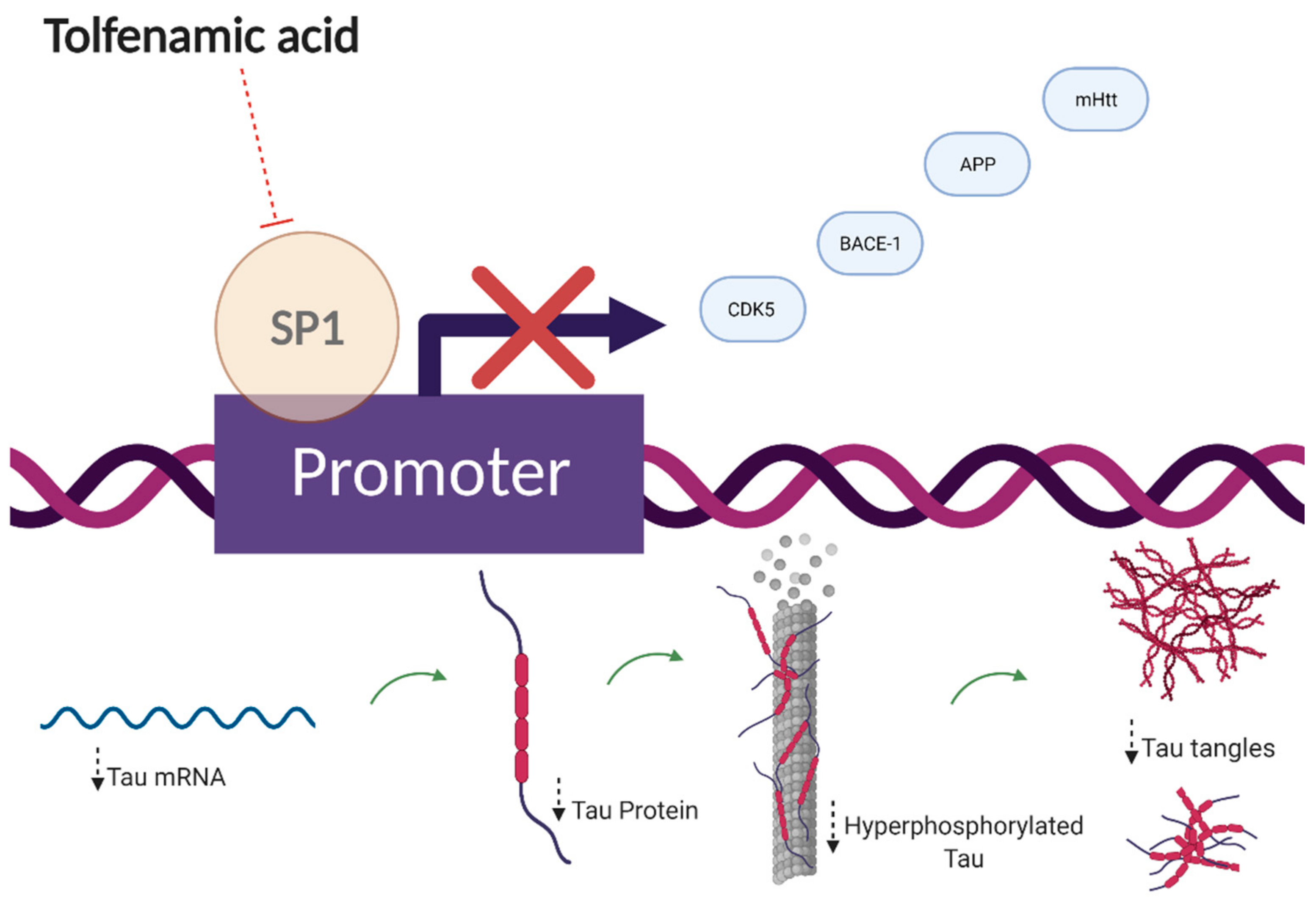
- Adwan, L.I.; Basha, R.; Abdelrahim, M.; Subaiea, G.M.; Zawia, N.H. Tolfenamic acid interrupts the de novo synthesis of the ?-amyloid precursor protein and lowers amyloid beta via a transcriptional pathway. Curr. Alzheimer Res. 2011, 8, 385–392. [Google Scholar] [CrossRef]
- Abdelrahim, M.; Baker, C.H.; Abbruzzese, J.L.; Safe, S. Tolfenamic Acid and Pancreatic Cancer Growth, Angiogenesis, and Sp Protein Degradation. J. Natl. Cancer Inst. 2006, 98, 855–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subaiea, G.M.; Adwan, L.I.; Ahmed, A.H.; Stevens, K.E.; Zawia, N.H. Short-Term Treatment with Tolfenamic Acid Improves Cognitive Functions in Alzheimer’s Disease Mice. Neurobiol. Aging 2013, 34, 2421–2430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adwan, L.; Subaiea, G.M.; Zawia, N.H. Tolfenamic Acid Downregulates BACE1 and Protects against Lead-Induced Upregulation of Alzheimer’s Disease Related Biomarkers. Neuropharmacology 2014, 79, 596–602. [Google Scholar] [CrossRef]
- Leso, A.; Bihaqi, S.W.; Masoud, A.; Chang, J.K.; Lahouel, A.; Zawia, N. Loss in Efficacy Measures of Tolfenamic Acid in a Tau Knock-out Model: Relevance to Alzheimer’s Disease. Exp. Biol. Med. 2019, 244, 1062–1069. [Google Scholar] [CrossRef]
- Adwan, L.; Subaiea, G.M.; Basha, R.; Zawia, N.H. Tolfenamic Acid Reduces Tau and CDK5 Levels: Implications for Dementia and Tauopathies. J. Neurochem. 2015, 133, 266–272. [Google Scholar] [CrossRef] [Green Version]
- Brock, B.; Basha, R.; DiPalma, K.; Anderson, A.; Harry, G.J.; Rice, D.C.; Maloney, B.; Lahiri, D.K.; Zawia, N.H. Co-Localization and Distribution of Cerebral APP and SP1 and Its Relationship to Amyloidogenesis. J. Alzheimer’s Dis. 2008, 13, 71–80. [Google Scholar] [CrossRef] [Green Version]
- Basha, M.R.; Wei, W.; Bakheet, S.A.; Benitez, N.; Siddiqi, H.K.; Ge, Y.W.; Lahiri, D.K.; Zawia, N.H. The Fetal Basis of Amyloidogenesis: Exposure to Lead and Latent Overexpression of Amyloid Precursor Protein and β-Amyloid in the Aging Brain. J. Neurosci. 2005, 25, 823–829. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, X.; Xu, P.; Ji, X.; Chi, T.; Liu, P.; Zou, L. Tolfenamic Acid Inhibits GSK-3β and PP2A Mediated Tau Hyperphosphorylation in Alzheimer’s Disease Models. J. Physiol. Sci. 2020, 70, 29. [Google Scholar] [CrossRef]
- Chang, J.K.; Leso, A.; Subaiea, G.M.; Lahouel, A.; Masoud, A.; Mushtaq, F.; Deeb, R.; Eid, A.; Dash, M.; Bihaqi, S.W.; et al. Tolfenamic Acid: A Modifier of the Tau Protein and Its Role in Cognition and Tauopathy. Curr. Alzheimer Res. 2018, 15, 655–663. [Google Scholar] [CrossRef]
- Nelson, P.T.; Alafuzoff, I.; Bigio, E.H.; Bouras, C.; Braak, H.; Cairns, N.J.; Castellani, R.J.; Crain, B.J.; Davies, P.; Del Tredici, K.; et al. Correlation of Alzheimer Disease Neuropathologic Changes With Cognitive Status: A Review of the Literature. J. Neuropathol. Exp. Neurol. 2012, 71, 362–381. [Google Scholar] [CrossRef]
- Pontecorvo, M.J.; Keene, C.D.; Beach, T.G.; Montine, T.J.; Arora, A.K.; Devous, M.D.; Navitsky, M.; Kennedy, I.; Joshi, A.D.; Lu, M.; et al. Comparison of Regional Flortaucipir PET with Quantitative Tau Immunohistochemistry in Three Subjects with Alzheimer’s Disease Pathology: A Clinicopathological Study. EJNMMI Res. 2020, 10, 65. [Google Scholar] [CrossRef]
- Caillet-Boudin, M.-L.; Buée, L.; Sergeant, N.; Lefebvre, B. Regulation of Human MAPT Gene Expression. Mol. Neurodegener. 2015, 10, 28. [Google Scholar] [CrossRef] [Green Version]
- Barbier, P.; Zejneli, O.; Martinho, M.; Lasorsa, A.; Belle, V.; Smet-Nocca, C.; Tsvetkov, P.O.; Devred, F.; Landrieu, I. Role of Tau as a Microtubule-Associated Protein: Structural and Functional Aspects. Front. Aging Neurosci. 2019, 11, 204. [Google Scholar] [CrossRef] [Green Version]
- Dolan, P.J.; Johnson, G.V.W. The Role of Tau Kinases in Alzheimer’s Disease. Curr. Opin. Drug. Discov. Devel. 2010, 13, 595–603. [Google Scholar]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 Inflammasome Activation Drives Tau Pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Fan, Y.; Chung, C.Y. Mefenamic Acid Can Attenuate Depressive Symptoms by Suppressing Microglia Activation Induced upon Chronic Stress. Brain Res. 2020, 1740, 146846. [Google Scholar] [CrossRef]
- Shao, H.J.; Lou, Z.; Jeong, J.B.; Kim, K.J.; Lee, J.; Lee, S.H. Tolfenamic Acid Suppresses Inflammatory Stimuli-Mediated Activation of NF-ΚB Signaling. Biomol. Ther. 2015, 23, 39–44. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Li, Z.; Liu, H.; Zhu, Y.; Xia, D.; Wang, S.; Gu, R.; Wu, W.; Zhang, P.; Liu, Y.; et al. Low Concentration Flufenamic Acid Enhances Osteogenic Differentiation of Mesenchymal Stem Cells and Suppresses Bone Loss by Inhibition of the NF-ΚB Signaling Pathway. Stem Cell Res. Ther. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Armagan, G.; Turunc, E.; Kanit, L.; Yalcin, A. Neuroprotection by Mefenamic Acid against D-Serine: Involvement of Oxidative Stress, Inflammation and Apoptosis. Free Radic. Res. 2012, 46, 726–739. [Google Scholar] [CrossRef] [PubMed]
- Jongsiriyanyong, S.; Limpawattana, P. Mild Cognitive Impairment in Clinical Practice: A Review Article. Am. J. Alzheimer’s Dis. Other Dement. 2018, 33, 500–507. [Google Scholar] [CrossRef]
- Gillis, C.; Mirzaei, F.; Potashman, M.; Ikram, M.A.; Maserejian, N. The incidence of mild cognitive impairment: A systematic review and data synthesis. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2019, 11, 248–256. [Google Scholar] [CrossRef]
- Eshkoor, S.A.; Mun, C.Y.; Ng, C.K.; Hamid, T.A. Mild cognitive impairment and its management in older people. Clin. Interv. Aging 2015, 10, 687–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, R.C. Mild Cognitive Impairment. Contin. Lifelong Learn. Neurol. 2016, 22, 404–418. [Google Scholar] [CrossRef]
- Petersen, R.C.; Lopez, O.; Armstrong, M.J.; Getchius, T.S.D.; Ganguli, M.; Gloss, D.; Gronseth, G.S.; Marson, D.; Pringsheim, T.; Day, G.S.; et al. Practice Guideline Update Summary: Mild Cognitive Impairment Report of Theguideline Development, Dissemination, and Implementation. Neurology 2018, 90, 126–135. [Google Scholar] [CrossRef]
- Chang, H.Y.; Sang, T.K.; Chiang, A.S. Untangling the Tauopathy for Alzheimer’s Disease and Parkinsonism. J. Biomed. Sci. 2018, 25, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Pleil, K.E.; Lowery-Gionta, E.G.; Crowley, N.A.; Li, C.; Marcinkiewcz, C.A.; Rose, J.H.; McCall, N.M.; Maldonado-Devincci, A.M.; Morrow, A.L.; Jones, S.R.; et al. Effects of Chronic Ethanol Exposure on Neuronal Function in the Prefrontal Cortex and Extended Amygdala. Neuropharmacology 2015, 99, 735–749. [Google Scholar] [CrossRef] [Green Version]
- Crews, F.T.; Sarkar, D.K.; Qin, L.; Zou, J.; Boyadjieva, N.; Vetreno, R.P. Neuroimmune Function and the Consequences of Alcohol Exposure. Alcohol Res. Curr. Rev. 2015, 37, 331–351. [Google Scholar]
- Rajesh, V.; Mridhulmohan, M.; Jayaseelan, S.; Sivakumar, P.; Ganesan, V. Mefenamic Acid Attenuates Chronic Alcohol Induced Cognitive Impairment in Zebrafish: Possible Role of Cholinergic Pathway. Neurochem. Res. 2018, 43, 1392–1404. [Google Scholar] [CrossRef]
- Li, Y.; Zhong, W.; Jiang, Z.; Tang, X. New progress in the approaches for blood–brain barrier protection in acute ischemic stroke. Brain Res. Bull. 2019, 144, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Olney, J.W.; Lukasiewicz, P.D.; Almli, T.; Romano, C. Fenamates Protect Neurons against Ischemic and Excitotoxic Injury in Chick Embryo Retina. Neurosci. Lett. 1998, 242, 163–166. [Google Scholar] [CrossRef]
- George, M.G.; Fischer, L.; Koroshetz, W.; Bushnell, C.; Frankel, M.; Foltz, J.; Thorpe, P.G. CDC Grand Rounds: Public Health Strategies to Prevent and Treat Strokes. MMWR. Morb. Mortal. Wkly. Rep. 2017, 66, 479–481. [Google Scholar] [CrossRef] [Green Version]
- Lopez, M.F.; Sarracino, D.A.; Prakash, A.; Athanas, M.; Krastins, B.; Rezai, T.; Sutton, J.N.; Peterman, S.; Gvozdyak, O.; Chou, S.; et al. Discrimination of Ischemic and Hemorrhagic Strokes Using a Multiplexed, Mass Spectrometry-Based Assay for Serum Apolipoproteins Coupled to Multi-Marker ROC Algorithm. Proteom. Clin. Appl. 2012, 6, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.W.; Lee, R.H.C.; Lee, M.H.H.; Wu, C.Y.C.; E Silva, A.C.; Possoit, H.E.; Hsieh, T.-H.; Minagar, A. Cerebral ischemia and neuroregeneration. Neural Regen. Res. 2018, 13, 373–385. [Google Scholar] [CrossRef]
- Khansari, P.S.; Halliwell, R.F. Mechanisms Underlying Neuroprotection by the NSAID Mefenamic Acid in an Experimental Model of Stroke. Front. Neurosci. 2019, 13, 64. [Google Scholar] [CrossRef] [Green Version]
- Khansari, P.S.; Halliwell, R.F. Evidence for Neuroprotection by the Fenamate NSAID, Mefenamic Acid. Neurochem. Int. 2009, 55, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Li, Y.; Yang, W.; Liu, D.; Ji, X.; Chi, T.; Guo, Z.; Li, L.; Zou, L. Prevention of Huntington’s Disease-like Behavioral Deficits in R6/1 Mouse by Tolfenamic Acid Is Associated with Decreases in Mutant Huntingtin and Oxidative Stress. Oxid. Med. Cell. Longev. 2019, 2019. [Google Scholar] [CrossRef] [Green Version]
- McColgan, P.; Tabrizi, S.J. Huntington’s Disease: A Clinical Review. Eur. J. Neurol. 2018, 25, 24–34. [Google Scholar] [CrossRef]
- Illarioshkin, S.N.; Klyushnikov, S.A.; Vigont, V.A.; Seliverstov, Y.A.; Kaznacheyeva, E.V. Molecular Pathogenesis in Huntington’s Disease. Biochemistry 2018, 83, 1030–1039. [Google Scholar] [CrossRef]
- Jimenez-Sanchez, M.; Licitra, F.; Underwood, B.R.; Rubinsztein, D.C. Huntington’s Disease: Mechanisms of Pathogenesis and Therapeutic Strategies. Cold Spring Harb. Perspect. Med. 2017, 7, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Thomson, S.B.; Leavitt, B.R. Transcriptional Regulation of the Huntingtin Gene. J. Huntingt. Dis. 2018, 7, 289–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beghi, E.; Giussani, G.; Sander, J.W. The Natural History and Prognosis of Epilepsy. Epileptic Disord. 2015, 17, 243–253. [Google Scholar] [CrossRef] [Green Version]
- Pearson-Smith, J.N.; Patel, M. Metabolic Dysfunction and Oxidative Stress in Epilepsy. Int. J. Mol. Sci. 2017, 18, 2365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helmstaedter, C.; Witt, J.-A. Epilepsy and cognition—A bidirectional relationship? Seizure 2017, 49, 83–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peretz, A.; Degani, N.; Nachman, R.; Uziyel, Y.; Gibor, G.; Shabat, D.; Attali, B. Meclofenamic Acid and Diclofenac, Novel Templates of KCNQ2/Q3 Potassium Channel Openers, Depress Cortical Neuron Activity and Exhibit Anticonvulsant Properties. Mol. Pharmacol. 2005, 67, 1053–1066. [Google Scholar] [CrossRef] [Green Version]
- Cooper, E.C.; Jan, L.Y. M-Channels: Neurological Diseases, Neuromodulation, and Drug Development. Arch. Neurol. 2003, 60, 496–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.-F.; Xu, Y.-J.; Kong, X.-H.; Su, Y.; Wang, Z.-Y. Fenamates inhibit human sodium channel Nav1.7 and Nav1.8. Neurosci. Lett. 2019, 696, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Yau, H.J.; Baranauskas, G.; Martina, M. Flufenamic Acid Decreases Neuronal Excitability through Modulation of Voltage-Gated Sodium Channel Gating. J. Physiol. 2010, 588, 3869–3882. [Google Scholar] [CrossRef]
- Wong, R.S.Y. Role of Nonsteroidal Anti-Inflammatory Drugs (NSAIDs) in Cancer Prevention and Cancer Promotion. Adv. Pharmacol. Sci. 2019, 2019, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Basha, R.; Ingersoll, S.B.; Sankpal, U.T.; Ahmad, S.; Baker, C.H.; Edwards, J.R.; Holloway, R.W.; Kaja, S.; Abdelrahim, M. Tolfenamic acid inhibits ovarian cancer cell growth and decreases the expression of c-Met and survivin through suppressing specificity protein transcription factors. Gynecol. Oncol. 2011, 122, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Sankpal, U.T.; Abdelrahim, M.; Connelly, S.F.; Lee, C.M.; Madero-Visbal, R.; Colon, J.; Smith, J.; Safe, S.; Maliakal, P.; Basha, R. Small Molecule Tolfenamic Acid Inhibits PC-3 Cell Proliferation and Invasion in Vitro, and Tumor Growth in Orthotopic Mouse Model for Prostate Cancer. Prostate 2012, 72, 1648–1658. [Google Scholar] [CrossRef] [PubMed]
- Eslin, D.; Sankpal, U.T.; Lee, C.; Sutphin, R.M.; Maliakal, P.; Currier, E.; Sholler, G.; Khan, M.; Basha, R. Tolfenamic Acid Inhibits Neuroblastoma Cell Proliferation and Induces Apoptosis: A Novel Therapeutic Agent for Neuroblastoma. Mol. Carcinog. 2013, 52, 377–386. [Google Scholar] [CrossRef]
- Li, D.; Hu, C.; Li, H. Survivin as a novel target protein for reducing the proliferation of cancer cells (Review). Biomed. Rep. 2018, 8, 399–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sleiman, S.F.; Langley, B.C.; Basso, M.; Berlin, J.; Xia, L.; Payappilly, J.B.; Kharel, M.K.; Guo, H.; Marsh, J.L.; Thompson, L.M.; et al. Mithramycin Is a Gene-Selective Sp1 Inhibitor That Identifies a Biological Intersection between Cancer and Neurodegeneration. J. Neurosci. 2011, 31, 6858–6870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shelake, S.; Sankpal, U.T.; Paul Bowman, W.; Wise, M.; Ray, A.; Basha, R. Targeting Specificity Protein 1 Transcription Factor and Survivin Using Tolfenamic Acid for Inhibiting Ewing Sarcoma Cell Growth. Investig. New Drugs 2017, 35, 158–165. [Google Scholar] [CrossRef]
- Ferrante, R.J.; Ryu, H.; Kubilus, J.K.; Mello, S.D.; Sugars, K.L.; Lee, J.; Lu, P.; Smith, K.; Browne, S.; Flint Beal, M.; et al. Neurobiology of Disease Chemotherapy for the Brain: The Antitumor Antibiotic Mithramycin Prolongs Survival in a Mouse Model of Huntington’s Disease. J. Neurosci. 2004. [Google Scholar] [CrossRef]
- Wei, C.; Zhang, W.; Zhou, Q.; Zhao, C.; Du, Y.; Yan, Q.; Li, Z.; Miao, J. Mithramycin A Alleviates Cognitive Deficits and Reduces Neuropathology in a Transgenic Mouse Model of Alzheimer’s Disease. Neurochem. Res. 2016, 41, 1924–1938. [Google Scholar] [CrossRef]
- Atluri, V.S.R.; Tiwari, S.; Rodriguez, M.; Kaushik, A.; Yndart, A.; Kolishetti, N.; Yatham, M.; Nair, M. Inhibition of Amyloid-Beta Production, Associated Neuroinflammation, and Histone Deacetylase 2-Mediated Epigenetic Modifications Prevent Neuropathology in Alzheimer’s Disease in Vitro Model. Front. Aging Neurosci. 2020, 11. [Google Scholar] [CrossRef] [Green Version]
- Woodling, N.S.; Colas, D.; Wang, Q.; Minhas, P.; Panchal, M.; Liang, X.; Mhatre, S.D.; Brown, H.; Ko, N.; Zagol-Ikapitte, I.; et al. Cyclooxygenase Inhibition Targets Neurons to Prevent Early Behavioural Decline in Alzheimer’s Disease Model Mice. Brain 2016, 139, 2063–2081. [Google Scholar] [CrossRef] [Green Version]
- Tyagi, A.; Kamal, M.A.; Poddar, N.K. Integrated Pathways of COX-2 and mTOR: Roles in Cell Sensing and Alzheimer’s Disease. Front. Neurosci. 2020, 14, 693. [Google Scholar] [CrossRef]
- Huang, L.-K.; Chao, S.-P.; Hu, C.-J. Clinical trials of new drugs for Alzheimer disease. J. Biomed. Sci. 2020, 27, 1–13. [Google Scholar] [CrossRef]
- Li, Z.Y.; Yin, Y.F.; Guo, Y.; Li, H.; Xu, M.Q.; Liu, M.; Wang, J.R.; Feng, Z.H.; Duan, X.C.; Zhang, S.; et al. Enhancing Anti-Tumor Activity of Sorafenib Mesoporous Silica Nanomatrix in Metastatic Breast Tumor and Hepatocellular Carcinoma via the Co-Administration with Flufenamic Acid. Int. J. Nanomed. 2020, 15, 1809–1821. [Google Scholar] [CrossRef] [Green Version]
- Zahid, A.; Li, B.; Kombe, A.J.K.; Jin, T.; Tao, J. Pharmacological Inhibitors of the NLRP3 Inflammasome. Front. Immunol. 2019, 10, 2538. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.-S.; Tan, Z.-X.; Wu, L.-Y.; Dong, F.; Zhang, F. The involvement of NLRP3 inflammasome in the treatment of Alzheimer’s disease. Ageing Res. Rev. 2020, 64, 101192. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W.; Potschka, H.; Sisodiya, S.M.; Vezzani, A. Drug Resistance in Epilepsy: Clinical Impact, Potential Mechanisms, and New Innovative Treatment Options. Pharmacol. Rev. 2020, 72, 606–638. [Google Scholar] [CrossRef] [PubMed]
- Sills, G.J.; Rogawski, M.A. Mechanisms of action of currently used antiseizure drugs. Neuropharmacology 2020, 168, 107966. [Google Scholar] [CrossRef]
- Manford, M. Recent Advances in Epilepsy. J. Neurol. 2017, 264, 1811–1824. [Google Scholar] [CrossRef] [Green Version]
- Castillo, J.; Loza, M.I.; Mirelman, D.; Brea, J.; Blanco, M.; Sobrino, T.; Campos, F. A novel mechanism of neuroprotection: Blood glutamate grabber. Br. J. Pharmacol. 2016, 36, 292–301. [Google Scholar] [CrossRef]
- Castillo, J.; Loza, M.I.; Mirelman, D.; Sobrino, T.; Campos, F. Beyond Glutamate Antagonists for Treatment of Ischemic Stroke: Blood Glutamate Grabbing. J. Neurol. Neuromed. 2016, 1, 31–34. [Google Scholar] [CrossRef] [Green Version]
- Abdelrahim, M.; Safe, S. Cyclooxygenase-2 Inhibitors Decrease Vascular Endothelial Growth Factor Expression in Colon Cancer Cells by Enhanced Degradation of Sp1 and Sp4 Proteins. Mol. Pharmacol. 2005, 68, 317–329. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hill, J.; Zawia, N.H. Fenamates as Potential Therapeutics for Neurodegenerative Disorders. Cells 2021, 10, 702. https://doi.org/10.3390/cells10030702
Hill J, Zawia NH. Fenamates as Potential Therapeutics for Neurodegenerative Disorders. Cells. 2021; 10(3):702. https://doi.org/10.3390/cells10030702
Chicago/Turabian StyleHill, Jaunetta, and Nasser H. Zawia. 2021. "Fenamates as Potential Therapeutics for Neurodegenerative Disorders" Cells 10, no. 3: 702. https://doi.org/10.3390/cells10030702
APA StyleHill, J., & Zawia, N. H. (2021). Fenamates as Potential Therapeutics for Neurodegenerative Disorders. Cells, 10(3), 702. https://doi.org/10.3390/cells10030702