
Energy Metabolism in IDH1 Wild-Type and IDH1-Mutated Glioblastoma Stem Cells: A Novel Target for Therapy?

 ,
,  and
and
Abstract
:1. Introduction
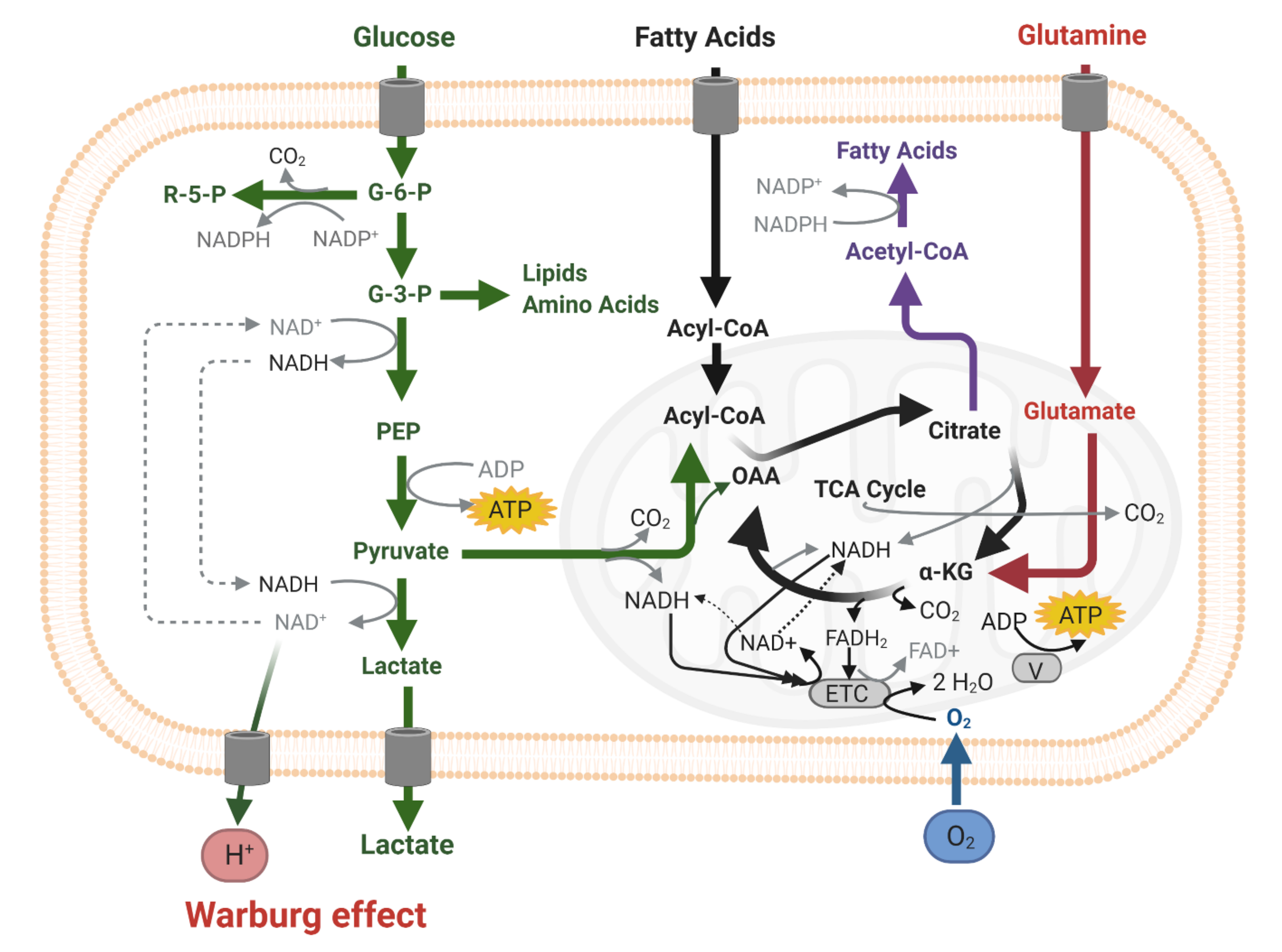
2. Energy Metabolism of IDH1wt versus IDH1mt Differentiated Glioblastoma Cells
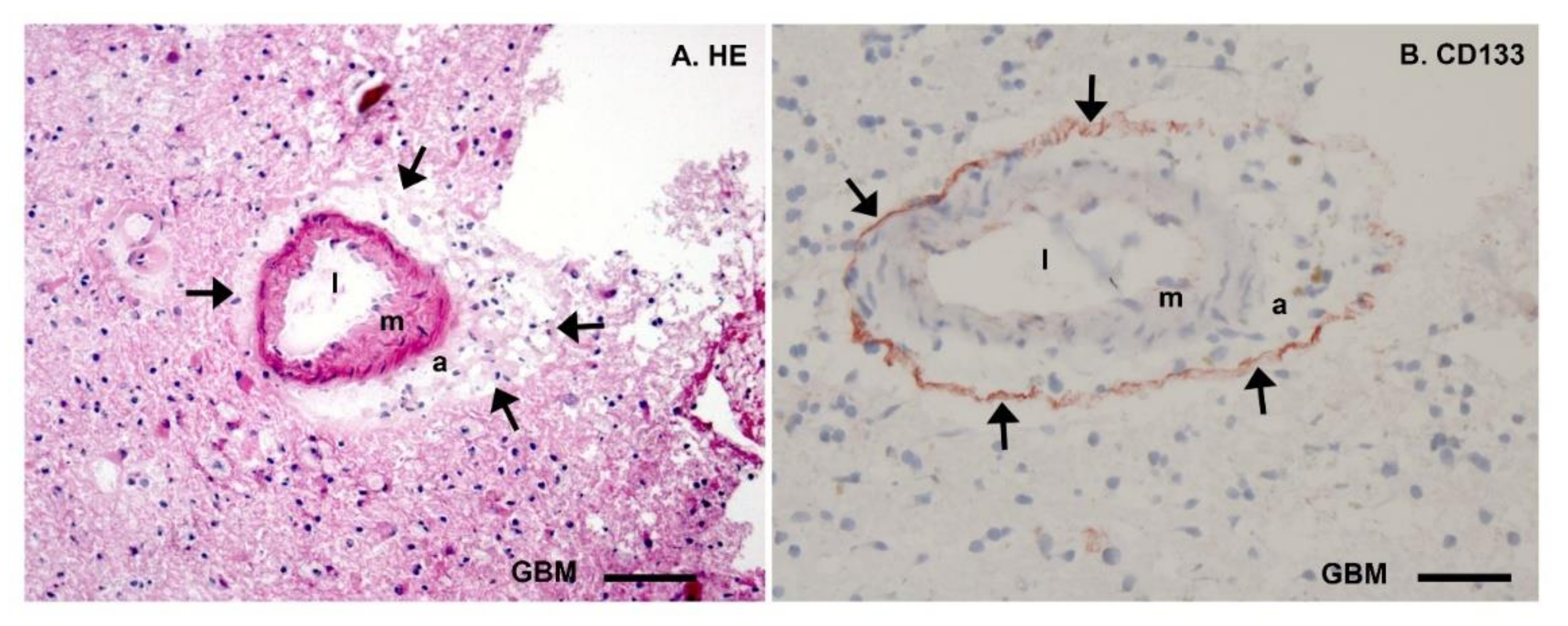
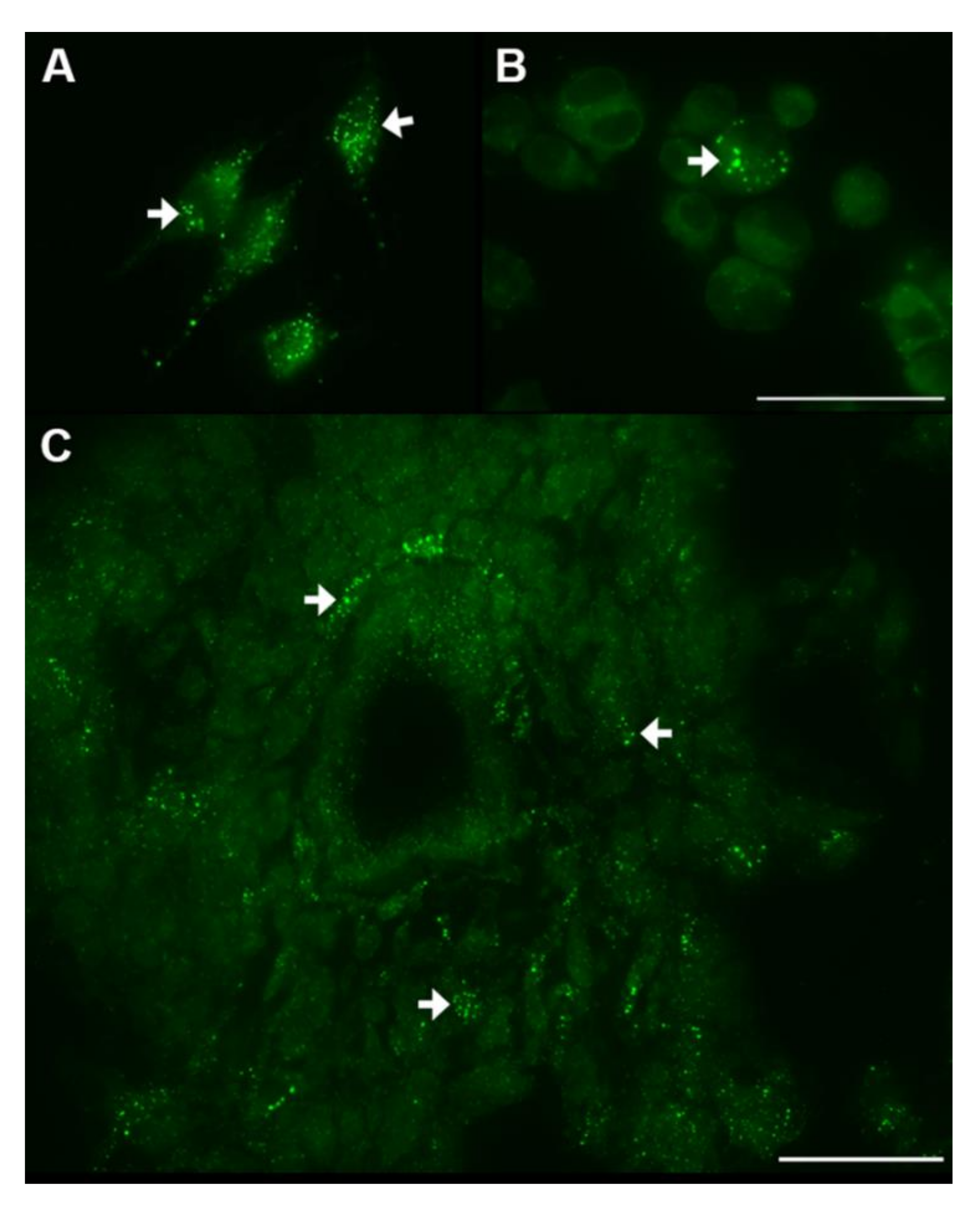
3. Energy Metabolism in IDH1wt GSCs
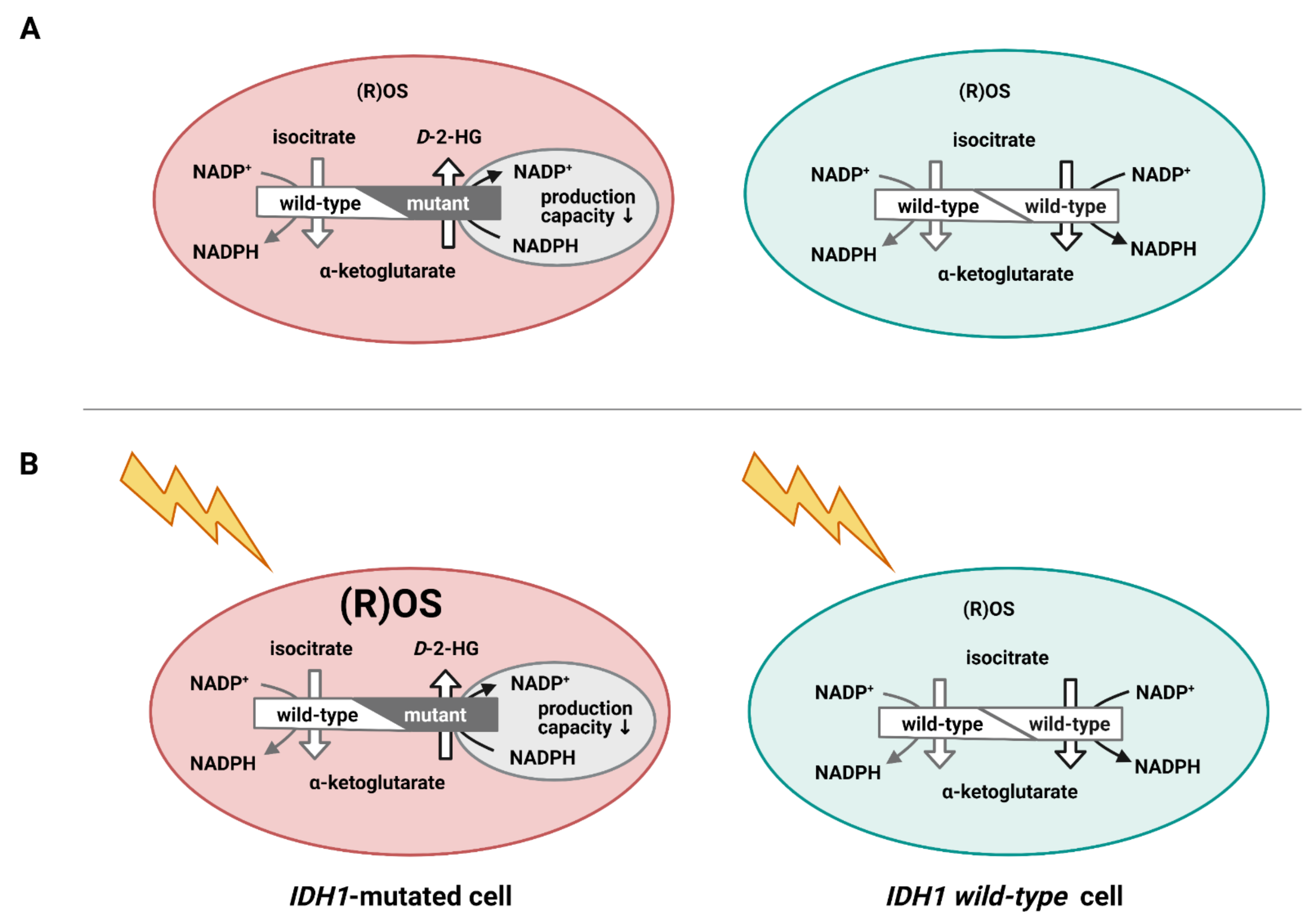
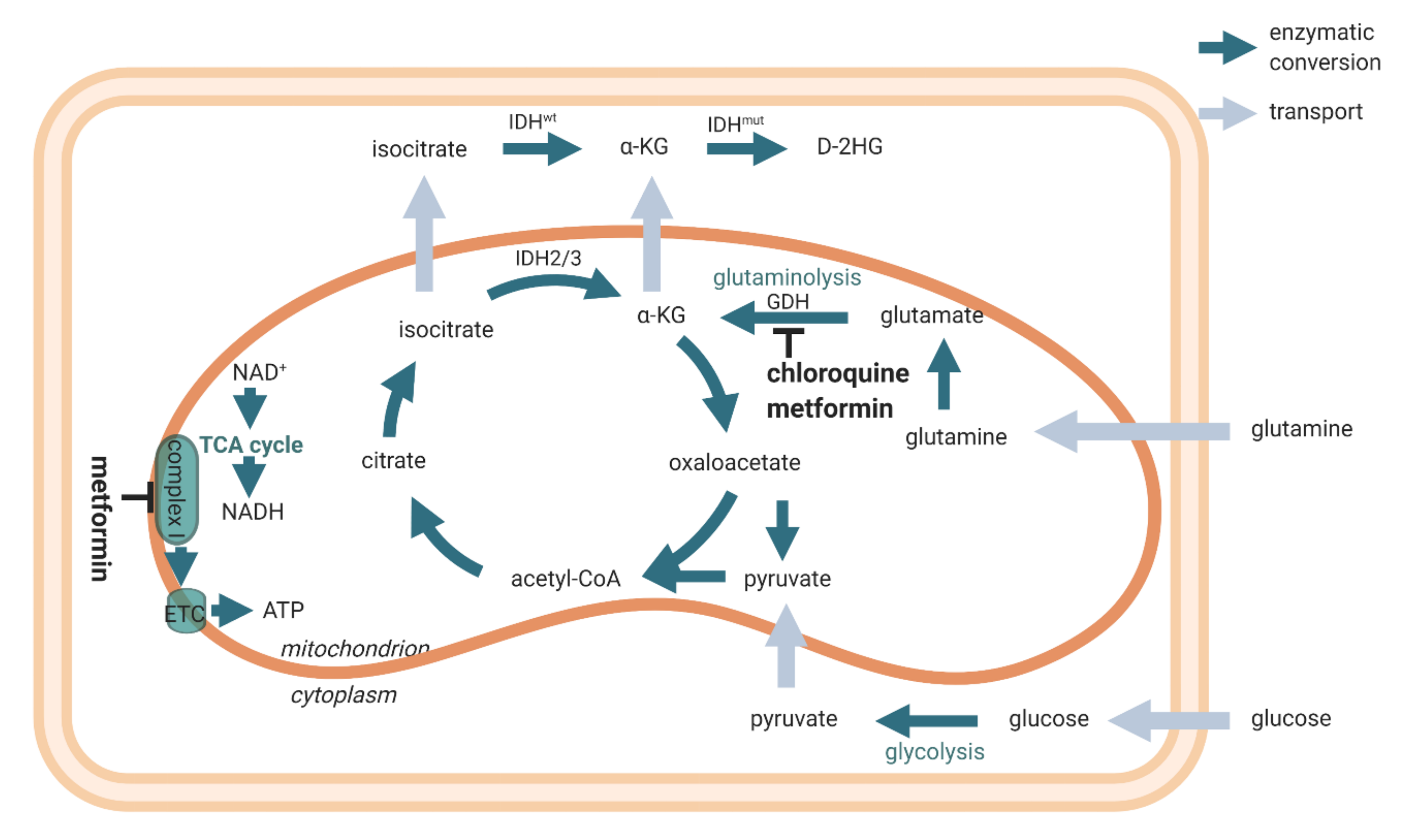
4. Energy Metabolism in IDH1mt GSCs
5. Concluding Remarks
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Watson, J.D. Type 2 diabetes as a redox disease. Lancet 2014, 383, 841–843. [Google Scholar] [CrossRef]
- Molenaar, R.J.; van Noorden, C.J.F. Type 2 diabetes and cancer as redox diseases? Lancet 2014, 384, 853. [Google Scholar] [CrossRef]
- Warburton, D.E.; Nicol, C.W.; Bredin, S.S. Health benefits of physical activity: The evidence. Can. Med. Assoc. J. 2006, 174, 801–909. [Google Scholar] [CrossRef] [Green Version]
- Irwin, M.L.; Smith, A.W.; McTiernan, A.; Ballard-Barbash, R.; Cronin, K.; Gilliland, F.D.; Baumgartner, R.N.; Baumgartner, K.B.; Bernstein, L. Influence of pre- and postdiagnosis physical activity on mortality in breast cancer survivors: The health, eating, activity, and lifestyle study. J. Clin. Oncol. 2008, 26, 3958–3964. [Google Scholar] [CrossRef]
- Thong, M.S.Y.; van Noorden, C.J.F.; Steindorf, K.; Arndt, V. Cancer-related fatigue: Causes and current treatment options. Curr. Treat. Options Oncol. 2020, 21, 17. [Google Scholar] [CrossRef]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even Warburg did not anticipate. Cancer Cell. 2012, 21, 297–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koehler, A.; Van Noorden, C.J.F. Reduced nicotinamide adenine dinucleotide phosphate and the higher incidence of pollution-induced liver cancer in female flounder. Environ. Toxicol. Chem. 2003, 22, 2703–2710. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Multhoff, G. Revisiting the Warburg effect: Historical dogma versus current understanding. J. Physiol. 2020. [Google Scholar] [CrossRef]
- Zhang, C.C.; Sadek, H.A. Hypoxia and metabolic properties of hematopoietic stem cells. Antioxid. Redox Signal. 2014, 20, 1891–1901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molenaar, R.J.; Maciejewski, J.P.; Wilmink, J.W.; van Noorden, C.J.F. Wild-type and mutated IDH1/2 enzymes and therapy responses. Oncogene 2018, 37, 1949–1960. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [Green Version]
- Sancho, P.; Barneda, D.; Heeschen, C. Hallmarks of cancer stem cell metabolism. Br. J. Cancer 2016, 114, 1305–1312. [Google Scholar] [CrossRef] [Green Version]
- Sica, V.; Bravo-San Pedro, J.M.; Stoll, G.; Kroemer, G. Oxidative phosphorylation as a potential therapeutic target for cancer therapy. Int. J. Cancer 2020, 146, 10–17. [Google Scholar] [CrossRef]
- Lleonart, M.E.; Abad, E.; Graifer, D.; Lyakhovich, A. Reactive oxygen species-mediated autophagy defines the fate of cancer stem cells. Antioxid. Redox Signal. 2018, 28, 1066–1079. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Ito, K. Metabolism and the control of cell fate decisions and stem cell renewal. Annu. Rev. Cell Dev. Biol. 2016, 32, 399–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hira, V.V.V.; Molenaar, R.J.; Breznik, B.; Lah, T.; Aronica, E.; van Noorden, C.J.F. Immunohistochemical detection of neural stem cells and glioblastoma stem cells in the subventricular zone of glioblastoma patients. J. Histochem. Cytochem. 2021. [Google Scholar] [CrossRef] [PubMed]
- Visweswaran, M.; Arfuso, F.; Warrier, S.; Dharmarajan, A. Aberrant lipid metabolism as an emerging therapeutic strategy to target cancer stem cells. Stem Cells 2020, 38, 6–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Feng, Z.; He, M.L. Lipid metabolism alteration contributes to and maintains the properties of cancer stem cells. Theranostics 2020, 10, 7053–7069. [Google Scholar] [CrossRef]
- Peiris-Pagès, M.; Martinez-Outschoorn, U.E.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer stem cell metabolism. Breast Cancer Res. 2016, 18, 55. [Google Scholar] [CrossRef]
- Snyder, V.; Reed-Newman, T.C.; Arnold, L.; Thomas, S.M.; Anant, S. Cancer stem cell metabolism and potential therapeutic targets. Front. Oncol. 2018, 8, 203. [Google Scholar] [CrossRef]
- Fiorillo, M.; Sotgia, F.; Lisanti, M.P. “Energetic” Cancer Stem Cells (e-CSCs): A new hyper-metabolic and proliferative tumor cell phenotype, driven by mitochondrial energy. Front. Oncol. 2019, 8, 677. [Google Scholar] [CrossRef] [Green Version]
- Hoang-Minh, L.B.; Siebzehnrubl, F.A.; Yang, C.; Suzuki-Hatano, S.; Dajac, K.; Loche, T.; Andrews, N.; Schmoll Massari, M.; Patel, J.; Amin, K.; et al. Infiltrative and drug-resistant slow-cycling cells support metabolic heterogeneity in glioblastoma. EMBO J. 2018, 37, e98772. [Google Scholar] [CrossRef]
- Erol, A. Type 2 diabetes and cancer as redox diseases? Lancet 2014, 384, 853–854. [Google Scholar] [CrossRef]
- Erol, A. Systemic DNA damage response and metabolic syndrome as a premalignant state. Curr. Mol. Med. 2010, 10, 321–334. [Google Scholar] [CrossRef]
- Hira, V.V.V.; Breznik, B.; Vittori, M.; Loncq de Jong, A.; Mlakar, J.; Oostra, R.J.; Khurshed, M.; Molenaar, R.J.; Lah, T.; van Noorden, C.J.F. Similarities between stem cell niches in glioblastoma and bone marrow: Rays of hope for novel treatment strategies. J. Histochem. Cytochem. 2020, 68, 33–57. [Google Scholar] [CrossRef] [PubMed]
- Hira, V.V.V.; van Noorden, C.J.F.; Molenaar, R.J. CXCR4 antagonists as stem cell mobilizers and therapy sensitizers for acute myeloid leukemia and glioblastoma? Biology 2020, 9, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hira, V.V.V.; Wormer, J.R.; Kakar, H.; Breznik, B.; van der Swaan, B.; Hulsbos, R.; Tigchelaar, W.; Tonar, Z.; Khurshed, M.; Molenaar, R.J.; et al. Periarteriolar glioblastoma stem cell niches express bone marrow hematopoietic stem cell niche proteins. J. Histochem. Cytochem. 2018, 66, 155–173. [Google Scholar] [CrossRef]
- Hira, V.V.V.; van Noorden, C.J.F.; Carraway, H.E.; Maciejewski, J.P.; Molenaar, R.J. Novel therapeutic strategies to target leukemic cells that hijack compartmentalized continuous hematopoietic stem cell niches. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 183–198. [Google Scholar] [CrossRef]
- Maffezzini, C.; Calvo-Garrido, J.; Wredenberg, A.; Freyer, C. Metabolic regulation of neurodifferentiation in the adult brain. Cell Mol. Life Sci. 2020, 77, 2483–2496. [Google Scholar] [CrossRef] [Green Version]
- Geribaldi-Doldan, N.; Fernandez-Ponce, C.; Navarro Quiroz, R.; Sanchez-Gomar, I.; Gomez-Escorcia, L.; Puentes Velasquez, E.; Navarro Quiroz, E. The role of microglia in glioblastoma. Front. Oncol. 2021, 10, 603495. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: A randomized clinical trial. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef] [Green Version]
- Mun, E.J.; Babiker, H.M.; Weinberg, U.; Kirson, E.D.; Von Hoff, D.D. Tumor-treating fields: A fourth modality in cancer treatment. Clin. Cancer Res. 2018, 24, 266–275. [Google Scholar] [CrossRef] [Green Version]
- Wong, E.T.; Lok, E.; Swanson, K.D. Alternating electric fields therapy for malignant gliomas: From bench observation to clinical reality. Prog. Neurol. Surg. 2018, 32, 180–195. [Google Scholar] [CrossRef] [PubMed]
- Patel, C.B.; Beinat, C.; Xie, Y.; Chang, E.; Gambhir, S.S. Tumor treating fields (TTFields) impairs aberrant glycolysis in glioblastoma as evaluated by [18F]DASA-23, a non-invasive probe of pyruvate kinase M2 (PKM2) expression. Neoplasia 2021, 23, 58–67. [Google Scholar] [CrossRef]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bleeker, F.E.; Atai, N.A.; Lamba, S.; Jonker, A.; Rijkeboer, D.; Bosch, K.S.; Tigchelaar, W.; Troost, D.; Vandertop, W.P.; Bardelli, A.; et al. The prognostic IDH1( R132 ) mutation is associated with reduced NADP+-dependent IDH activity in glioblastoma. Acta Neuropathol. 2010, 119, 487–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dewhirst, M.W.; Lee, C.T.; Ashcraft, K.A. The future of biology in driving the field of hyperthermia. Int. J. Hyperth. 2016, 32, 4–13. [Google Scholar] [CrossRef]
- Jagust, P.; de Luxán-Delgado, B.; Parejo-Alonso, B.; Sancho, P. Metabolism-based therapeutic strategies targeting cancer stem cells. Front. Pharmacol. 2019, 10, 203. [Google Scholar] [CrossRef] [Green Version]
- El Hout, M.; Cosialls, E.; Mehrpour, M.; Hamaï, A. Crosstalk between autophagy and metabolic regulation of cancer stem cells. Mol. Cancer 2020, 19, 27. [Google Scholar] [CrossRef]
- Koch, K.; Hartmann, R.; Tsiampali, J.; Uhlmann, C.; Nickel, A.C.; He, X.; Kamp, M.A.; Sabel, M.; Barker, R.A.; Steiger, H.J.; et al. A comparative pharmaco-metabolomic study of glutaminase inhibitors in glioma stem-like cells confirms biological effectiveness but reveals differences in target-specificity. Cell Death Discov. 2020, 6, 20. [Google Scholar] [CrossRef] [Green Version]
- Cheng, G.; Zhang, Q.; Pan, J.; Lee, Y.; Ouari, O.; Hardy, M.; Zielonka, M.; Myers, C.R.; Zielonka, J.; Weh, K.; et al. Targeting lonidamine to mitochondria mitigates lung tumorigenesis and brain metastasis. Nat. Commun. 2019, 10, 2205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khurshed, M.; Molenaar, R.J.; Lenting, K.; Leenders, W.P.; van Noorden, C.J.F. In silico gene expression analysis reveals glycolysis and acetate anaplerosis in IDH1 wild-type glioma and lactate and glutamate anaplerosis in IDH1-mutated glioma. Oncotarget 2017, 8, 49165–49177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, S.; Lin, Y.; Xu, W.; Jiang, W.; Zha, Z.; Wang, P.; Yu, W.; Li, Z.; Gong, L.; Peng, Y.; et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science 2009, 324, 261–265. [Google Scholar] [CrossRef] [Green Version]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef] [Green Version]
- Fack, F.; Tardito, S.; Hochart, G.; Oudin, A.; Zheng, L.; Fritah, S.; Golebiewska, A.; Nazarov, P.V.; Bernard, A.; Hau, A.C.; et al. Altered metabolic landscape in IDH-mutant gliomas affects phospholipid, energy, and oxidative stress pathways. EMBO Mol. Med. 2017, 9, 1681–1695. [Google Scholar] [CrossRef]
- Kessler, J.; Hohmann, T.; Güttler, A.; Petrenko, M.; Ostheimer, C.; Hohmann, U.; Bache, M.; Dehghani, F.; Vordermark, D. Radiosensitization and a less aggressive phenotype of human malignant glioma cells expressing isocitrate dehydrogenase 1 (IDH1) mutant protein: Dissecting the mechanisms. Cancers 2019, 11, 889. [Google Scholar] [CrossRef] [Green Version]
- Park, J.S.; Burckhardt, C.J.; Lazcano, R.; Solis, L.M.; Isogai, T.; Li, L.; Chen, C.S.; Gao, B.; Minna, J.D.; Bachoo, R.; et al. Mechanical regulation of glycolysis via cytoskeleton architecture. Nature 2020, 578, 621–626. [Google Scholar] [CrossRef]
- Miroshnikova, Y.A.; Mouw, J.K.; Barnes, J.M.; Pickup, M.W.; Lakins, J.N.; Kim, Y.; Lobo, K.; Persson, A.I.; Reis, G.F.; McKnight, T.R.; et al. Tissue mechanics promote IDH1-dependent HIF1α-tenascin C feedback to regulate glioblastoma aggression. Nat. Cell Biol. 2016, 18, 1336–1345. [Google Scholar] [CrossRef]
- Navis, A.C.; Niclou, S.P.; Fack, F.; Stieber, D.; van Lith, S.; Verrijp, K.; Wright, A.; Stauber, J.; Tops, B.; Otte-Holler, I.; et al. Increased mitochondrial activity in a novel IDH1-R132H mutant human oligodendroglioma xenograft model: In situ detection of 2-HG and α-KG. Acta Neuropathol. Commun. 2013, 1, 18. [Google Scholar] [CrossRef] [Green Version]
- Sperry, J.; Condro, M.C.; Guo, L.; Braas, D.; Vanderveer-Harris, N.; Kim, K.K.O.; Pope, W.B.; Divakaruni, A.S.; Lai, A.; Christofk, H.; et al. Glioblastoma utilizes fatty acids and ketone bodies for growth allowing progression during ketogenic diet therapy. Science 2020, 23, 101453. [Google Scholar] [CrossRef]
- Kant, S.; Kesarwani, P.; Prabhu, A.; Graham, S.F.; Buelow, K.L.; Nakano, I.; Chinnaiyan, P. Enhanced fatty acid oxidation provides glioblastoma cells metabolic plasticity to accommodate to its dynamic nutrient microenvironment. Cell Death Dis. 2020, 11, 253. [Google Scholar] [CrossRef] [Green Version]
- Dekker, L.J.M.; Wu, S.; Jurriëns, C.; Mustafa, D.A.N.; Grevers, F.; Burgers, P.C.; Sillevis Smitt, P.; Kros, J.M.; Luider, T.M. Metabolic changes related to the IDH1 mutation in gliomas preserve TCA-cycle activity: An investigation at the protein level. FASEB J. 2020, 34, 3646–3657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grassian, A.R.; Parker, S.J.; Davidson, S.M.; Divakaruni, A.S.; Green, C.R.; Zhang, X.; Slocum, K.L.; Pu, M.; Lin, F.; Vickers, C.; et al. IDH1 mutations alter citric acid cycle metabolism and increase dependence on oxidative mitochondrial metabolism. Cancer Res. 2014, 74, 3317–3331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mustafa, D.A.; Swagemakers, S.M.; Buise, L.; van der Spek, P.J.; Kros, J.M. Metabolic alterations due to IDH1 mutation in glioma: Opening for therapeutic opportunities? Acta Neuropathol. Commun. 2014, 2, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenting, K.; Khurshed, M.; Peeters, T.H.; van den Heuvel, C.N.A.M.; van Lith, S.A.M.; de Bitter, T.; Hendriks, W.; Span, P.N.; Molenaar, R.J.; Botman, D.; et al. Isocitrate dehydrogenase 1-mutated human gliomas depend on lactate and glutamate to alleviate metabolic stress. FASEB J. 2019, 33, 557–571. [Google Scholar] [CrossRef]
- Frattini, V.; Pagnotta, S.M.; Tala Fan, J.J.; Russo, M.V.; Lee, S.B.; Garofano, L.; Zhang, J.; Shi, P.; Lewis, G.; Sanson, H.; et al. A metabolic function of FGFR3-TACC3 gene fusions in cancer. Nature 2018, 553, 222–227. [Google Scholar] [CrossRef]
- Atai, N.A.; Renkema-Mills, N.A.; Bosman, J.; Schmidt, N.; Rijkeboer, D.; Tigchelaar, W.; Bosch, K.S.; Troost, D.; Jonker, A.; Bleeker, F.E.; et al. Differential activity of NADPH-producing dehydrogenases renders rodents unsuitable models to study IDH1R132 mutation effects in human glioblastoma. J. Histochem. Cytochem. 2011, 59, 489–503. [Google Scholar] [CrossRef] [Green Version]
- Molenaar, R.J.; Radivoyevitch, T.; Nagata, Y.; Khurshed, M.; Przychodzen, B.; Makishima, H.; Xu, M.; Bleeker, F.E.; Wilmink, J.W.; Carraway, H.E.; et al. IDH1/2 mutations sensitize acute myeloid leukemia to PARP inhibition and this is reversed by IDH1/2-mutant inhibitors. Clin. Cancer Res. 2018, 24, 1705–1715. [Google Scholar] [CrossRef] [Green Version]
- Biedermann, J.; Preussler, M.; Conde, M.; Peitzsch, M.; Richter, S.; Wiedemuth, R.; Abou-El-Ardat, K.; Krüger, A.; Meinhardt, M.; Schackert, G.; et al. Mutant IDH1 differently affects redox state and metabolism in glial cells of normal and tumor origin. Cancers 2019, 11, 2028. [Google Scholar] [CrossRef] [Green Version]
- Gelman, S.J.; Naser, F.; Mahieu, N.G.; McKenzie, L.D.; Dunn, G.P.; Chheda, M.G.; Patti, G.J. Consumption of NADPH for 2-HG synthesis increases pentose phosphate pathway flux and sensitizes cells to oxidative stress. Cell Rep. 2018, 22, 512–522. [Google Scholar] [CrossRef] [Green Version]
- Molenaar, R.J.; Coelen, R.J.S.; Khurshed, M.; Roos, E.; Caan, M.W.A.; van Linde, M.E.; Kouwenhoven, M.; Bramer, J.; Bovée, J.; Mathôt, R.A.; et al. Study protocol of a phase IB/II clinical trial of metformin and chloroquine in patients with IDH1-mutated or IDH2-mutated solid tumours. BMJ Open 2017, 7, e014961. [Google Scholar] [CrossRef] [Green Version]
- Vlashi, E.; Lagadec, C.; Vergnes, L.; Matsutani, T.; Masui, K.; Poulou, M.; Popescu, R.; Della Donna, L.; Evers, P.; Dekmezian, C.; et al. Metabolic state of glioma stem cells and nontumorigenic cells. Proc. Natl. Acad. Sci. USA 2011, 108, 16062–16067. [Google Scholar] [CrossRef] [Green Version]
- Badr, C.E.; Silver, D.J.; Siebzehnrubl, F.A.; Deleyrolle, L.P. Metabolic heterogeneity and adaptability in brain tumors. Cell Mol. Life Sci. 2020, 77, 5101–5119. [Google Scholar] [CrossRef]
- Duraj, T.; Garcia-Romero, N.; Carrion-Navarro, J.; Madurga, R.; Ortiz de Mendivil, A.; Prat-Acin, R.; Garcia-Cañamaque, L.; Ayuso-Sacido, A. Beyond the Warburg effect: Oxidative and glycolytic phenotypes coexist within the metabolic heterogeneity of glioblastoma. Cells 2021, 10, 202. [Google Scholar] [CrossRef]
- Lee, Y.; Park, Y.; Nam, H.; Lee, J.W.; Yu, S.W. Translocator protein (TSPO): The new story of the old protein in neuroinflammation. BMB Rep. 2020, 53, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Wang, D.; Wang, H.; Cai, M.; Li, C.; Zhang, X.; Ying, M.; He, W.; Zhang, J. TSPO deficiency induces mitochondrial dysfunction, leading to hypoxia, angiogenesis, and a growth-promoting metabolic shift toward glycolysis in glioblastoma. Neuro Oncol. 2020, 22, 240–252. [Google Scholar] [CrossRef]
- Cao, J.; Mu, Q.; Huang, H. The roles of insulin-like growth factor 2 mRNA-binding protein 2 in cancer and cancer stem cells. Stem Cells Int. 2018, 2018, 4217259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janiszewska, M.; Suvà, M.L.; Riggi, N.; Houtkooper, R.H.; Auwerx, J.; Clément-Schatlo, V.; Radovanovic, I.; Rheinbay, E.; Provero, P.; Stamenkovic, I. Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes Dev. 2012, 26, 1926–1944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rusu, P.; Shao, C.; Neuerburg, A.; Acikgöz, A.A.; Wu, Y.; Zou, P.; Phapale, P.; Shankar, T.S.; Döring, K.; Dettling, S.; et al. GPD1 specifically marks dormant glioma stem cells with a distinct metabolic profile. Cell Stem Cell 2019, 25, 241–257. [Google Scholar] [CrossRef]
- Houben, E.; Hellings, N.; Broux, B. Oncostatin M, an underestimated player in the central nervous system. Front. Immunol. 2019, 10, 1165. [Google Scholar] [CrossRef] [PubMed]
- Sharanek, A.; Burban, A.; Laaper, M.; Heckel, E.; Joyal, J.S.; Soleimani, V.D.; Jahani-Asl, A. OSMR controls glioma stem cell respiration and confers resistance of glioblastoma to ionizing radiation. Nat. Commun. 2020, 11, 4116. [Google Scholar] [CrossRef]
- Wei, X.; Chen, Y.; Jiang, X.; Peng, M.; Liu, Y.; Mo, Y.; Ren, D.; Hua, Y.; Yu, B.; Zhou, Y.; et al. Mechanisms of vasculogenic mimicry in hypoxic tumor microenvironments. Mol. Cancer 2021, 20, 7. [Google Scholar] [CrossRef]
- Libby, C.J.; Tran, A.N.; Scott, S.E.; Griguer, C.; Hjelmeland, A.B. The pro-tumorigenic effects of metabolic alterations in glioblastoma including brain tumor initiating cells. Biochim. Biophys. Acta Rev. Cancer 2018, 1869, 175–188. [Google Scholar] [CrossRef]
- Zhang, J.; Hang, C.; Jiang, T.; Yi, S.; Shao, W.; Li, W.; Lin, D. Nuclear magnetic resonance-based metabolomic analysis of the anticancer effect of metformin treatment on cholangiocarcinoma cells. Front. Oncol. 2020, 10, 570516. [Google Scholar] [CrossRef]
- Mudassar, F.; Shen, H.; O’Neill, G.; Hau, E. Targeting tumor hypoxia and mitochondrial metabolism with anti-parasitic drugs to improve radiation response in high-grade gliomas. J. Exp. Clin. Cancer Res. 2020, 39, 208. [Google Scholar] [CrossRef] [PubMed]
- Kuramoto, K.; Yamamoto, M.; Suzuki, S.; Sanomachi, T.; Togashi, K.; Seino, S.; Kitanaka, C.; Okada, M. Verteporfin inhibits oxidative phosphorylation and induces cell death specifically in glioma stem cells. FEBS J. 2020, 287, 2023–2036. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.C.; Hu, Y.Y.; Cheng, G.; Liang, L.; Gao, B.; Ren, Y.P.; Liu, J.T.; Cao, X.L.; Zheng, M.H.; Li, S.Z.; et al. A ketogenic diet attenuates proliferation and stemness of glioma stem‑like cells by altering metabolism resulting in increased ROS production. Int. J. Oncol. 2020, 56, 606–617. [Google Scholar] [CrossRef] [PubMed]
- Le, C.T.; Leenders, W.P.J.; Molenaar, R.J.; van Noorden, C.J.F. Effects of the green tea polyphenol epigallocatechin-3-gallate on Glioma: A critical evaluation of the literature. Nutr. Cancer 2018, 70, 317–333. [Google Scholar] [CrossRef] [PubMed]
- Peeters, T.H.; Lenting, K.; Breukels, V.; van Lith, S.A.M.; van den Heuvel, C.N.A.M.; Molenaar, R.; van Rooij, A.; Wevers, R.; Span, P.N.; Heerschap, A.; et al. Isocitrate dehydrogenase 1-mutated cancers are sensitive to the green tea polyphenol epigallocatechin-3-gallate. Cancer Metab. 2019, 7, 4. [Google Scholar] [CrossRef] [PubMed]
- Van Lith, S.A.; Navis, A.C.; Verrijp, K.; Niclou, S.P.; Bjerkvig, R.; Wesseling, P.; Tops, B.; Molenaar, R.; van Noorden, C.J.F.; Leenders, W.P. Glutamate as chemotactic fuel for diffuse glioma cells: Are they glutamate suckers? Biochim. Biophys. Acta 2014, 1846, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Van Lith, S.A.; Molenaar, R.; van Noorden, C.J.F.; Leenders, W.P. Tumor cells in search for glutamate: An alternative explanation for increased invasiveness of IDH1 mutant gliomas. Neuro Oncol. 2014, 16, 1669–1670. [Google Scholar] [CrossRef] [Green Version]
- Vasconcelos Esá, J.; Simão, D.; Terrasso, A.P.; Silva, M.M.; Brito, C.; Isidro, I.A.; Alves, P.M.; Carrondo, M. Unveiling dynamic metabolic signatures in human induced pluripotent and neural stem cells. PLoS Comput. Biol. 2020, 16, 1007780. [Google Scholar] [CrossRef] [Green Version]
- Breznik, B.; Limbaeck Stokin, C.; Kos, J.; Khurshed, M.; Hira, V.V.V.; Bošnjak, R.; Lah, T.T.; van Noorden, C.J.F. Cysteine cathepsins B, X and K expression in peri-arteriolar glioblastoma stem cell niches. J. Mol. Histol. 2018, 49, 481–497. [Google Scholar] [CrossRef] [Green Version]
- Breznik, B.; Limback, C.; Porcnik, A.; Blejec, A.; Krajnc, M.K.; Bosnjak, R.; Kos, J.; van Noorden, C.J.F.; Lah, T.T. Localization patterns of cathepsins K and X and their predictive value in glioblastoma. Radiol. Oncol. 2018, 52, 433–442. [Google Scholar] [CrossRef] [Green Version]
- Verbovšek, U.; van Noorden, C.J.F.; Lah, T.T. Complexity of cancer protease biology: Cathepsin K expression and function in cancer progression. Semin. Cancer Biol. 2015, 35, 71–84. [Google Scholar] [CrossRef]
- DeBerardinis, R.J. Tumor microenvironment, metabolism, and immunotherapy. N. Engl. J. Med. 2020, 382, 869–871. [Google Scholar] [CrossRef]
- McBrayer, S.K.; Mayers, J.R.; DiNatale, G.J.; Shi, D.D.; Khanal, J.; Chakraborty, A.A.; Sarosiek, K.A.; Briggs, K.J.; Robbins, A.K.; Sewastianik, T.; et al. Transaminase Inhibition by 2-hydroxyglutarate impairs glutamate biosynthesis and redox homeostasis in Glioma. Cell 2018, 175, 101–116. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Yu, J.; Tu, L.; Huang, N.; Li, H.; Luo, Y. Isocitrate dehydrogenase mutations in glioma: From basic discovery to therapeutics development. Front. Oncol. 2019, 9, 506. [Google Scholar] [CrossRef] [Green Version]
- Molenaar, R.J.; Botman, D.; Smits, M.A.; Hira, V.V.V.; van Lith, S.A.; Stap, J.; Henneman, P.; Khurshed, M.; Lenting, K.; Mul, A.N.; et al. Radioprotection of IDH1-mutated cancer cells by the IDH1-mutant inhibitor AGI-5198. Cancer Res. 2015, 75, 4790–4802. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Function | Reference |
|---|---|---|
| Translocator protein (TSPO) | Mitochondrial transmembrane cholesterol transporter | [66] |
| Insulin-like growth factor 2 mRNA-binding protein 2 (IGF2BP2) | Delivering of mRNAs to mitochondria | [68] |
| Glycerol-3-phosphate dehydrogenase I (GPDI) | Prevention of osmotic stress | [69] |
| Oncostatin M | Cytokine in immunosurveillance | [71] |
| Inhibitor | FDA-Approved? | Indication | Reference |
|---|---|---|---|
| Atovaquone | Approved | Pneumocystis jiroveci pneumonia treatment, malaria prophylaxis | [75] |
| Mito-Lonidamine (Mito-LND) | Not approved | N/A | [41] |
| Metformin | Approved | Type 2 diabetes mellitus | [61] |
| Phenformin | Not approved | N/A | [61] |
| Verteporfin | Approved | Macular degeneration | [76] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Noorden, C.J.F.; Hira, V.V.V.; van Dijck, A.J.; Novak, M.; Breznik, B.; Molenaar, R.J. Energy Metabolism in IDH1 Wild-Type and IDH1-Mutated Glioblastoma Stem Cells: A Novel Target for Therapy? Cells 2021, 10, 705. https://doi.org/10.3390/cells10030705
van Noorden CJF, Hira VVV, van Dijck AJ, Novak M, Breznik B, Molenaar RJ. Energy Metabolism in IDH1 Wild-Type and IDH1-Mutated Glioblastoma Stem Cells: A Novel Target for Therapy? Cells. 2021; 10(3):705. https://doi.org/10.3390/cells10030705
Chicago/Turabian Stylevan Noorden, Cornelis J.F., Vashendriya V.V. Hira, Amber J. van Dijck, Metka Novak, Barbara Breznik, and Remco J. Molenaar. 2021. "Energy Metabolism in IDH1 Wild-Type and IDH1-Mutated Glioblastoma Stem Cells: A Novel Target for Therapy?" Cells 10, no. 3: 705. https://doi.org/10.3390/cells10030705
APA Stylevan Noorden, C. J. F., Hira, V. V. V., van Dijck, A. J., Novak, M., Breznik, B., & Molenaar, R. J. (2021). Energy Metabolism in IDH1 Wild-Type and IDH1-Mutated Glioblastoma Stem Cells: A Novel Target for Therapy? Cells, 10(3), 705. https://doi.org/10.3390/cells10030705