
High Glucose Treatment Limits Drosha Protein Expression and Alters AngiomiR Maturation in Microvascular Primary Endothelial Cells via an Mdm2-dependent Mechanism



Abstract
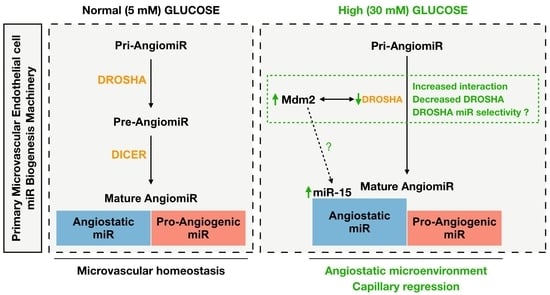
:
1. Introduction
2. Materials and Methods
2.1. Mouse Model
2.2. Primary Microvascular Endothelial Cell Cultures
2.3. D-Glucose and D-Mannitol Cell Treatment
2.4. Inhibition of MDM2 Activity
2.5. Immunoblotting
2.6. Protein Co-Immunoprecipitation Assay
2.7. Isolation of pri-miR, miR, and total RNA. RT-qPCR
2.8. Statistical Analysis
3. Results
3.1. High Glucose Treatment Alters the miR Biogenesis Machinery by Decreasing DROSHA Protein Expression in Human Primary Dermal Microvascular Endothelial Cells
3.2. High Glucose-Mediated Decrease in DROSHA Protein Expression Is Dose- and Time-Dependent but Is Not Affected by Osmotic Stress in Primary Microvascular Endothelial Cells
3.2.1. Dose-Effect of Glucose Treatment on DROSHA and DICER Protein Expression
3.2.2. Time-Effect of High Glucose Treatment on DROSHA Protein Expression
3.2.3. Mannitol Treatment Does Not Affect DROSHA Protein Expression
3.3. High Glucose Treatment Shifts Endothelial-Derived Mature angiomiR Expression towards an Anti-Angiogenic Profile without Altering Primary pri-miR Expression
3.3.1. High Glucose Treatment Did Not Alter the Expression of pri-angiomiRs pri-miR-126, pri-miR-15 and pri-miR-17-92 in Human Primary Dermal Microvascular Cells
3.3.2. High Glucose Treatment Upregulated the Expression of the Angiostatic Mature miR-15a without Altering the Expression of the Pro-Angiogenic Mature miR-126-3p, miR-126-5p, and miR-18a
3.3.3. High Glucose Treatment Results in Decreased VEGF-A Protein Expression but No Change in THBS-1 Protein Levels
3.4. Reduced Endothelial DROSHA Expression in Response to High Glucose Treatment Is Regulated by Murine Double Minute-2/DROSHA Interaction and MDM2 Activity
3.4.1. High Glucose Treatment Increased MDM2 mRNA and Protein Expression in Human Primary Dermal Microvascular Endothelial Cells
3.4.2. High Glucose Treatment Enhanced MDM2 Binding to DROSHA in HDMECs
3.4.3. Inhibition of MDM2 Activity Prevented the Decrease in DROSHA Protein Levels
3.4.4. Inhibition of MDM2 Activity by Nutlin-3 Prevented the Increase in the Mature Form of miR-15a
3.5. DROSHA Protein Expression Is Reduced in High Glucose-Treated Murine Primary Skeletal Muscle Endothelial Cells and In Vivo in Skeletal Muscles from Diabetic Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Wagenmakers, A.J.M.; Strauss, J.A.; Shepherd, S.O.; Keske, M.A.; Cocks, M. Increased muscle blood supply and transendo-thelial nutrient and insulin transport induced by food intake and exercise: Effect of obesity and ageing. J. Physiol. 2016, 594, 2207–2222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murrant, C.L.; Sarelius, I.H. Local control of blood flow during active hyperaemia: What kinds of integration are important? J. Physiol. 2015, 593, 4699–4711. [Google Scholar] [CrossRef] [Green Version]
- Reglin, B.; Pries, A.R. Metabolic Control of Microvascular Networks: Oxygen Sensing and Beyond. J. Vasc. Res. 2014, 51, 376–392. [Google Scholar] [CrossRef] [PubMed]
- Varricchi, G.; Granata, F.; Loffredo, S.; Genovese, A.; Marone, G. Angiogenesis and lymphangiogenesis in inflammatory skin disorders. J. Am. Acad. Dermatol. 2015, 73, 144–153. [Google Scholar] [CrossRef]
- Haas, T.L.; Nwadozi, E. Regulation of skeletal muscle capillary growth in exercise and disease. Appl. Physiol. Nutr. Metab. 2015, 40, 1221–1232. [Google Scholar] [CrossRef] [PubMed]
- Olfert, I.M.; Birot, O. Importance of Anti-angiogenic Factors in the Regulation of Skeletal Muscle Angiogenesis. Microcirc. 2011, 18, 316–330. [Google Scholar] [CrossRef]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Olson, E.N. AngiomiRs--key regulators of angiogenesis. Curr. Opin. Genet. Dev. 2009, 19, 205–211. [Google Scholar] [CrossRef] [Green Version]
- Fish, J.E.; Srivastava, D. MicroRNAs: Opening a New Vein in Angiogenesis Research. Sci. Signal. 2009, 2, pe1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fish, J.E.; Santoro, M.M.; Morton, S.U.; Yu, S.; Yeh, R.-F.; Wythe, J.D.; Ivey, K.N.; Bruneau, B.G.; Stainier, D.Y.; Srivastava, D. miR-126 Regulates Angiogenic Signaling and Vascular Integrity. Dev. Cell 2008, 15, 272–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhnert, F.; Mancuso, M.R.; Hampton, J.; Stankunas, K.; Asano, T.; Chen, C.-Z.; Kuo, C.J. Attribution of vascular phenotypes of the murine Egfl7 locus to the microRNA miR-126. Dev. 2008, 135, 3989–3993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Aurora, A.B.; Johnson, B.A.; Qi, X.; McAnally, J.; Hill, J.A.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. The Endothelial-Specific MicroRNA miR-126 Governs Vascular Integrity and Angiogenesis. Dev. Cell 2008, 15, 261–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mogilyansky, E.; Rigoutsos, I. The miR-17/92 cluster: A comprehensive update on its genomics, genetics, functions and in-creasingly important and numerous roles in health and disease. Cell Death Differ. 2013, 20, 1603–1614. [Google Scholar] [CrossRef] [PubMed]
- Dews, M.; Homayouni, A.; Yu, D.; Murphy, D.; Sevignani, C.; A Wentzel, E.; E Furth, E.; Lee, W.M.; Enders, G.H.; Mendell, J.T.; et al. Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nat. Genet. 2006, 38, 1060–1065. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.-Y.; Wang, W.; Li, F.-F.; Chen, Y.-C.; Jiang, D.; Chen, Y.-D.; Yang, H.; Liu, L.; Lu, M.; Sun, J.-S.; et al. Maggot excretions/secretions promote diabetic wound angiogenesis via miR18a/19a—TSP-1 axis. Diabetes Res. Clin. Pr. 2020, 165, 108140. [Google Scholar] [CrossRef]
- Besnier, M.; Shantikumar, S.; Anwar, M.; Dixit, P.; Chamorro-Jorganes, A.; Sweaad, W.; Sala-Newby, G.; Madeddu, P.; Thomas, A.C.; Howard, L.; et al. miR-15a/-16 Inhibit Angiogenesis by Targeting the Tie2 Coding Sequence: Therapeutic Po-tential of a miR-15a/16 Decoy System in Limb Ischemia. Mol. Nucleic Acids 2019, 17, 49–62. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.-Y.; She, X.-M.; Qin, Y.; Chu, Z.-B.; Chen, L.; Ai, L.-S.; Zhang, L.; Hu, Y. miR-15a and miR-16 affect the angiogenesis of multiple myeloma by targeting VEGF. Carcinog. 2013, 34, 426–435. [Google Scholar] [CrossRef] [Green Version]
- Sun, P.; Zhang, K.; Hassan, S.H.; Zhang, X.; Tang, X.; Pu, H.; Stetler, R.A.; Chen, J.; Yin, K.-J. Endothelium-Targeted Deletion of microRNA-15a/16-1 Promotes Poststroke Angiogenesis and Improves Long-Term Neurological Recovery. Circ. Res. 2020, 126, 1040–1057. [Google Scholar] [CrossRef]
- Karaca, Ü.; Schram, M.T.; Houben, A.J.H.M.; Muris, D.M.J.; Stehouwer, C.D.A. Microvascular dysfunction as a link between obesity, insulin resistance and hypertension. Diabetes Res. Clin. Pr. 2014, 103, 382–387. [Google Scholar] [CrossRef]
- Muris, D.M.J.; Houben, A.J.H.M.; Schram, M.T.; Stehouwer, C.D.A. Microvascular dysfunction is associated with a higher incidence of type 2 diabetes mellitus: A systematic review and meta-analysis. Arter. Thromb. Vasc. Biol. 2012, 32, 3082–3094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stehouwer, C.D. Microvascular Dysfunction and Hyperglycemia: A Vicious Cycle With Widespread Consequences. Diabetes 2018, 67, 1729–1741. [Google Scholar] [CrossRef] [Green Version]
- Roudier, E.; Chapados, N.; Décary, S.; Gineste, C.; Le Bel, C.; Lavoie, J.-M.; Bergeron, R.; Birot, O. Angiomotin p80/p130 ratio: A new indicator of exercise-induced angiogenic activity in skeletal muscles from obese and non-obese rats? J. Physiol. 2009, 587, 4105–4119. [Google Scholar] [CrossRef]
- Roudier, E.; Forn, P.; Perry, M.E.; Birot, O. Murine double minute-2 expression is required for capillary maintenance and exercise-induced angiogenesis in skeletal muscle. Faseb J. 2012, 26, 4530–4539. [Google Scholar] [CrossRef] [Green Version]
- Martens, R.J.H.; Henry, R.M.A.; Houben, A.J.; Van Der Kallen, C.J.H.; Kroon, A.A.; Schalkwijk, C.G.; Schram, M.T.; Sep, S.J.S.; Schaper, N.C.; Dagnelie, P.C.; et al. Capillary Rarefaction Associates with Albuminuria: The Maastricht Study. J. Am. Soc. Nephrol. 2016, 27, 3748–3757. [Google Scholar] [CrossRef] [PubMed]
- Jaap, A.J.; Shore, A.C.; Stockman, A.J.; Tooke, J.E. Skin capillary density in subjects with impaired glucose tolerance and patients with type 2 diabetes. Diabet. Med. 1996, 13, 160–164. [Google Scholar] [CrossRef]
- Sörensen, B.M.; Houben, A.J.H.M.; Berendschot, T.T.J.M.; Schouten, J.S.A.G.; Kroon, A.A.; van der Kallen, C.J.H.; Henry, R.M.A.; Koster, A.; Sep, S.J.S.; Dagnelie, P.C.; et al. Prediabetes and Type 2 Diabetes Are Associated With Generalized Mi-crovascular Dysfunction: The Maastricht Study. Circulation 2016, 134, 1339–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aiken, J.; Mandel, E.R.; Riddell, M.C.; Birot, O. Hyperglycaemia correlates with skeletal muscle capillary regression and is associated with alterations in the murine double minute-2/forkhead box O1/thrombospondin-1 pathway in type 1 diabetic BioBreeding rats. Diab. Vasc. Dis. Res. 2019, 16, 28–37. [Google Scholar] [CrossRef] [Green Version]
- Dunford, E.C.; LeClair, E.; Aiken, J.; Mandel, E.R.; Haas, T.L.; Birot, O.; Riddell, M.C. The effects of voluntary exercise and prazosin on capillary rarefaction and metabolism in streptozotocin-induced diabetic male rats. J. Appl. Physiol. 2017, 122, 492–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kivelä, R.; Silvennoinen, M.; Touvra, A.-M.; Lehti, T.M.; Kainulainen, H.; Vihko, V. Effects of experimental type 1 diabetes and exercise training on angiogenic gene expression and capillarization in skeletal muscle. Faseb J. 2006, 20, 1570–1572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lillioja, S.; A Young, A.; Culter, C.L.; Ivy, J.L.; Abbott, W.G.; Zawadzki, J.K.; Yki-Järvinen, H.; Christin, L.; Secomb, T.W.; Bogardus, C. Skeletal muscle capillary density and fiber type are possible determinants of in vivo insulin resistance in man. J. Clin. Investig. 1987, 80, 415–424. [Google Scholar] [CrossRef]
- Silvennoinen, M.; Rinnankoski-Tuikka, R.; Vuento, M.; Hulmi, J.J.; Torvinen, S.; Lehti, M.; Kivelä, R.; Kainulainen, H. High-fat feeding induces angiogenesis in skeletal muscle and activates angiogenic pathways in capillaries. Angiogenesis 2012, 16, 297–307. [Google Scholar] [CrossRef]
- Groen, B.B.L.; Hamer, H.M.; Snijders, T.; Van Kranenburg, J.; Frijns, D.; Vink, H.; Van Loon, L.J.C. Skeletal muscle capillary density and microvascular function are compromised with aging and type 2 diabetes. J. Appl. Physiol. 2014, 116, 998–1005. [Google Scholar] [CrossRef] [Green Version]
- Baum, O.; Bigler, M. Pericapillary basement membrane thickening in human skeletal muscles. Am. J. Physiol. Circ. Physiol. 2016, 311, H654–H666. [Google Scholar] [CrossRef] [Green Version]
- Mortensen, S.P.; Winding, K.M.; Iepsen, U.W.; Munch, G.W.; Marcussen, N.; Hellsten, Y.; Pedersen, B.K.; Baum, O. The effect of two exercise modalities on skeletal muscle capillary ultrastructure in individuals with type 2 diabetes. Scand. J. Med. Sci. Sports 2018, 29, 360–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínek, J.; Krausová, Z. A morphometric analysis of the capillary basement membrane as a diagnostic parameter in diabetes mellitus. Funct. Dev. Morphol. 1991, 1, 35–38. [Google Scholar]
- Stallmeyer, B.; Pfeilschifter, J.; Frank, S. Systemically and topically supplemented leptin fails to reconstitute a normal angiogenic response during skin repair in diabetic ob/ob mice. Diabetol. 2001, 44, 471–479. [Google Scholar] [CrossRef] [Green Version]
- Sawada, N.; Arany, Z. Metabolic Regulation of Angiogenesis in Diabetes and Aging. Physiol. 2017, 32, 290–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aiken, J.; Roudier, E.; Ciccone, J.; Drouin, G.; Stromberg, A.; Vojnovic, J.; Olfert, I.M.; Haas, T.; Gustafsson, T.; Grenier, G.; et al. Phosphorylation of murine double minute-2 on Ser 166 is downstream of VEGF-A in exercised skeletal muscle and regulates primary endothelial cell migration and FoxO gene expression. Faseb J. 2015, 30, 1120–1134. [Google Scholar] [CrossRef]
- Nwadozi, E.; Roudier, E.; Rullman, E.; Tharmalingam, S.; Liu, H.; Gustafsson, T.; Haas, T.L. Endothelial FoxO proteins impair insulin sensitivity and restrain muscle angiogenesis in response to a high-fat diet. Faseb J. 2016, 30, 3039–3052. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Narayanan, S.; Sunkari, V.G.; Eliasson, S.; Botusan, I.R.; Grünler, J.; Catrina, A.I.; Radtke, F.; Xu, C.; Zhao, A.; et al. Triggering of a Dll4–Notch1 loop impairs wound healing in diabetes. Proc. Natl. Acad. Sci. USA 2019, 116, 6985–6994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, M.; LoGerfo, F.W.; Guthrie, P.; Pradhan, L. Effect of hyperglycemia and neuropeptides on interleukin-8 expression and angiogenesis in dermal microvascular endothelial cells. J. Vasc. Surg. 2011, 53, 1654–1660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Lockhart, S.M.; Rathjen, T.; Albadawi, H.; Sørensen, D.; O’Neill, B.T.; Dwivedi, N.; Preil, S.R.; Beck, H.C.; Dunwoodie, S.L.; et al. Insulin Downregulates the Transcriptional Coregulator CITED2, an Inhibitor of Proangiogenic Function in Endothelial Cells. Diabetes 2016, 65, 3680–3690. [Google Scholar] [CrossRef] [Green Version]
- Kuehbacher, A.; Urbich, C.; Zeiher, A.M.; Dimmeler, S. Role of Dicer and Drosha for Endothelial MicroRNA Expression and Angiogenesis. Circ. Res. 2007, 101, 59–68. [Google Scholar] [CrossRef] [Green Version]
- Suárez, Y.; Fernández-Hernando, C.; Pober, J.S.; Sessa, W.C. Dicer Dependent MicroRNAs Regulate Gene Expression and Functions in Human Endothelial Cells. Circ. Res. 2007, 100, 1164–1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shilo, S.; Roy, S.; Khanna, S.; Sen, C.K. Evidence for the Involvement of miRNA in Redox Regulated Angiogenic Response of Human Microvascular Endothelial Cells. Arter. Thromb. Vasc. Biol. 2008, 28, 471–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Han, J.; Yeom, K.-H.; Jin, H.; Kim, V. Drosha in Primary MicroRNA Processing. Cold Spring Harb. Symp. Quant. Biol. 2006, 71, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Macrae, I.; Li, F.; Zhou, K.; Cande, W.; Doudna, J. Structure of Dicer and Mechanistic Implications for RNAi. Cold Spring Harb. Symp. Quant. Biol. 2006, 71, 73–80. [Google Scholar] [CrossRef] [Green Version]
- Sen, C.K.; Gordillo, G.M.; Khanna, S.; Roy, S. Micromanaging Vascular Biology: Tiny MicroRNAs Play Big Band. J. Vasc. Res. 2009, 46, 527–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kriegel, A.J.; Baker, M.A.; Liu, Y.; Liu, P.; Cowley, A.W.; Liang, M. Endogenous microRNAs in human microvascular endo-thelial cells regulate mRNAs encoded by hypertension-related genes. Hypertension 2015, 66, 793–799. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Chen, S.; Biswas, S.; Nagrani, N.; Chu, Y.; Chakrabarti, S.; Feng, B. Glucose-induced oxidative stress and accelerated aging in endothelial cells are mediated by the depletion of mitochondrial SIRTs. Physiol. Rep. 2020, 8, e14331. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Sul, K.; Krukovets, I.; Nestor, C.; Li, J.; Adognravi, O.S. Novel Tissue-Specific Mechanism of Regulation of Angiogenesis and Cancer Growth in Response to Hyperglycemia. J. Am. Hear. Assoc. 2012, 1, e005967. [Google Scholar] [CrossRef] [Green Version]
- Ye, E.-A.; Steinle, J.J. miR-15b/16 protects primary human retinal microvascular endothelial cells against hyperglyce-mia-induced increases in tumor necrosis factor alpha and suppressor of cytokine signaling 3. J. Neuroinflamm. 2015, 12, 44. [Google Scholar] [CrossRef] [Green Version]
- Ye, E.-A.; Liu, L.; Steinle, J.J. miR-15a/16 inhibits TGF-beta3/VEGF signaling and increases retinal endothelial cell barrier pro-teins. Vis. Res. 2017, 139, 23–29. [Google Scholar] [CrossRef]
- Ye, P.; Liu, Y.; Chen, C.; Tang, F.; Wu, Q.; Wang, X.; Liu, C.-G.; Liu, X.; Liu, R.; Liu, Y.; et al. An mTORC1-Mdm2-Drosha Axis for miRNA Biogenesis in Response to Glucose- and Amino Acid-Deprivation. Mol. Cell 2015, 57, 708–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, K.P.; Leong, W.F.; Chau, J.F.L.; Jia, D.; Zeng, L.; Liu, H.; He, L.; Hao, A.; Zhang, H.; Meek, D.; et al. S6K1 is a multifaceted regulator of Mdm2 that connects nutrient status and DNA damage response. Embo J. 2010, 29, 2994–3006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nwadozi, E.; Ng, A.; Strömberg, A.; Liu, H.-Y.; Olsson, K.; Gustafsson, T.; Haas, T.L. Leptin is a physiological regulator of skeletal muscle angiogenesis and is locally produced by PDGFRα and PDGFRβ expressing perivascular cells. Angiogenesis 2019, 22, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Rudnicki, M.; Abdifarkosh, G.; Nwadozi, E.; Ramos, S.V.; Makki, A.; Sepa-Kishi, D.M.; Ceddia, R.B.; Perry, C.G.; Roudier, E.; Haas, T.L. Endothelial-specific FoxO1 depletion prevents obesity-related disorders by increasing vascular metabolism and growth. eLife 2018, 7. [Google Scholar] [CrossRef]
- Chen, J.; Marechal, V.; Levine, A.J. Mapping of the p53 and mdm-2 interaction domains. Mol. Cell. Biol. 1993, 13, 4107–4114. [Google Scholar] [CrossRef] [PubMed]
- Wagner-Ecker, M.; Schwager, C.; Wirkner, U.; Abdollahi, A.; E Huber, P. MicroRNA expression after ionizing radiation in human endothelial cells. Radiat. Oncol. 2010, 5, 25. [Google Scholar] [CrossRef] [Green Version]
- Fåhraeus, R.; Olivares-Illana, V. MDM2′s social network. Oncogene 2014, 33, 4365–4376. [Google Scholar] [CrossRef]
- Gu, L.; Zhang, H.; Liu, T.; Zhou, S.; Du, Y.; Xiong, J.; Yi, S.; Qu, C.-K.; Fu, H.; Zhou, M. Discovery of Dual Inhibitors of MDM2 and XIAP for Cancer Treatment. Cancer Cell 2016, 30, 623–636. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Zatloukalova, P.; Muller, P.; Gil-Mir, M.; Kote, S.; Wilkinson, S.; Kemp, A.J.; Hernychova, L.; Wang, Y.; Ball, K.L.; et al. The MDM2 ligand Nutlin-3 differentially alters expression of the immune blockade receptors PD-L1 and CD276. Cell. Mol. Biol. Lett. 2020, 25, 1–21. [Google Scholar] [CrossRef]
- Dandona, P. Minimizing Glycemic Fluctuations in Patients with Type 2 Diabetes: Approaches and Importance. Diabetes Technol. Ther. 2017, 19, 498–506. [Google Scholar] [CrossRef] [Green Version]
- Torimoto, K.; Okada, Y.; Mori, H.; Tanaka, Y. Relationship between fluctuations in glucose levels measured by continuous glucose monitoring and vascular endothelial dysfunction in type 2 diabetes mellitus. Cardiovasc. Diabetol. 2013, 12, 1. [Google Scholar] [CrossRef] [Green Version]
- Popov, D. Endothelial cell dysfunction in hyperglycemia: Phenotypic change, intracellular signaling modification, ultrastructural alteration, and potential clinical outcomes. Int. J. Diabetes Mellit. 2010, 2, 189–195. [Google Scholar] [CrossRef] [Green Version]
- Silambarasan, M.; Tan, J.R.; Karolina, D.S.; Armugam, A.; Kaur, C.; Jeyaseelan, K. MicroRNAs in Hyperglycemia Induced Endothelial Cell Dysfunction. Int. J. Mol. Sci. 2016, 17, 518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Wang, H.; Liu, C.; Mu, X.; Cheng, S. Hyperglycemia inhibition of endothelial miR-140-3p mediates angiogenic dysfunction in diabetes mellitus. J. Diabetes Its Complicat. 2019, 33, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.I.; Yamagata, K.; Sugimoto, K.; Iwamoto, T.; Kato, S.; Miyazono, K. Modulation of microRNA processing by p53. Nat. Cell Biol. 2009, 460, 529–533. [Google Scholar] [CrossRef]
- Kawai, S.; Amano, A. BRCA1 regulates microRNA biogenesis via the DROSHA microprocessor complex. J. Cell Biol. 2012, 197, 201–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soufi, F.G.; Zonouzi, A.A.P.; Eftekhar, E.; Kamali, K.; Chegeni, S.A.; Zonouzi, A.P. The expression of Drosha, DGCR8, Dicer and Ago-2 genes are upregulated in human umbilical vein endothelial cells under hyperglycemic condition. Endocr. Regul. 2018, 52, 123–127. [Google Scholar] [CrossRef] [Green Version]
- Nakato, R.; Wada, Y.; Nakaki, R.; Nagae, G.; Katou, Y.; Tsutsumi, S.; Nakajima, N.; Fukuhara, H.; Iguchi, A.; Kohro, T.; et al. Comprehensive epigenome characterization reveals diverse transcriptional regulation across human vascular endothelial cells. Epigenetics Chromatin 2019, 12, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pong, S.K.; Gullerova, M. Noncanonical functions of microRNA pathway enzymes—Drosha, DGCR8, Dicer and Ago proteins. Febs Lett. 2018, 592, 2973–2986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Havens, M.A.; Reich, A.A.; Hastings, M.L. Drosha Promotes Splicing of a Pre-microRNA-like Alternative Exon. Plos Genet. 2014, 10, e1004312. [Google Scholar] [CrossRef]
- Stavast, C.; Erkeland, S. The Non-Canonical Aspects of MicroRNAs: Many Roads to Gene Regulation. Cells 2019, 8, 1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pendzialek, S.M.; Knelangen, J.M.; Schindler, M.; Gürke, J.; Grybel, K.J.; Gocza, E.; Fischer, B.; Navarrete Santos, A. Tropho-blastic microRNAs are downregulated in a diabetic pregnancy through an inhibition of Drosha. Mol. Cell. Endocrinol. 2019, 480, 167–179. [Google Scholar] [CrossRef]
- Feng, Z.; Zhang, C.; Wu, R.; Hu, W. Tumor suppressor p53 meets microRNAs. J. Mol. Cell Biol. 2011, 3, 44–50. [Google Scholar] [CrossRef]
- Santiago, A.R.; Boia, R.; Aires, I.D.; Ambrósio, A.F.; Fernandes, R. Sweet Stress: Coping With Vascular Dysfunction in Diabetic Retinopathy. Front. Physiol. 2018, 9, 820. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Navitskaya, S.; Chakravarthy, H.; Huang, C.; Kady, N.; Lydic, T.A.; Chen, Y.E.; Yin, K.-J.; Powell, F.L.; Martin, P.M.; et al. Dual Anti-Inflammatory and Anti-Angiogenic Action of miR-15a in Diabetic Retinopathy. EBioMedicine 2016, 11, 138–150. [Google Scholar] [CrossRef] [Green Version]
- Spinetti, G.; Fortunato, O.; Caporali, A.; Shantikumar, S.; Marchetti, M.; Meloni, M.; Descamps, B.; Floris, I.; Sangalli, E.; Vono, R.; et al. MicroRNA-15a and MicroRNA-16 Impair Human Circulating Proangiogenic Cell Functions and Are Increased in the Proangiogenic Cells and Serum of Patients With Critical Limb Ischemia. Circ. Res. 2013, 112, 335–346. [Google Scholar] [CrossRef] [Green Version]
- Gnanasundram, S.V.; Malbert-Colas, L.; Chen, S.; Fusée, L.; Daskalogianni, C.; Muller, P.; Salomao, N.; Fåhraeus, R. MDM2′s dual mRNA binding domains co-ordinate its oncogenic and tumour suppressor activities. Nucleic Acids Res. 2020, 48, 6775–6787. [Google Scholar] [CrossRef]
- Elenbaas, B.; Dobbelstein, M.; Roth, J.; Shenk, T.; Levine, A.J. The MDM2 Oncoprotein Binds Specifically to RNA through its RING Finger Domain. Mol. Med. 1996, 2, 439–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, S.; Gu, L.; He, J.; Zhang, H.; Zhou, M. MDM2 Regulates Vascular Endothelial Growth Factor mRNA Stabilization in Hypoxia. Mol. Cell. Biol. 2011, 31, 4928–4937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forbes, J.M.; Cooper, M.E. Mechanisms of Diabetic Complications. Physiol. Rev. 2013, 93, 137–188. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assay ID | Target | Sequences |
|---|---|---|
| Hs03303230_pri | hsa-mir-126 | CGCUGGCGACGGGACAUUAUUACUUUUGGUACGC GCUGUGACACUUCAAACUCGUACCGUGAGUAAUA AUGCGCCGUCCACGGCA |
| Hs03302582_pri | hsa-mir-15a | CCUUGGAGUAAAGUAGCAGCACAUAAUGGUUUGU GGAUUUUGAAAAGGUGCAGGCCAUAUUGUGCUGC CUCAAAAAUACAAGG |
| Hs03295901_pri | hsa-mir-17 | GUCAGAAUAAUGUCAAAGUGCUUACAGUGCAGGU AGUGAUAUGUGCAUCUACUGCAGUGAAGGCACUU GUAGCAUUAUGGUGAC |
| 477887_mir | hsa-miR-126-3p | UCGUACCGUGAGUAAUAAUGCG |
| 477888_mir | hsa-miR-126-5p | CAUUAUUACUUUUGGUACGCG |
| 477858_mir | hsa-miR-15a-5p | UAGCAGCACAUAAUGGUUUGUG |
| 478551_mir | hsa-miR-18a-5p | UAAGGUGCAUCUAGUGCAGAUAG |
| 478293_mir | cel-miR-39-3p | UCACCGGGUGUAAAUCAGCUUG |
| 482962_mir | mmu-miR-15a-5p | UAGCAGCACAUAAUGGUUUGUG |
| Hs02800695_m1 | HPRT | GGACTAATTATGGACAGGACTGAAC |
| Hs01066930_m1 | MDM2 | GAACAAGAGACCCTGGTTAGACCAA |
| Hs00900055_m1 | VEGF-A | ACATCACCATGCAGATTATGCGGAT |
| Hs00962908_m1 | THBS-1 | ACACAATCCGGATCAGCTGGACTCT |
| Hs00229023_m1 | DICER1 | GTGCTACCCAAAAGCAATTCCAGAG |
| Hs00203008_m1 | DROSHA | AATGATCCGGACCTGCGCGAAGTCT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lam, B.; Nwadozi, E.; Haas, T.L.; Birot, O.; Roudier, E. High Glucose Treatment Limits Drosha Protein Expression and Alters AngiomiR Maturation in Microvascular Primary Endothelial Cells via an Mdm2-dependent Mechanism. Cells 2021, 10, 742. https://doi.org/10.3390/cells10040742
Lam B, Nwadozi E, Haas TL, Birot O, Roudier E. High Glucose Treatment Limits Drosha Protein Expression and Alters AngiomiR Maturation in Microvascular Primary Endothelial Cells via an Mdm2-dependent Mechanism. Cells. 2021; 10(4):742. https://doi.org/10.3390/cells10040742
Chicago/Turabian StyleLam, Brian, Emmanuel Nwadozi, Tara L. Haas, Olivier Birot, and Emilie Roudier. 2021. "High Glucose Treatment Limits Drosha Protein Expression and Alters AngiomiR Maturation in Microvascular Primary Endothelial Cells via an Mdm2-dependent Mechanism" Cells 10, no. 4: 742. https://doi.org/10.3390/cells10040742
APA StyleLam, B., Nwadozi, E., Haas, T. L., Birot, O., & Roudier, E. (2021). High Glucose Treatment Limits Drosha Protein Expression and Alters AngiomiR Maturation in Microvascular Primary Endothelial Cells via an Mdm2-dependent Mechanism. Cells, 10(4), 742. https://doi.org/10.3390/cells10040742