
Parkinson’s Disease Causative Mutation in Vps35 Disturbs Tetherin Trafficking to Cell Surfaces and Facilitates Virus Spread

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
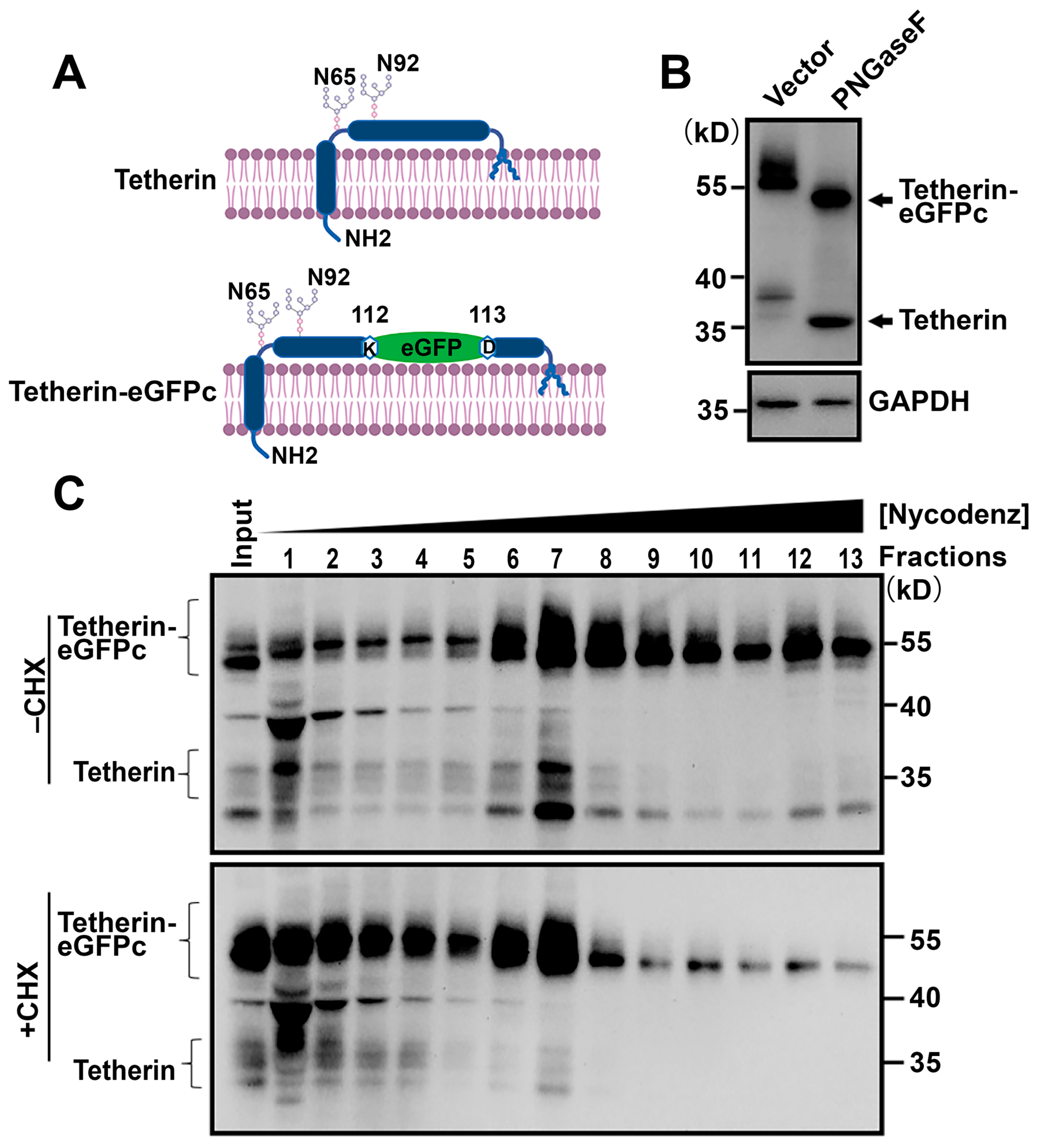{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmids
2.2. Cell Culture and Transfection
2.3. Virus Titer Determination
2.4. HSV-1 Entry, Replication, and Release Assays
2.5. Subcellular Fractionation
2.6. Western Blot
2.7. Immunofluorescence Microscopy and Time-Lapse Live Cell Imaging
2.8. Image Analysis
2.9. Statistical Analysis
3. Results
3.1. The D620N Mutation Dampens Vps35 in Constraining HSV-1 Propagation
3.2. Tetherin Mediates the Inhibitory Effect of Vps35 on HSV-1 Propagation
3.3. Tetherin Traffics Together with Vps35
3.4. Tetherin Recycles Mainly through the Vps35 Dependent Trafficking Pathway
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Simon, D.K.; Tanner, C.M.; Brundin, P. Parkinson Disease Epidemiology, Pathology, Genetics, Pathophysiology. Clin. Geriatr. Med. 2020, 36, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.E.; Healy, M.D.; Collins, B.M. Towards a Molecular Understanding of Endosomal Trafficking by Retromer and Retriever. Traffic 2019, 20, 465–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cullen, P.J.; Steinberg, F. To Degrade or Not to Degrade: Mechanisms and Significance of Endocytic Recycling. Nat. Rev. Mol. Cell Biol. 2018, 19, 679–696. [Google Scholar] [CrossRef]
- Pang, S.Y.; Ho, P.W.; Liu, H.F.; Leung, C.T.; Li, L.; Chang, E.E.S.; Ramsden, D.B.; Ho, S.L. The Interplay of Aging, Genetics and Environmental Factors in the Pathogenesis of Parkinson’s Disease. Transl. Neurodegener. 2019, 8, 23. [Google Scholar] [CrossRef]
- Vilarino-Guell, C.; Wider, C.; Ross, O.A.; Dachsel, J.C.; Kachergus, J.M.; Lincoln, S.J.; Soto-Ortolaza, A.I.; Cobb, S.A.; Wilhoite, G.J.; Bacon, J.A.; et al. Vps35 Mutations in Parkinson Disease. Am. J. Hum. Genet. 2011, 89, 162–167. [Google Scholar] [CrossRef] [Green Version]
- Zimprich, A.; Benet-Pages, A.; Struhal, W.; Graf, E.; Eck, S.H.; Offman, M.N.; Haubenberger, D.; Spielberger, S.; Schulte, E.C.; Lichtner, P.; et al. A Mutation in Vps35, Encoding a Subunit of the Retromer Complex, Causes Late-Onset Parkinson Disease. Am. J. Hum. Genet. 2011, 89, 168–175. [Google Scholar] [CrossRef] [Green Version]
- Williams, E.T.; Chen, X.; Moore, D.J. Vps35, the Retromer Complex and Parkinson’s Disease. J. Parkinsons Dis. 2017, 7, 219–233. [Google Scholar] [CrossRef] [Green Version]
- Sassone, J.; Reale, C.; Dati, G.; Regoni, M.; Pellecchia, M.T.; Garavaglia, B. The Role of Vps35 in the Pathobiology of Parkinson’s Disease. Cell Mol. Neurobiol. 2020, 41, 199–227. [Google Scholar] [CrossRef]
- Small, S.A.; Kent, K.; Pierce, A.; Leung, C.; Kang, M.S.; Okada, H.; Honig, L.; Vonsattel, J.P.; Kim, T.W. Model-Guided Microarray Implicates the Retromer Complex in Alzheimer’s Disease. Ann. Neurol. 2005, 58, 909–919. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Tang, F.L.; Hong, Y.; Luo, S.W.; Wang, C.L.; He, W.; Shen, C.; Jung, J.U.; Xiong, F.; Lee, D.H.; et al. Vps35 Haploinsufficiency Increases Alzheimer’s Disease Neuropathology. J. Cell Biol. 2011, 195, 765–779. [Google Scholar] [CrossRef] [Green Version]
- Li, J.G.; Chiu, J.; Pratico, D. Full Recovery of the Alzheimer’s Disease Phenotype by Gain of Function of Vacuolar Protein Sorting 35. Mol. Psychiatry 2020, 25, 2630–2640. [Google Scholar] [CrossRef]
- Temkin, P.; Lauffer, B.; Jager, S.; Cimermancic, P.; Krogan, N.J.; von Zastrow, M. Snx27 Mediates Retromer Tubule Entry and Endosome-to-Plasma Membrane Trafficking of Signalling Receptors. Nat. Cell Biol. 2011, 13, 715–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seaman, M.N.; McCaffery, J.M.; Emr, S.D. A Membrane Coat Complex Essential for Endosome-to-Golgi Retrograde Transport in Yeast. J. Cell Biol. 1998, 142, 665–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peter, B.J.; Kent, H.M.; Mills, I.G.; Vallis, Y.; Butler, P.J.; Evans, P.R.; McMahon, H.T. Bar Domains as Sensors of Membrane Curvature: The Amphiphysin Bar Structure. Science 2004, 303, 495499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovtun, O.; Leneva, N.; Bykov, Y.S.; Ariotti, N.; Teasdale, R.D.; Schaffer, M.; Engel, B.D.; Owen, D.J.; Briggs, J.A.G.; Collins, B.M. Structure of the Membrane-Assembled Retromer Coat Determined by Cryo-Electron Tomography. Nature 2018, 561, 561–564. [Google Scholar] [CrossRef]
- Hierro, A.; Rojas, A.L.; Rojas, R.; Murthy, N.; Effantin, G.; Kajava, A.V.; Steven, A.C.; Bonifacino, J.S.; Hurley, J.H. Functional Architecture of the Retromer Cargo-Recognition Complex. Nature 2007, 449, 1063–1067. [Google Scholar] [CrossRef]
- Follett, J.; Norwood, S.J.; Hamilton, N.A.; Mohan, M.; Kovtun, O.; Tay, S.; Zhe, Y.; Wood, S.A.; Mellick, G.D.; Silburn, P.A.; et al. The Vps35 D620n Mutation Linked to Parkinson’s Disease Disrupts the Cargo Sorting Function of Retromer. Traffic 2014, 15, 230–244. [Google Scholar] [CrossRef]
- Tang, F.L.; Erion, J.R.; Tian, Y.; Liu, W.; Yin, D.M.; Ye, J.; Tang, B.; Mei, L.; Xiong, W.C. Vps35 in Dopamine Neurons Is Required for Endosome-to-Golgi Retrieval of Lamp2a, a Receptor of Chaperone-Mediated Autophagy That Is Critical for Alpha-Synuclein Degradation and Prevention of Pathogenesis of Parkinson’s Disease. J. Neurosci. 2015, 35, 10613–10628. [Google Scholar] [CrossRef]
- Wang, C.; Niu, M.; Zhou, Z.; Zheng, X.; Zhang, L.; Tian, Y.; Yu, X.; Bu, G.; Xu, H.; Ma, Q.; et al. Vps35 Regulates Cell Surface Recycling and Signaling of Dopamine Receptor D1. Neurobiol. Aging 2016, 46, 22–31. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Fagan, R.R.; Uttamapinant, C.; Lifshitz, L.M.; Fogarty, K.E.; Ting, A.Y.; Melikian, H.E. The Dopamine Transporter Recycles Via a Retromer-Dependent Postendocytic Mechanism: Tracking Studies Using a Novel Fluorophore-Coupling Approach. J. Neurosci. 2017, 37, 9438–9452. [Google Scholar] [CrossRef] [Green Version]
- Cui, Y.; Yang, Z.; Flores-Rodriguez, N.; Follett, J.; Ariotti, N.; Wall, A.A.; Parton, R.G.; Teasdale, R.D. Formation of Retromer Transport Carriers Is Disrupted by the Parkinson Disease-Linked Vps35 D620n Variant. Traffic 2021, 22, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Zavodszky, E.; Seaman, M.N.; Moreau, K.; Jimenez-Sanchez, M.; Breusegem, S.Y.; Harbour, M.E.; Rubinsztein, D.C. Mutation in Vps35 Associated with Parkinson’s Disease Impairs Wash Complex Association and Inhibits Autophagy. Nat. Commun. 2014, 5, 3828. [Google Scholar] [CrossRef]
- Dhungel, N.; Eleuteri, S.; Li, L.B.; Kramer, N.J.; Chartron, J.W.; Spencer, B.; Kosberg, K.; Fields, J.A.; Stafa, K.; Adame, A.; et al. Parkinson’s Disease Genes Vps35 and Eif4g1 Interact Genetically and Converge on Alpha-Synuclein. Neuron 2015, 85, 76–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Kordich, J.K.; Williams, E.T.; Levine, N.; Cole-Strauss, A.; Marshall, L.; Labrie, V.; Ma, J.; Lipton, J.W.; Moore, D.J. Parkinson’s Disease-Linked D620n Vps35 Knockin Mice Manifest Tau Neuropathology and Dopaminergic Neurodegeneration. Proc. Natl. Acad. Sci. USA 2019, 116, 5765–5774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brundin, P.; Nath, A.; Beckham, J.D. Is Covid-19 a Perfect Storm for Parkinson’s Disease? Trends Neurosci. 2020, 43, 931–933. [Google Scholar] [CrossRef] [PubMed]
- Monje, M.H.G.; Martinez-Fernandez, R. Severe Acute Respiratory Syndrome Coronavirus 2 Infection and Parkinsonism: Is There Evidence for Concern? Mov. Disord. 2020, 35, 1725. [Google Scholar] [CrossRef]
- Cohen, M.E.; Eichel, R.; Steiner-Birmanns, B.; Janah, A.; Ioshpa, M.; Bar-Shalom, R.; Paul, J.J.; Gaber, H.; Skrahina, V.; Bornstein, N.M.; et al. A Case of Probable Parkinson’s Disease after Sars-Cov-2 Infection. Lancet Neurol. 2020, 19, 804–805. [Google Scholar] [CrossRef]
- Hawkes, C.H.; Del Tredici, K.; Braak, H. Parkinson’s Disease: A Dual-Hit Hypothesis. Neuropathol. Appl. Neurobiol. 2007, 33, 599–614. [Google Scholar] [CrossRef]
- Johnson, M.E.; Stecher, B.; Labrie, V.; Brundin, L.; Brundin, P. Triggers, Facilitators, Aggravators: Redefining Parkinson’s Disease Pathogenesis. Trends Neurosci. 2019, 42, 4–13. [Google Scholar] [CrossRef] [Green Version]
- Olsen, L.K.; Dowd, E.; McKernan, D.P. A Role for Viral Infections in Parkinson’s Etiology? Neuronal Signal. 2018, 2, NS20170166. [Google Scholar] [CrossRef]
- Jang, H.; Boltz, D.; Sturm-Ramirez, K.; Shepherd, K.R.; Jiao, Y.; Webster, R.; Smeyne, R.J. Highly Pathogenic H5n1 Influenza Virus Can Enter the Central Nervous System and Induce Neuroinflammation and Neurodegeneration. Proc. Natl. Acad. Sci. USA 2009, 106, 14063–14068. [Google Scholar] [CrossRef] [Green Version]
- Bantle, C.M.; Phillips, A.T.; Smeyne, R.J.; Rocha, S.M.; Olson, K.E.; Tjalkens, R.B. Infection with Mosquito-Borne Alphavirus Induces Selective Loss of Dopaminergic Neurons, Neuroinflammation and Widespread Protein Aggregation. NPJ Parkinsons Dis. 2019, 5, 20. [Google Scholar] [CrossRef] [Green Version]
- Marreiros, R.; Müller-Schiffmann, A.; Trossbach, S.V.; Prikulis, I.; Hänsch, S.; Weidtkamp-Peters, S.; Moreira, A.R.; Sahu, S.; Soloviev, I.; Selvarajah, S.; et al. Disruption of Cellular Proteostasis by H1n1 Influenza a Virus Causes Alpha-Synuclein Aggregation. Proc. Natl. Acad. Sci. USA 2020, 117, 6741–6751. [Google Scholar] [CrossRef]
- Smeyne, R.J.; Noyce, A.J.; Byrne, M.; Savica, R.; Marras, C. Infection and Risk of Parkinson’s Disease. J. Parkinsons Dis. 2021, 11, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Kupzig, S.; Korolchuk, V.; Rollason, R.; Sugden, A.; Wilde, A.; Banting, G. Bst-2/Hm1.24 Is a Raft-Associated Apical Membrane Protein with an Unusual Topology. Traffic 2003, 4, 694–709. [Google Scholar] [CrossRef]
- Neil, S.J.; Zang, T.; Bieniasz, P.D. Tetherin Inhibits Retrovirus Release and Is Antagonized by Hiv-1 Vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef] [Green Version]
- Sauter, D. Counteraction of the Multifunctional Restriction Factor Tetherin. Front. Microbiol. 2014, 5, 163. [Google Scholar] [CrossRef] [PubMed]
- Rollason, R.; Korolchuk, V.; Hamilton, C.; Schu, P.; Banting, G. Clathrin-Mediated Endocytosis of a Lipid-Raft-Associated Protein Is Mediated through a Dual Tyrosine Motif. J. Cell Sci. 2007, 120, 3850–3858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arighi, C.N.; Hartnell, L.M.; Aguilar, R.C.; Haft, C.R.; Bonifacino, J.S. Role of the Mammalian Retromer in Sorting of the Cation-Independent Mannose 6-Phosphate Receptor. J. Cell Biol. 2004, 165, 123–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Weng, M.; Ke, Y.; Sapp, E.; DiFiglia, M.; Li, X. Kalirin Interacts with Trapp and Regulates Rab11 and Endosomal Recycling. Cells 2020, 9, 1132. [Google Scholar] [CrossRef]
- Groppelli, E.; Len, A.C.; Granger, L.A.; Jolly, C. Retromer Regulates Hiv-1 Envelope Glycoprotein Trafficking and Incorporation into Virions. PLoS Pathog. 2014, 10, e1004518. [Google Scholar] [CrossRef]
- Lipovsky, A.; Popa, A.; Pimienta, G.; Wyler, M.; Bhan, A.; Kuruvilla, L.; Guie, M.A.; Poffenberger, A.C.; Nelson, C.D.; Atwood, W.J.; et al. Genome-Wide Sirna Screen Identifies the Retromer as a Cellular Entry Factor for Human Papillomavirus. Proc. Natl. Acad. Sci. USA 2013, 110, 7452–7457. [Google Scholar] [CrossRef] [Green Version]
- Popa, A.; Zhang, W.; Harrison, M.S.; Goodner, K.; Kazakov, T.; Goodwin, E.C.; Lipovsky, A.; Burd, C.G.; DiMaio, D. Direct Binding of Retromer to Human Papillomavirus Type 16 Minor Capsid Protein L2 Mediates Endosome Exit During Viral Infection. PLoS Pathog. 2015, 11, e1004699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, P.; Hong, Z.; Yang, X.; Chung, R.T.; Zhang, L. A Role for Retromer in Hepatitis C Virus Replication. Cell Mol. Life Sci. 2016, 73, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Duarte, L.F.; Farias, M.A.; Alvarez, D.M.; Bueno, S.M.; Riedel, C.A.; Gonzalez, P.A. Herpes Simplex Virus Type 1 Infection of the Central Nervous System: Insights into Proposed Interrelationships with Neurodegenerative Disorders. Front. Cell Neurosci. 2019, 13, 46. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Kaloyanova, D.; van Eijk, M.; Eerland, R.; van der Goot, G.; Oorschot, V.; Klumperman, J.; Lottspeich, F.; Starkuviene, V.; Wieland, F.T.; et al. Involvement of a Golgi-Resident Gpi-Anchored Protein in Maintenance of the Golgi Structure. Mol. Biol. Cell 2007, 18, 1261–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Sullivan, M.J.; Lindsay, A.J. The endosomal recycling pathway-at the crossroads of the cell. Int. J. Mol. Sci. 2020, 21, 6074. [Google Scholar] [CrossRef]
- D’Souza-Schorey, C.; Chavrier, P. Arf proteins: Roles in membrane traffic and beyond. Nat. Rev. Mol. Cell Biol. 2006, 7, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, O.; Reinsch, S.; Urbe, S.; Zerial, M.; Parton, R.G. Rab11 Regulates Recycling through the Pericentriolar Recycling Endosome. J. Cell Biol. 1996, 135, 913–924. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Bembenek, J.N. Protease Dead Separase Inhibits Chromosome Segregation and Rab-11 Vesicle Trafficking. Cell Cycle 2017, 16, 1902–1917. [Google Scholar] [CrossRef] [Green Version]
- Gambardella, S.; Biagioni, F.; Ferese, R.; Busceti, C.L.; Frati, A.; Novelli, G.; Ruggieri, S.; Fornai, F. vacuolar protein sorting genes in parkinson’s disease: A re-appraisal of mutations detection rate and neurobiology of disease. Front. Neurosci. 2016, 10, 532. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Ioannidis, J.P.; Aasly, J.O.; Annesi, G.; Brice, A.; Bertram, L.; Bozi, M.; Barcikowska, M.; Crosiers, D.; Clarke, C.E.; et al. Geopd consortium, A Multi-Centre Clinico-Genetic Analysis of the Vps35 Gene in Parkinson Disease Indicates Reduced Penetrance for Disease-Associated Variants. J. Med. Genet. 2012, 49, 721–726. [Google Scholar] [CrossRef] [Green Version]
- Dafa-Berger, A.; Kuzmina, A.; Fassler, M.; Yitzhak-Asraf, H.; Shemer-Avni, Y.; Taube, R. Modulation of Hepatitis C Virus Release by the Interferon-Induced Protein Bst-2/Tetherin. Virology 2012, 428, 98–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Sancho, L.; Lewinski, M.K.; Pache, L.; Stoneham, C.A.; Yin, X.; Pratt, D.; Churas, C.; Rosenthal, S.B.; Liu, S.; De Jesus, P.D.; et al. Functional Landscape of Sars-Cov-2 Cellular Restriction. bioRxiv 2020. [Google Scholar] [CrossRef]
- Blondeau, C.; Pelchen-Matthews, A.; Mlcochova, P.; Marsh, M.; Milne, R.S.; Towers, G.J. Tetherin Restricts Herpes Simplex Virus 1 and Is Antagonized by Glycoprotein M. J. Virol. 2013, 87, 13124–13133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, I.; Wilson, D.W. HSV-1 Cytoplasmic Envelopment and Egress. Int. J. Mol Sci. 2020, 21, 5969. [Google Scholar] [CrossRef]
- Beitia Ortiz de Zarate, I.; Cantero-Aguilar, L.; Longo, M.; Berlioz-Torrent, C.; Rozenberg, F. Contribution of Endocytic Motifs in the Cytoplasmic Tail of Herpes Simplex Virus Type 1 Glycoprotein B to Virus Replication and Cell-Cell Fusion. J. Virol. 2007, 81, 13889–13903. [Google Scholar] [CrossRef] [Green Version]
- Munsie, L.N.; Milnerwood, A.J.; Seibler, P.; Beccano-Kelly, D.A.; Tatarnikov, I.; Khinda, J.; Volta, M.; Kadgien, C.; Cao, L.P.; Tapia, L.; et al. Retromer-Dependent Neurotransmitter Receptor Trafficking to Synapses Is Altered by the Parkinson’s Disease Vps35 Mutation P.D620n. Hum. Mol. Genet. 2015, 24, 1691–1703. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, Y.; Li, Y.; Chhetri, G.; Peng, X.; Wu, J.; Wang, Z.; Zhao, B.; Zhao, W.; Li, X. Parkinson’s Disease Causative Mutation in Vps35 Disturbs Tetherin Trafficking to Cell Surfaces and Facilitates Virus Spread. Cells 2021, 10, 746. https://doi.org/10.3390/cells10040746
Ding Y, Li Y, Chhetri G, Peng X, Wu J, Wang Z, Zhao B, Zhao W, Li X. Parkinson’s Disease Causative Mutation in Vps35 Disturbs Tetherin Trafficking to Cell Surfaces and Facilitates Virus Spread. Cells. 2021; 10(4):746. https://doi.org/10.3390/cells10040746
Chicago/Turabian StyleDing, Yingzhuo, Yan Li, Gaurav Chhetri, Xiaoxin Peng, Jing Wu, Zejian Wang, Bo Zhao, Wenjuan Zhao, and Xueyi Li. 2021. "Parkinson’s Disease Causative Mutation in Vps35 Disturbs Tetherin Trafficking to Cell Surfaces and Facilitates Virus Spread" Cells 10, no. 4: 746. https://doi.org/10.3390/cells10040746
APA StyleDing, Y., Li, Y., Chhetri, G., Peng, X., Wu, J., Wang, Z., Zhao, B., Zhao, W., & Li, X. (2021). Parkinson’s Disease Causative Mutation in Vps35 Disturbs Tetherin Trafficking to Cell Surfaces and Facilitates Virus Spread. Cells, 10(4), 746. https://doi.org/10.3390/cells10040746