
Assembly and Entry of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV2): Evaluation Using Virus-Like Particles


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Culture Conditions
2.2. Reagents and Antibodies
2.3. Generation of Expression Plasmids
2.4. VLP Production and Purification
2.5. Western Immunoblot Analysis
2.6. Binding and Entry Assay of SARS-CoV2 VLPs
2.7. Statistical Analysis
3. Results
3.1. Determinants of SARS-CoV2 VLP Assembly
3.2. Nucleocapsid Drives Efficient SARS-CoV2 VLP Production
3.3. Nucleocapsid Carboxyl-Terminal Domains (CTDs) Drive Efficient VLP Production
3.4. A SARS-CoV2 Nucleocapsid Fragment Interferes with VLP Production
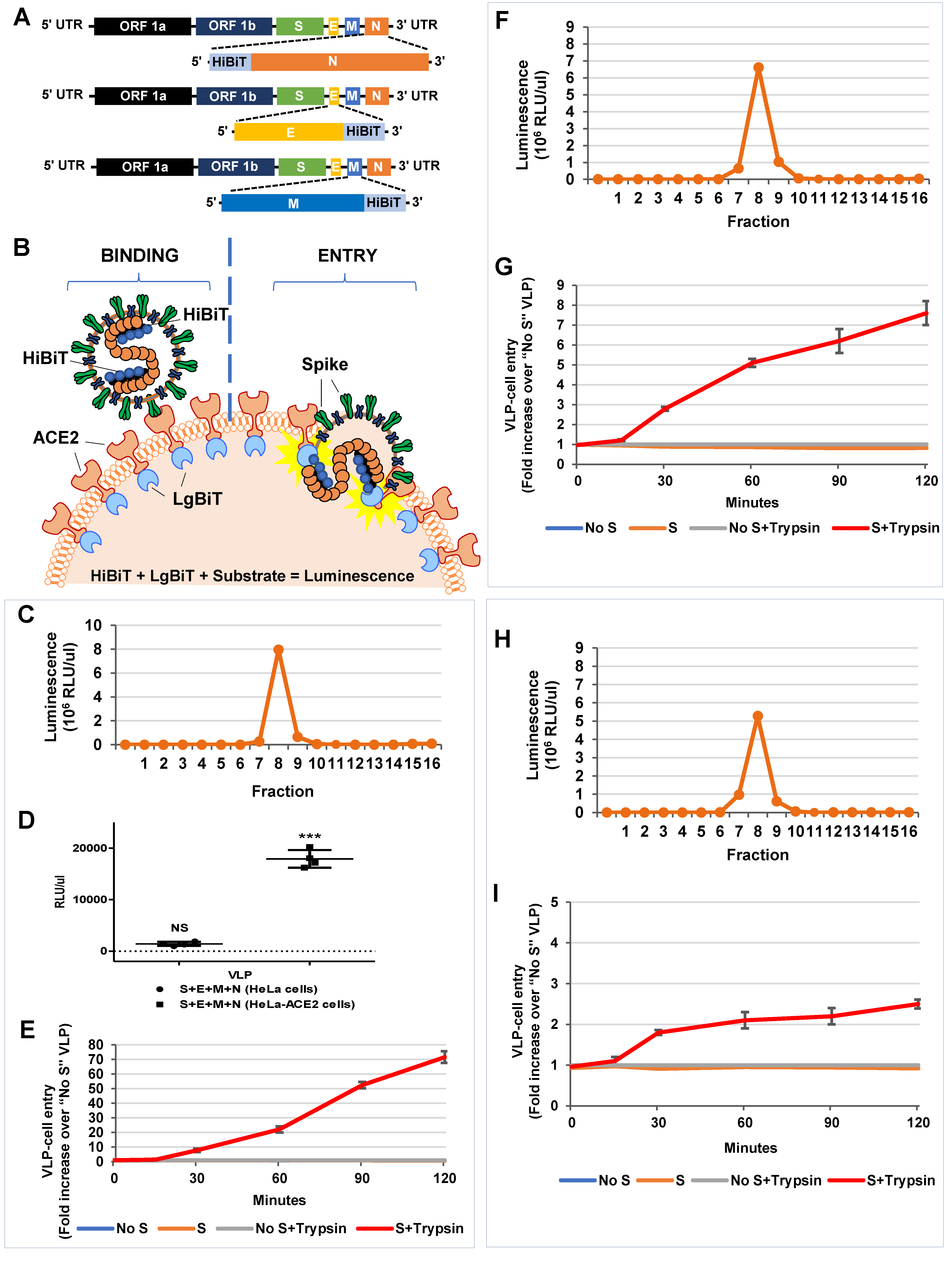
3.5. HiBiT-Tagged SARS-CoV2 VLPs Assess Virus Binding and Entry Events
4. Discussion and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gerna, G.; Campanini, G.; Rovida, F.; Percivalle, E.; Sarasini, A.; Marchi, A.; Baldanti, F. Genetic variability of human coronavirus OC43-, 229E-, and NL63-like strains and their association with lower respiratory tract infections of hospitalized infants and immunocompromised patients. J. Med. Virol. 2006, 78, 938–949. [Google Scholar] [CrossRef]
- Woo, P.C.; Lau, S.K.; Huang, Y.; Yuen, K.Y. Coronavirus diversity, phylogeny and interspecies jumping. Exp. Biol. Med. (Maywood) 2009, 234, 1117–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, J.; Li, F.; Shi, Z.-L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ksiazek, T.G.; Erdman, D.; Goldsmith, C.S.; Zaki, S.R.; Peret, T.; Emery, S.; Tong, S.; Urbani, C.; Comer, J.A.; Lim, W.; et al. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1953–1966. [Google Scholar] [CrossRef] [PubMed]
- Zaki, A.M.; van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.; Fouchier, R.A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.; Lau, S.K.; Li, K.S.; Poon, R.W.; Wong, B.H.; Tsoi, H.W.; Yip, B.C.; Huang, Y.; Chan, K.H.; Yuen, K.Y. Molecular diversity of coronaviruses in bats. Virology 2006, 351, 180–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. WHO Coronavirus Disease (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 28 January 2021).
- De Haan, C.A.M.; Vennema, H.; Rottier, P.J.M. Assembly of the Coronavirus Envelope: Homotypic Interactions between the M Proteins. J. Virol. 2000, 74, 4967–4978. [Google Scholar] [CrossRef]
- Krijnse-Locker, J.; Ericsson, M.; Rottier, P.J.; Griffiths, G. Characterization of the budding compartment of mouse hepatitis virus: Evidence that transport from the RER to the Golgi complex requires only one vesicular transport step. J. Cell Biol. 1994, 124, 55–70. [Google Scholar] [CrossRef] [Green Version]
- Fischer, F.; Stegen, C.F.; Masters, P.S.; Samsonoff, W.A. Analysis of Constructed E Gene Mutants of Mouse Hepatitis Virus Confirms a Pivotal Role for E Protein in Coronavirus Assembly. J. Virol. 1998, 72, 7885–7894. [Google Scholar] [CrossRef] [Green Version]
- DeDiego, M.L.; Alvarez, E.; Almazán, F.; Rejas, M.T.; Lamirande, E.; Roberts, A.; Shieh, W.J.; Zaki, S.R.; Subbarao, K.; Enjuanes, L. A severe acute respiratory syndrome coronavirus that lacks the E gene is attenuated in vitro and in vivo. J. Virol. 2007, 81, 1701–1713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortego, J.; Escors, D.; Laude, H.; Enjuanes, L. Generation of a replication-competent, propagation-deficient virus vector based on the transmissible gastroenteritis coronavirus genome. J. Virol. 2002, 76, 11518–11529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madan, V.; García Mde, J.; Sanz, M.A.; Carrasco, L. Viroporin activity of murine hepatitis virus E protein. FEBS Lett. 2005, 579, 3607–3612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, L.; McKinlay, C.; Gage, P.; Ewart, G. SARS coronavirus E protein forms cation-selective ion channels. Virology 2004, 330, 322–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boscarino, J.A.; Logan, H.L.; Lacny, J.J.; Gallagher, T.M. Envelope protein palmitoylations are crucial for murine coronavirus assembly. J. Virol. 2008, 82, 2989–2999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westerbeck, J.W.; Machamer, C.E. The Infectious Bronchitis Coronavirus Envelope Protein Alters Golgi pH To Protect the Spike Protein and Promote the Release of Infectious Virus. J. Virol. 2019, 93, e00015–e00019. [Google Scholar] [CrossRef] [Green Version]
- Venkatagopalan, P.; Daskalova, S.M.; Lopez, L.A.; Dolezal, K.A.; Hogue, B.G. Coronavirus envelope (E) protein remains at the site of assembly. Virology 2015, 478, 75–85. [Google Scholar] [CrossRef]
- Bos, E.C.; Luytjes, W.; van der Meulen, H.V.; Koerten, H.K.; Spaan, W.J. The production of recombinant infectious DI-particles of a murine coronavirus in the absence of helper virus. Virology 1996, 218, 52–60. [Google Scholar] [CrossRef]
- Corse, E.; Machamer, C.E. The cytoplasmic tails of infectious bronchitis virus E and M proteins mediate their interaction. Virology 2003, 312, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Corse, E.; Machamer, C.E. Infectious Bronchitis Virus E Protein Is Targeted to the Golgi Complex and Directs Release of Virus-Like Particles. J. Virol. 2000, 74, 4319–4326. [Google Scholar] [CrossRef] [Green Version]
- Baudoux, P.; Carrat, C.; Besnardeau, L.; Charley, B.; Laude, H. Coronavirus Pseudoparticles Formed with Recombinant M and E Proteins Induce Alpha Interferon Synthesis by Leukocytes. J. Virol. 1998, 72, 8636–8643. [Google Scholar] [CrossRef] [Green Version]
- Kuo, L.; Masters, P.S. Genetic evidence for a structural interaction between the carboxy termini of the membrane and nucleocapsid proteins of mouse hepatitis virus. J. Virol. 2002, 76, 4987–4999. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-K.; Hou, M.-H.; Chang, C.-F.; Hsiao, C.-D.; Huang, T.-H. The SARS coronavirus nucleocapsid protein--forms and functions. Antivir. Res. 2014, 103, 39–50. [Google Scholar] [CrossRef]
- Chang, C.K.; Sue, S.C.; Yu, T.H.; Hsieh, C.M.; Tsai, C.K.; Chiang, Y.C.; Lee, S.J.; Hsiao, H.H.; Wu, W.J.; Chang, W.L.; et al. Modular organization of SARS coronavirus nucleocapsid protein. J. Biomed. Sci. 2006, 13, 59–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayaram, H.; Fan, H.; Bowman, B.R.; Ooi, A.; Jayaram, J.; Collisson, E.W.; Lescar, J.; Prasad, B.V. X-ray structures of the N- and C-terminal domains of a coronavirus nucleocapsid protein: Implications for nucleocapsid formation. J. Virol. 2006, 80, 6612–6620. [Google Scholar] [CrossRef] [Green Version]
- Kuo, L.; Koetzner, C.A.; Hurst, K.R.; Masters, P.S. Recognition of the murine coronavirus genomic RNA packaging signal depends on the second RNA-binding domain of the nucleocapsid protein. J. Virol. 2014, 88, 4451–4465. [Google Scholar] [CrossRef] [Green Version]
- Schwinn, M.K.; Machleidt, T.; Zimmerman, K.; Eggers, C.T.; Dixon, A.S.; Hurst, R.; Hall, M.P.; Encell, L.P.; Binkowski, B.F.; Wood, K.V. CRISPR-Mediated Tagging of Endogenous Proteins with a Luminescent Peptide. ACS Chem. Biol. 2018, 13, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Qing, E.; Hantak, M.P.; Galpalli, G.G.; Gallagher, T. Evaluating MERS-CoV Entry Pathways. Methods Mol. Biol. 2020, 2099, 9–20. [Google Scholar]
- Siu, Y.L.; Teoh, K.T.; Lo, J.; Chan, C.M.; Kien, F.; Escriou, N.; Tsao, S.W.; Nicholls, J.M.; Altmeyer, R.; Peiris, J.S.M.; et al. The M, E, and N Structural Proteins of the Severe Acute Respiratory Syndrome Coronavirus Are Required for Efficient Assembly, Trafficking, and Release of Virus-Like Particles. J. Virol. 2008, 82, 11318–11330. [Google Scholar] [CrossRef] [Green Version]
- Xu, R.; Shi, M.; Li, J.; Song, P.; Li, N. Construction of SARS-CoV-2 Virus-Like Particles by Mammalian Expression System. Front. Bioeng. Biotechnol. 2020, 8, 862. [Google Scholar] [CrossRef] [PubMed]
- Plescia, C.B.; David, E.A.; Patra, D.; Sengupta, R.; Amiar, S.; Su, Y.; Stahelin, R.V. SARS-CoV-2 viral budding and entry can be modeled using BSL-2 level virus-like particles. J. Biol. Chem. 2021, 296, 100103. [Google Scholar] [CrossRef] [PubMed]
- Mortola, E.; Roy, P. Efficient assembly and release of SARS coronavirus-like particles by a heterologous expression system. FEBS Lett. 2004, 576, 174–178. [Google Scholar] [CrossRef] [Green Version]
- Qing, E.; Hantak, M.; Perlman, S.; Gallagher, T. Distinct Roles for Sialoside and Protein Receptors in Coronavirus Infection. mBio 2020, 11, e02764-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, H.; Chen, J.; Chen, K.; Shen, X.; Jiang, H. Carboxyl terminus of severe acute respiratory syndrome coronavirus nucleocapsid protein: Self-association analysis and nucleic acid binding characterization. Biochemistry 2006, 45, 11827–11835. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-Y.; Chang, C.-K.; Chang, Y.-W.; Sue, S.-C.; Bai, H.-I.; Riang, L.; Hsiao, C.-D.; Huang, T.-H. Structure of the SARS coronavirus nucleocapsid protein RNA-binding dimerization domain suggests a mechanism for helical packaging of viral RNA. J. Mol. Biol. 2007, 368, 1075–1086. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Anindita, P.D.; Phongphaew, W.; Carr, M.; Kobayashi, S.; Orba, Y.; Sawa, H. Development of a rapid and quantitative method for the analysis of viral entry and release using a NanoLuc luciferase complementation assay. Virus Res. 2018, 243, 69–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- McBride, R.; van Zyl, M.; Fielding, B.C. The coronavirus nucleocapsid is a multifunctional protein. Viruses 2014, 6, 2991–3018. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Pan, J.a.; Tao, J.; Guo, D. SARS-CoV nucleocapsid protein antagonizes IFN-β response by targeting initial step of IFN-β induction pathway, and its C-terminal region is critical for the antagonism. Virus Genes 2011, 42, 37–45. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Hauns, K.; Langland, J.O.; Jacobs, B.L.; Hogue, B.G. Mouse hepatitis coronavirus A59 nucleocapsid protein is a type I interferon antagonist. J. Virol. 2007, 81, 2554–2563. [Google Scholar] [CrossRef] [Green Version]
- Surjit, M.; Liu, B.; Chow, V.T.; Lal, S.K. The nucleocapsid protein of severe acute respiratory syndrome-coronavirus inhibits the activity of cyclin-cyclin-dependent kinase complex and blocks S phase progression in mammalian cells. J. Biol. Chem. 2006, 281, 10669–10681. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Zhang, H.; Zhang, Q.; Huang, Y.; Dong, J.; Liang, Y.; Liu, H.-J.; Tong, D. Porcine epidemic diarrhea virus N protein prolongs S-phase cell cycle, induces endoplasmic reticulum stress, and up-regulates interleukin-8 expression. Vet. Microbiol. 2013, 164, 212–221. [Google Scholar] [CrossRef] [PubMed]
- De Haan, C.A.M.; Rottier, P.J.M. Molecular interactions in the assembly of coronaviruses. Adv. Virus Res. 2005, 64, 165–230. [Google Scholar] [PubMed]
- Vennema, H.; Godeke, G.J.; Rossen, J.W.; Voorhout, W.F.; Horzinek, M.C.; Opstelten, D.J.; Rottier, P.J. Nucleocapsid-independent assembly of coronavirus-like particles by co-expression of viral envelope protein genes. EMBO J. 1996, 15, 2020–2028. [Google Scholar] [CrossRef] [PubMed]
- Takeda, M.; Chang, C.-k.; Ikeya, T.; Güntert, P.; Chang, Y.-h.; Hsu, Y.-l.; Huang, T.-h.; Kainosho, M. Solution Structure of the C-terminal Dimerization Domain of SARS Coronavirus Nucleocapsid Protein Solved by the SAIL-NMR Method. J. Mol. Biol. 2008, 380, 608–622. [Google Scholar] [CrossRef]
- Peng, T.-Y.; Lee, K.-R.; Tarn, W.-Y. Phosphorylation of the arginine/serine dipeptide-rich motif of the severe acute respiratory syndrome coronavirus nucleocapsid protein modulates its multimerization, translation inhibitory activity and cellular localization. FEBS J. 2008, 275, 4152–4163. [Google Scholar] [CrossRef] [Green Version]
- Hurst, K.R.; Kuo, L.; Koetzner, C.A.; Ye, R.; Hsue, B.; Masters, P.S. A major determinant for membrane protein interaction localizes to the carboxy-terminal domain of the mouse coronavirus nucleocapsid protein. J. Virol. 2005, 79, 13285–13297. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Wu, D.; Shen, C.; Chen, K.; Shen, X.; Jiang, H. Severe acute respiratory syndrome coronavirus membrane protein interacts with nucleocapsid protein mostly through their carboxyl termini by electrostatic attraction. Int. J. Biochem. Cell Biol. 2006, 38, 589–599. [Google Scholar] [CrossRef]
- Zhou, R.; Zeng, R.; von Brunn, A.; Lei, J. Structural characterization of the C-terminal domain of SARS-CoV-2 nucleocapsid protein. Mol. Biomed. 2020, 1, 2. [Google Scholar] [CrossRef]
- Kuo, L.; Hurst-Hess, K.R.; Koetzner, C.A.; Masters, P.S. Analyses of Coronavirus Assembly Interactions with Interspecies Membrane and Nucleocapsid Protein Chimeras. J. Virol. 2016, 90, 4357–4368. [Google Scholar] [CrossRef] [Green Version]
- Huang, Q.; Yu, L.; Petros, A.M.; Gunasekera, A.; Liu, Z.; Xu, N.; Hajduk, P.; Mack, J.; Fesik, S.W.; Olejniczak, E.T. Structure of the N-terminal RNA-binding domain of the SARS CoV nucleocapsid protein. Biochemistry 2004, 43, 6059–6063. [Google Scholar] [CrossRef] [PubMed]
- Rolta, R.; Yadav, R.; Salaria, D.; Trivedi, S.; Imran, M.; Sourirajan, A.; Baumler, D.J.; Dev, K. In silico screening of hundred phytocompounds of ten medicinal plants as potential inhibitors of nucleocapsid phosphoprotein of COVID-19: An approach to prevent virus assembly. J. Biomol. Struct. Dyn. 2020, 1–18. [Google Scholar] [CrossRef]
- Gupta, M.K.; Vemula, S.; Donde, R.; Gouda, G.; Behera, L.; Vadde, R. In-silico approaches to detect inhibitors of the human severe acute respiratory syndrome coronavirus envelope protein ion channel. J. Biomol. Struct. Dyn. 2020, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Debica, M.; Upasana, R. SARS-CoV-2 Nucleocapsid Assembly Inhibitors: Repurposing Antiviral and Antimicrobial Drugs Targeting Nucleocapsid-RNA Interaction. ChemRxiv 2020. Available online: https://europepmc.org/article/ppr/ppr186727 (accessed on 8 April 2021). [CrossRef]
- Bhowmik, D.; Nandi, R.; Jagadeesan, R.; Kumar, N.; Prakash, A.; Kumar, D. Identification of potential inhibitors against SARS-CoV-2 by targeting proteins responsible for envelope formation and virion assembly using docking based virtual screening, and pharmacokinetics approaches. Infection, genetics and evolution. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2020, 84, 104451. [Google Scholar] [CrossRef]
- Liang, X.Y.; Zhu, Q.C.; Liang, J.Q.; Liu, S.Y.; Liu, D.X.; Fung, T.S. Development of HiBiT-Tagged Recombinant Infectious Bronchitis Coronavirus for Efficient in vitro and in vivo Viral Quantification. Front. Microbiol. 2020, 11, 2100. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, B.; Hawkins, G.M.; Kicmal, T.; Qing, E.; Timm, E.; Gallagher, T. Assembly and Entry of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV2): Evaluation Using Virus-Like Particles. Cells 2021, 10, 853. https://doi.org/10.3390/cells10040853
Kumar B, Hawkins GM, Kicmal T, Qing E, Timm E, Gallagher T. Assembly and Entry of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV2): Evaluation Using Virus-Like Particles. Cells. 2021; 10(4):853. https://doi.org/10.3390/cells10040853
Chicago/Turabian StyleKumar, Binod, Grant M. Hawkins, Tom Kicmal, Enya Qing, Emily Timm, and Tom Gallagher. 2021. "Assembly and Entry of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV2): Evaluation Using Virus-Like Particles" Cells 10, no. 4: 853. https://doi.org/10.3390/cells10040853
APA StyleKumar, B., Hawkins, G. M., Kicmal, T., Qing, E., Timm, E., & Gallagher, T. (2021). Assembly and Entry of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV2): Evaluation Using Virus-Like Particles. Cells, 10(4), 853. https://doi.org/10.3390/cells10040853