
Transcriptional Regulation of Thrombin-Induced Endothelial VEGF Induction and Proangiogenic Response

 ,
,  , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Endothelial Cell Culture, Tube Formation Assay, and VEGF Protein and Gene Expression
2.3. DNA Construct, Transient Transfection, and Analysis of the VEGF Promoter
2.4. Nuclear Extracts and Electrophoretic Mobility Shift Assay
2.5. Western Blotting Assays and Flow Cytometry on PAR-1 Expression and Internalization
2.6. Statistics
3. Results
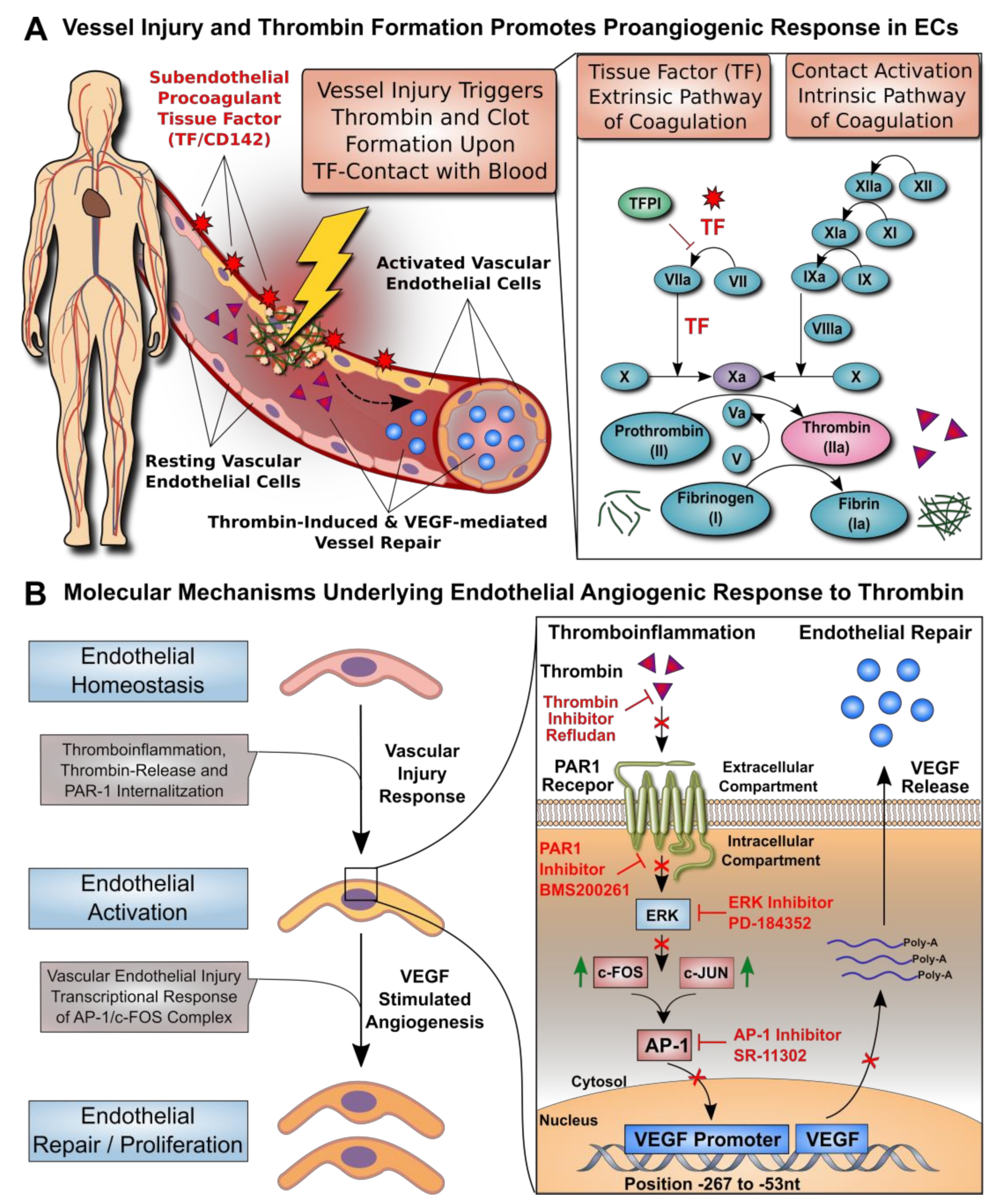
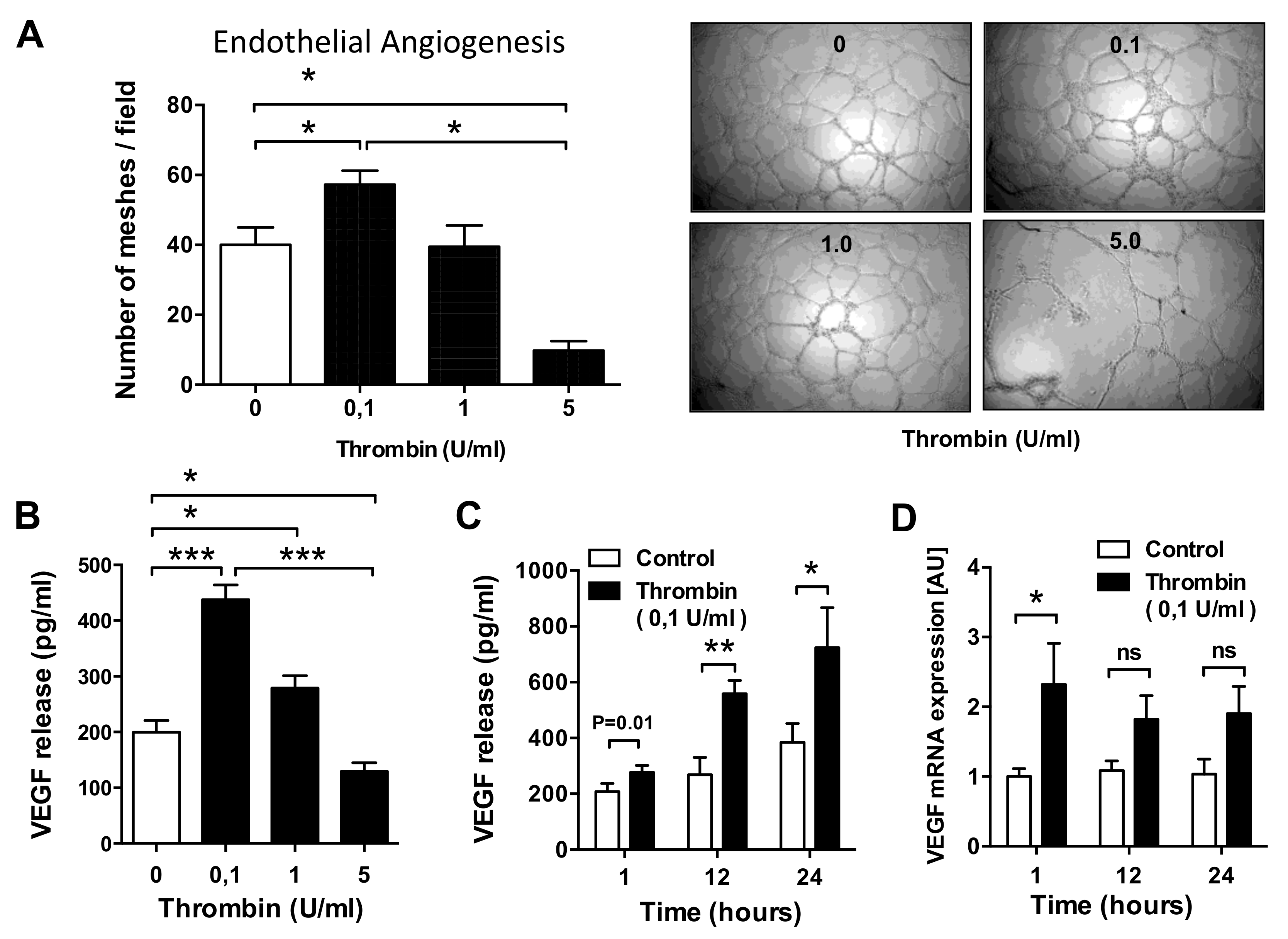
3.1. Dose–Response Effect of Thrombin on EC Angiogenesis and VEGF Production
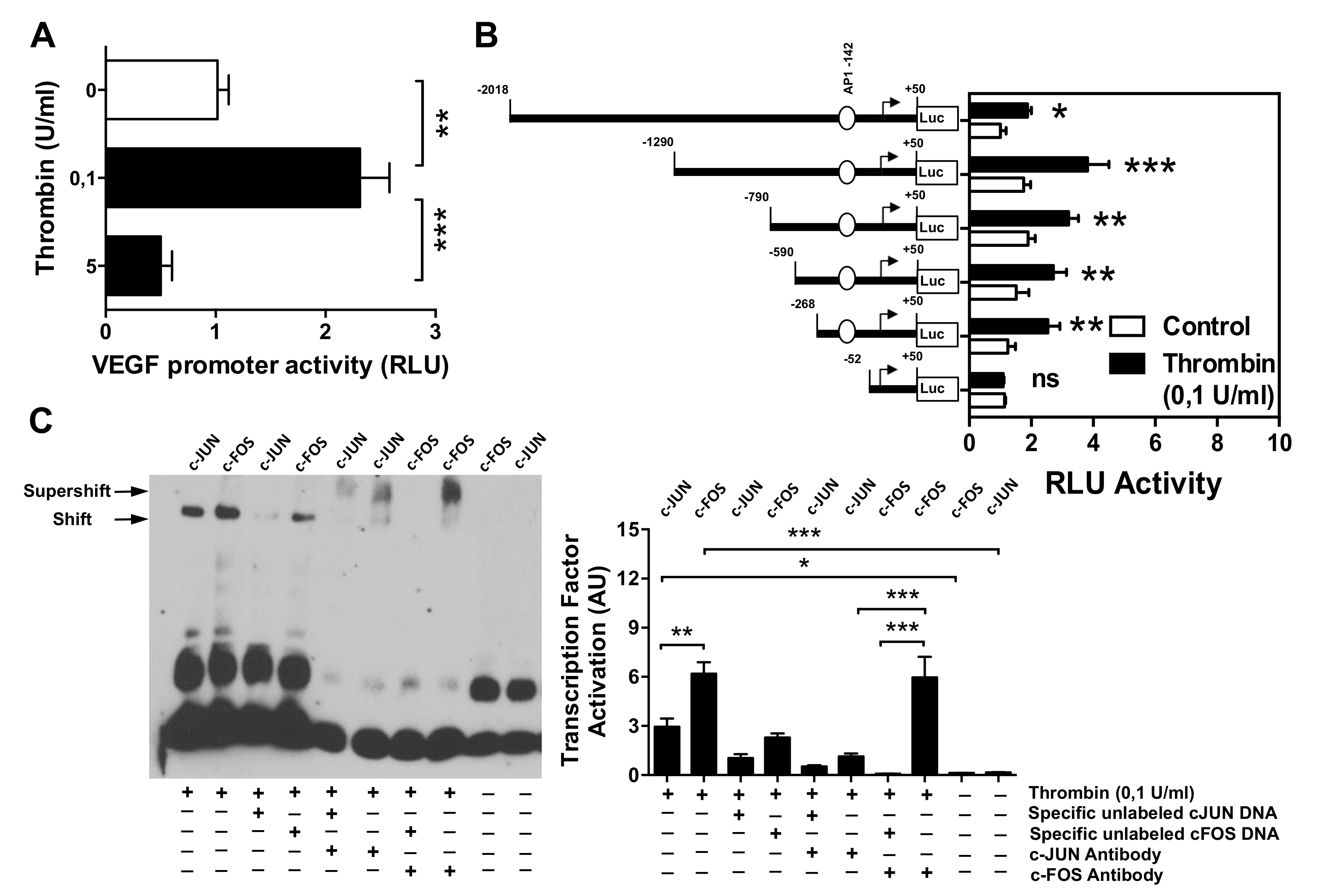
3.2. Effect of Thrombin on the VEGF Promoter Activity in Microvascular Endothelial Cells
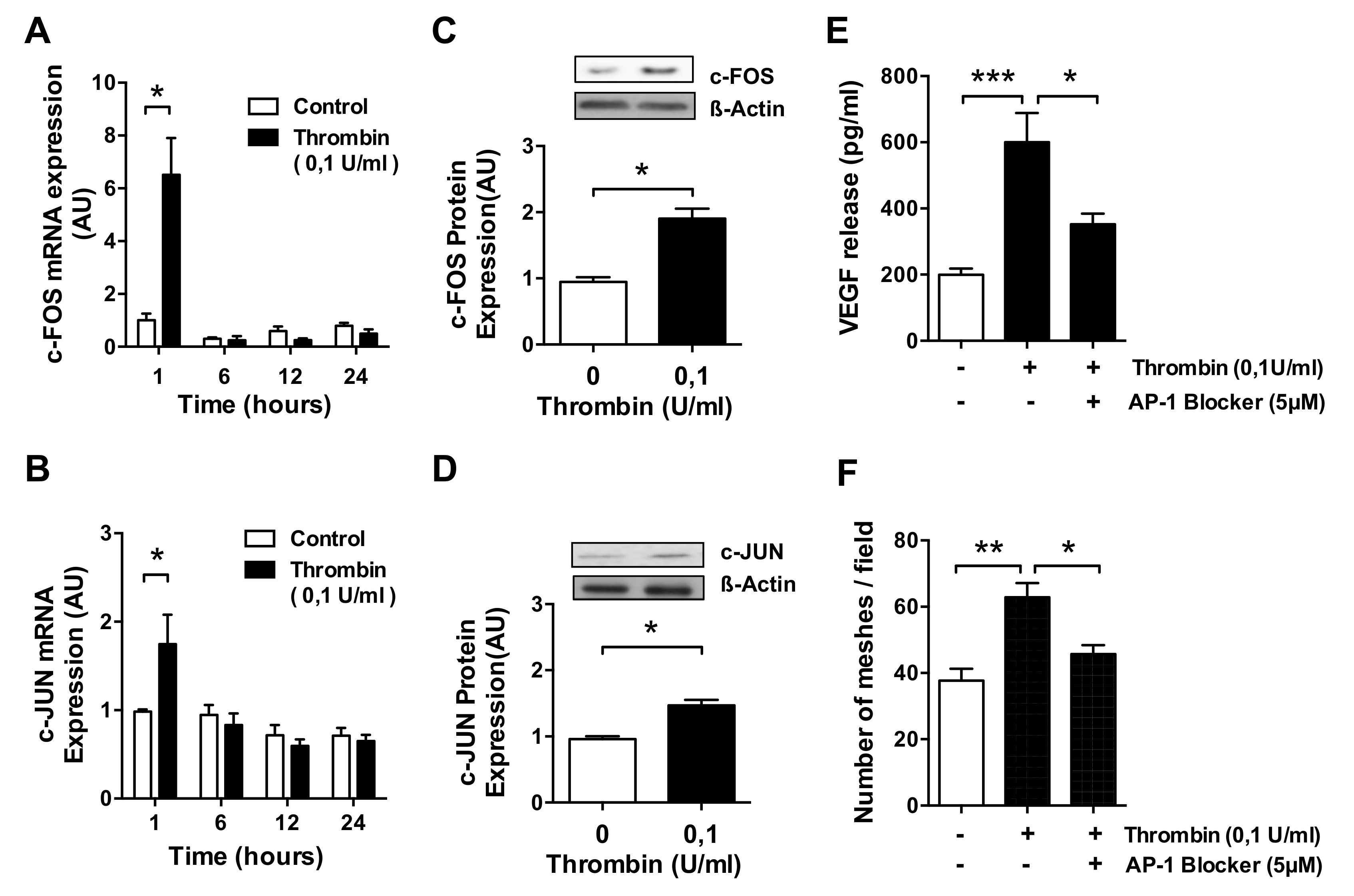
3.3. AP-1 Transcription Factor Mediates Thrombin-Induced EC Angiogenesis
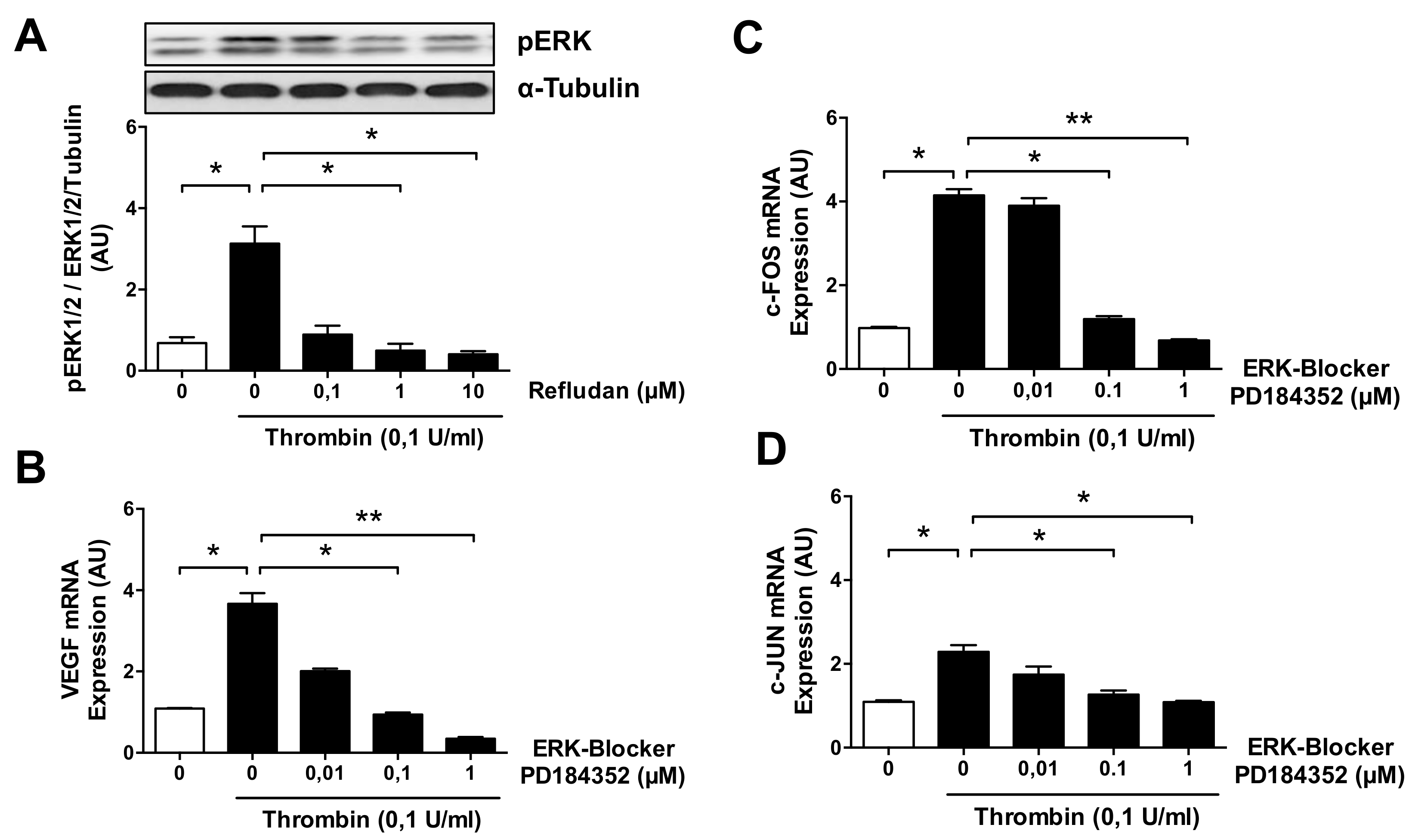
3.4. ERK Blockade Impairs Thrombin-Induced VEGF Expression by ECs
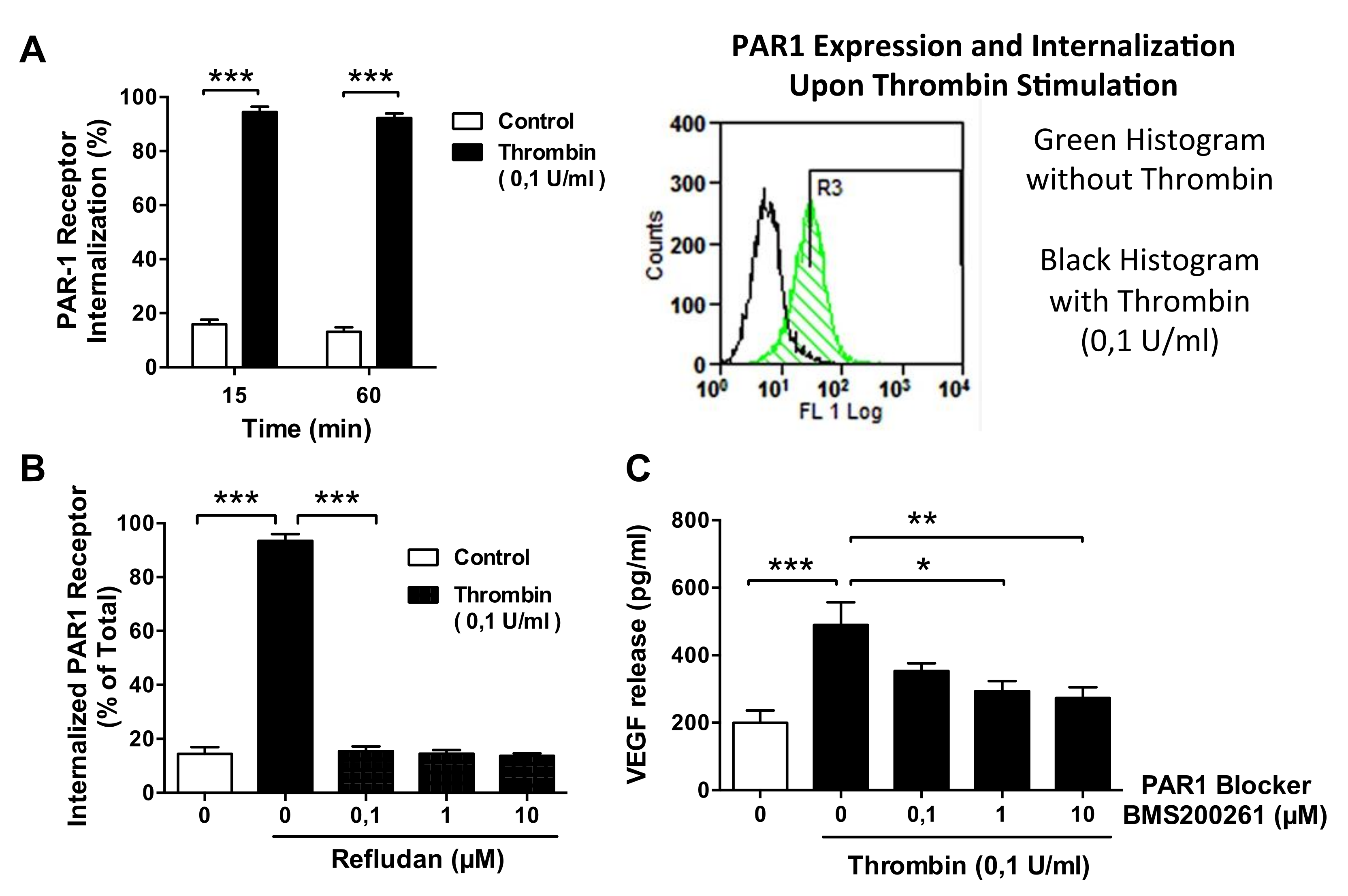
3.5. Thrombin-Induced PAR-1 Activation Modulates VEGF Expression by ECs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ARDS | Acute respiratory distress syndrome |
| COVID-19 | Coronavirus-induced disease 2019 |
| ECs / HMECs | Endothelial cells / Human microvascular endothelial cells |
| AP-1 | Activator protein 1 transcription factor complex composed of subunits c-FOS and c-JUN |
| ERK1/2 | Extracellular signal-regulated kinases 1 and 2 |
| PAR-1 | Protease activated receptor 1, thrombin receptor |
| TF/CD142 | Tissue factor, highly procoagulant molecule triggers the TF pathway of coagulation |
| FI-XII / I-XIIa | Native and activated coagulation factors I-XII, respectively. |
| VEGF | Vascular endothelial growth factor |
References
- Collins, D.R.J.; Tompson, A.C.; Onakpoya, I.J.; Roberts, N.; Ward, A.M.; Heneghan, C.J. Global cardiovascular risk assessment in the primary prevention of cardiovascular disease in adults: Systematic review of systematic reviews. BMJ Open 2017, 7, e013650. [Google Scholar] [CrossRef]
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019: Update from the GBD 2019 Study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef]
- Vanhoutte, P.M.; Shimokawa, H.; Tang, E.H.C.; Feletou, M. Endothelial dysfunction and vascular disease. Acta Physiol. 2009, 196, 193–222. [Google Scholar] [CrossRef] [Green Version]
- Yau, J.W.; Teoh, H.; Verma, S. Endothelial cell control of thrombosis. BMC Cardiovasc. Disord. 2015, 15, 130. [Google Scholar] [CrossRef] [Green Version]
- Bermejo-Martin, J.F.; Martín-Fernandez, M.; López-Mestanza, C.; Duque, P.; Almansa, R. Shared Features of Endothelial Dysfunction between Sepsis and Its Preceding Risk Factors (Aging and Chronic Disease). J. Clin. Med. 2018, 7, 400. [Google Scholar] [CrossRef] [Green Version]
- Terenzi, D.C.; Trac, J.Z.; Teoh, H.; Gerstein, H.C.; Bhatt, D.L.; Al-Omran, M.; Verma, S.; Hess, D.A. Vascular Regenerative Cell Exhaustion in Diabetes: Translational Opportunities to Mitigate Cardiometabolic Risk. Trends Mol. Med. 2019, 25, 640–655. [Google Scholar] [CrossRef] [PubMed]
- Morrissey, J.H. Tissue factor: A key molecule in hemostatic and nonhemostatic systems. Int. J. Hematol. 2004, 79, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Moll, G.; Ankrum, J.A.; Kamhieh-Milz, J.; Bieback, K.; Ringden, O.; Volk, H.D.; Geissler, S.; Reinke, P. Intravascular Mesenchymal Stromal/Stem Cell Therapy Product Diversification: Time for New Clinical Guidelines. Trends Mol. Med. 2019, 25, 149–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moll, G.; Drzeniek, N.; Kamhieh-Milz, J.; Geissler, S.; Volk, H.-D.; Reinke, P. MSC Therapies for COVID-19: Importance of Patient Coagulopathy, Thromboprophylaxis, Cell Product Quality and Mode of Delivery for Treatment Safety and Efficacy. Front. Immunol. 2020, 11, 1091. [Google Scholar] [CrossRef]
- Van Hinsbergh, V.W.M. Endothelium—Role in regulation of coagulation and inflammation. Semin. Immunopathol. 2012, 34, 93–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minami, T.; Sugiyama, A.; Wu, S.-Q.; Abid, R.; Kodama, T.; Aird, W.C. Thrombin and Phenotypic Modulation of the Endothelium. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 41–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrlova, J.; Petruk, G.; Huber, R.G.; McBurnie, E.W.; van der Plas, M.J.A.; Bond, P.J.; Puthia, M.; Schmidtchen, A. Thrombin-derived C-terminal fragments aggregate and scavenge bacteria and their proinflammatory products. J. Biol. Chem. 2020, 295, 3417–3430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saravanan, R.; Choong, Y.K.; Lim, C.H.; Lim, L.M.; Petrlova, J.; Schmidtchen, A. Cell-Free DNA Promotes Thrombin Autolysis and Generation of Thrombin-Derived C-Terminal Fragments. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef]
- Maragoudakis, M.E.; Tsopanoglou, N.E.; Andriopoulou, P. Mechanism of thrombin-induced angiogenesis. Biochem. Soc. Trans. 2002, 30, 173–177. [Google Scholar] [CrossRef]
- Herbert, J.M.; Dupuy, E.; Laplace, M.C.; Zini, J.M.; Bar Shavit, R.; Tobelem, G. Thrombin induces endothelial cell growth via both a proteolytic and a non-proteolytic pathway. Biochem. J. 1994, 303 Pt 1, 227–231. [Google Scholar] [CrossRef] [Green Version]
- Tsopanoglou, N.E.; Maragoudakis, M.E. Thrombin’s central role in angiogenesis and pathophysiological processes. Eur. Cytokine Netw. 2009, 20, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Qadura, M.; Terenzi, D.C.; Verma, S.; Al-Omran, M.; Hess, D.A. Concise Review: Cell Therapy for Critical Limb Ischemia: An Integrated Review of Preclinical and Clinical Studies. Stem Cells 2018, 36, 161–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsopanoglou, N.E.; Andriopoulou, P.; Maragoudakis, M.E. On the mechanism of thrombin-induced angiogenesis: Involvement of alphavbeta3-integrin. Am. J. Physiol. Cell Physiol. 2002, 283, C1501–C1510. [Google Scholar] [CrossRef] [Green Version]
- Haralabopoulos, G.C.; Grant, D.S.; Kleinman, H.K.; Maragoudakis, M.E. Thrombin promotes endothelial cell alignment in Matrigel in vitro and angiogenesis in vivo. Am. J. Physiol. 1997, 273, C239–C245. [Google Scholar] [CrossRef]
- Tsopanoglou, N.E.; Maragoudakis, M.E. On the mechanism of thrombin-induced angiogenesis. Potentiation of vascular endothelial growth factor activity on endothelial cells by up-regulation of its receptors. J. Biol. Chem. 1999, 274, 23969–23976. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Morita, I.; Onodera, M.; Murota, S.I. Induction of KDR expression in bovine arterial endothelial cells by thrombin: Involvement of nitric oxide. J. Cell Physiol. 2002, 190, 238–250. [Google Scholar] [CrossRef]
- Huang, Y.Q.; Li, J.J.; Hu, L.; Lee, M.; Karpatkin, S. Thrombin induces increased expression and secretion of VEGF from human FS4 fibroblasts, DU145 prostate cells and CHRF megakaryocytes. Thromb. Haemost. 2001, 86, 1094–1098. [Google Scholar]
- Möhle, R.; Green, D.; Moore, M.A.; Nachman, R.L.; Rafii, S. Constitutive production and thrombin-induced release of vascular endothelial growth factor by human megakaryocytes and platelets. Proc. Natl. Acad. Sci. USA 1997, 94, 663–668. [Google Scholar] [CrossRef] [Green Version]
- Hu, S.; Wu, G.; Zheng, J.; Liu, X.; Zhang, Y. Astrocytic thrombin-evoked VEGF release is dependent on p44/42 MAPKs and PAR1. Biochem. Biophys. Res. Commun. 2019, 509, 585–589. [Google Scholar] [CrossRef]
- Bian, Z.M.; Elner, S.G.; Elner, V.M. Thrombin-induced VEGF expression in human retinal pigment epithelial cells. Investig. Ophthalmol. Vis. Sci. 2007, 48, 2738–2746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terasaki, H.; Shirasawa, M.; Otsuka, H.; Yamashita, T.; Uchino, E.; Hisatomi, T.; Sonoda, S.; Sakamoto, T. Different Effects of Thrombin on VEGF Secretion, Proliferation, and Permeability in Polarized and Non-polarized Retinal Pigment Epithelial Cells. Curr. Eye Res. 2015, 40, 936–945. [Google Scholar] [CrossRef] [PubMed]
- Hollborn, M.; Petto, C.; Steffen, A.; Trettner, S.; Bendig, A.; Wiedemann, P.; Bringmann, A.; Kohen, L. Effects of thrombin on RPE cells are mediated by transactivation of growth factor receptors. Investig. Ophthalmol. Vis. Sci. 2009, 50, 4452–4459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arisato, T.; Sarker, K.P.; Kawahara, K.; Nakata, M.; Hashiguchi, T.; Osame, M.; Kitajima, I.; Maruyama, I. The agonist of the protease-activated receptor-1 (PAR) but not PAR3 mimics thrombin-induced vascular endothelial growth factor release in human smooth muscle cells. Cell. Mol. Life Sci. 2003, 60, 1716–1724. [Google Scholar] [CrossRef] [PubMed]
- Bassus, S.; Herkert, O.; Kronemann, N.; Görlach, A.; Bremerich, D.; Kirchmaier, C.M.; Busse, R.; Schini-Kerth, V.B. Thrombin causes vascular endothelial growth factor expression in vascular smooth muscle cells: Role of reactive oxygen species. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1550–1555. [Google Scholar] [CrossRef] [Green Version]
- Strande, J.L.; Phillips, S.A. Thrombin increases inflammatory cytokine and angiogenic growth factor secretion in human adipose cells in vitro. J. Inflamm. 2009, 6, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belling, F.; Ribeiro, A.; Wörnle, M.; Ladurner, R.; Mussack, T.; Sitter, T.; Sauter, M. PAR-1 mediates the thrombin-induced mesothelial cell overproduction of VEGF and PAI-1. Int. J. Artif. Organs 2013, 36, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Sarker, K.P.; Yamahata, H.; Nakata, M.; Arisato, T.; Nakajima, T.; Kitajima, I.; Maruyama, I. Recombinant thrombomodulin inhibits thrombin-induced vascular endothelial growth factor production in neuronal cells. Haemostasis 1999, 29, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Yamahata, H.; Takeshima, H.; Kuratsu, J.; Sarker, K.P.; Tanioka, K.; Wakimaru, N.; Nakata, M.; Kitajima, I.; Maruyama, I. The role of thrombin in the neo-vascularization of malignant gliomas: An intrinsic modulator for the up-regulation of vascular endothelial growth factor. Int. J. Oncol. 2002, 20, 921–928. [Google Scholar] [CrossRef] [PubMed]
- Sarker, K.P.; Biswas, K.K.; Yamaji, K.; Yamakuchi, M.; Hashiguchi, T.; Lee, K.Y.; Maruyama, I. Inhibition of thrombin-induced vascular endothelial growth factor production in human neuroblastoma (NB-1) cells by argatroban. Pathophysiol. Haemost. Thromb. 2005, 34, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Schuff-Werner, P.; Steiner, M. Thrombin/thrombin receptor (PAR-1)-mediated induction of IL-8 and VEGF expression in prostate cancer cells. Biochem. Biophys. Res. Commun. 2006, 343, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Dupuy, E.; Habib, A.; Lebret, M.; Yang, R.; Levy-Toledano, S.; Tobelem, G. Thrombin induces angiogenesis and vascular endothelial growth factor expression in human endothelial cells: Possible relevance to HIF-1α. J. Thromb. Haemost. JTH 2003, 1, 1096–1102. [Google Scholar] [CrossRef]
- Pagès, G.; Pouysségur, J. Transcriptional regulation of the Vascular Endothelial Growth Factor gene—A concert of activating factors. Cardiovasc. Res. 2005, 65, 564–573. [Google Scholar] [CrossRef]
- Colling, M.E.; Kanthi, Y. COVID-19-associated coagulopathy: An exploration of mechanisms. Vasc. Med. 2020, 25, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef]
- Nagy, J.A.; Dvorak, A.M.; Dvorak, H.F. VEGF-A and the induction of pathological angiogenesis. Annu. Rev. Pathol. 2007, 2, 251–275. [Google Scholar] [CrossRef]
- Barratt, S.; Medford, A.R.; Millar, A.B. Vascular endothelial growth factor in acute lung injury and acute respiratory distress syndrome. Respir. Int. Rev. Thorac. Dis. 2014, 87, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Catar, R.; Witowski, J.; Zhu, N.; Lucht, C.; Derrac Soria, A.; Uceda Fernandez, J.; Chen, L.; Jones, S.A.; Fielding, C.A.; Rudolf, A.; et al. IL-6 Trans-Signaling Links Inflammation with Angiogenesis in the Peritoneal Membrane. J. Am. Soc. Nephrol. 2017, 28, 1188–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrzejewska, A.; Catar, R.; Schoon, J.; Qazi, T.H.; Sass, F.A.; Jacobi, D.; Blankenstein, A.; Reinke, S.; Kruger, D.; Streitz, M.; et al. Multi-Parameter Analysis of Biobanked Human Bone Marrow Stromal Cells Shows Little Influence for Donor Age and Mild Comorbidities on Phenotypic and Functional Properties. Front. Immunol. 2019, 10, 2474. [Google Scholar] [CrossRef]
- Korybalska, K.; Pyda, M.; Kawka, E.; Grajek, S.; Bręborowicz, A.; Witowski, J. Interpretation of elevated serum VEGF concentrations in patients with myocardial infarction. Cytokine 2011, 54, 74–78. [Google Scholar] [CrossRef]
- Catar, R.; Witowski, J.; Wagner, P.; Annett Schramm, I.; Kawka, E.; Philippe, A.; Dragun, D.; Jorres, A. The proto-oncogene c-Fos transcriptionally regulates VEGF production during peritoneal inflammation. Kidney Int. 2013, 84, 1119–1128. [Google Scholar] [CrossRef] [Green Version]
- Moll, G.; Rasmusson-Duprez, I.; von Bahr, L.; Connolly-Andersen, A.M.; Elgue, G.; Funke, L.; Hamad, O.A.; Lonnies, H.; Magnusson, P.U.; Sanchez, J.; et al. Are therapeutic human mesenchymal stromal cells compatible with human blood? Stem Cells 2012, 30, 1565–1574. [Google Scholar] [CrossRef] [Green Version]
- Moll, G.; Alm, J.J.; Davies, L.C.; von Bahr, L.; Heldring, N.; Stenbeck-Funke, L.; Hamad, O.A.; Hinsch, R.; Ignatowicz, L.; Locke, M.; et al. Do cryopreserved mesenchymal stromal cells display impaired immunomodulatory and therapeutic properties? Stem Cells 2014, 32, 2430–2442. [Google Scholar] [CrossRef] [Green Version]
- Zickler, D.; Luecht, C.; Willy, K.; Chen, L.; Witowski, J.; Girndt, M.; Fiedler, R.; Storr, M.; Kamhieh-Milz, J.; Schoon, J.; et al. Tumour necrosis factor-alpha in uraemic serum promotes osteoblastic transition and calcification of vascular smooth muscle cells via extracellular signal-regulated kinases and activator protein 1/c-FOS-mediated induction of interleukin 6 expression. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc. Eur. Ren. Assoc. 2018, 33, 574–585. [Google Scholar] [CrossRef]
- Finkenzeller, G.; Sparacio, A.; Technau, A.; Marmé, D.; Siemeister, G. Sp1 recognition sites in the proximal promoter of the human vascular endothelial growth factor gene are essential for platelet-derived growth factor-induced gene expression. Oncogene 1997, 15, 669–676. [Google Scholar] [CrossRef] [Green Version]
- Brass, L.F.; Pizarro, S.; Ahuja, M.; Belmonte, E.; Blanchard, N.; Stadel, J.M.; Hoxie, J.A. Changes in the structure and function of the human thrombin receptor during receptor activation, internalization, and recycling. J. Biol. Chem. 1994, 269, 2943–2952. [Google Scholar] [CrossRef]
- Sadeghi, B.; Moretti, G.; Arnberg, F.; Samen, E.; Kohein, B.; Catar, R.; Kamhieh-Milz, J.; Geissler, S.; Moll, G.; Holmin, S.; et al. Preclinical Toxicity Evaluation of Clinical Grade Placenta-Derived Decidua Stromal Cells. Front. Immunol. 2019, 10, 2685. [Google Scholar] [CrossRef]
- Bonder, C.S.; Ebert, L.M. Fos-icking for control of angiogenesis: Increasing the longevity of peritoneal dialysis. Kidney Int. 2013, 84, 1065–1067. [Google Scholar] [CrossRef] [Green Version]
- Caunt, M.; Huang, Y.Q.; Brooks, P.C.; Karpatkin, S. Thrombin induces neoangiogenesis in the chick chorioallantoic membrane. J. Thromb. Haemost. 2003, 1, 2097–2102. [Google Scholar] [CrossRef] [Green Version]
- Moll, G.; Ignatowicz, L.; Catar, R.; Luecht, C.; Sadeghi, B.; Hamad, O.; Jungebluth, P.; Dragun, D.; Schmidtchen, A.; Ringden, O. Different Procoagulant Activity of Therapeutic Mesenchymal Stromal Cells Derived from Bone Marrow and Placental Decidua. Stem Cells Dev. 2015, 24, 2269–2279. [Google Scholar] [CrossRef]
- Delerive, P.; Martin-Nizard, F.; Chinetti, G.; Trottein, F.; Fruchart, J.C.; Najib, J.; Duriez, P.; Staels, B. Peroxisome proliferator-activated receptor activators inhibit thrombin-induced endothelin-1 production in human vascular endothelial cells by inhibiting the activator protein-1 signaling pathway. Circ. Res. 1999, 85, 394–402. [Google Scholar] [CrossRef] [Green Version]
- Takata, M.; Urakaze, M.; Temaru, R.; Yamazaki, K.; Nakamura, N.; Nobata, Y.; Kishida, M.; Sato, A.; Kobayashi, M. Pravastatin suppresses the interleukin-8 production induced by thrombin in human aortic endothelial cells cultured with high glucose by inhibiting the p44/42 mitogen activated protein kinase. Br. J. Pharmacol. 2001, 134, 753–762. [Google Scholar] [CrossRef] [Green Version]
- Pendurthi, U.R.; Williams, J.T.; Rao, L.V. Inhibition of tissue factor gene activation in cultured endothelial cells by curcumin. Suppression of activation of transcription factors Egr-1, AP-1, and NF-κB. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 3406–3413. [Google Scholar] [CrossRef]
- Heo, K.; Park, K.A.; Kim, Y.H.; Kim, S.H.; Oh, Y.S.; Kim, I.H.; Ryu, S.H.; Suh, P.G. Sphingosine 1-phosphate induces vascular endothelial growth factor expression in endothelial cells. BMB Rep. 2009, 42, 685–690. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Wang, S.; Sun, Y.; Matt, Y.; Colburn, N.H.; Shu, Y.; Han, X. JNK/AP-1 pathway is involved in tumor necrosis factor-alpha induced expression of vascular endothelial growth factor in MCF7 cells. Biomed. Pharmacother. 2009, 63, 429–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleiner, J.; Hollborn, M.; Wiedemann, P.; Bringmann, A. Activator protein-1 contributes to the NaCl-induced expression of VEGF and PlGF in RPE cells. Mol. Vis. 2018, 24, 647–666. [Google Scholar]
- Lee, C.C.; Chen, S.C.; Tsai, S.C.; Wang, B.W.; Liu, Y.C.; Lee, H.M.; Shyu, K.G. Hyperbaric oxygen induces VEGF expression through ERK, JNK and c-Jun/AP-1 activation in human umbilical vein endothelial cells. J. Biomed. Sci. 2006, 13, 143–156. [Google Scholar] [CrossRef] [Green Version]
- Bancroft, C.C.; Chen, Z.; Yeh, J.; Sunwoo, J.B.; Yeh, N.T.; Jackson, S.; Jackson, C.; Van Waes, C. Effects of pharmacologic antagonists of epidermal growth factor receptor, PI3K and MEK signal kinases on NF-kappaB and AP-1 activation and IL-8 and VEGF expression in human head and neck squamous cell carcinoma lines. Int. J. Cancer 2002, 99, 538–548. [Google Scholar] [CrossRef]
- Cho, M.L.; Jung, Y.O.; Moon, Y.M.; Min, S.Y.; Yoon, C.H.; Lee, S.H.; Park, S.H.; Cho, C.S.; Jue, D.M.; Kim, H.Y. Interleukin-18 induces the production of vascular endothelial growth factor (VEGF) in rheumatoid arthritis synovial fibroblasts via AP-1-dependent pathways. Immunol. Lett. 2006, 103, 159–166. [Google Scholar] [CrossRef]
- Chang, H.J.; Park, J.S.; Kim, M.H.; Hong, M.H.; Kim, K.M.; Kim, S.M.; Shin, B.A.; Ahn, B.W.; Jung, Y.D. Extracellular signal-regulated kinases and AP-1 mediate the up-regulation of vascular endothelial growth factor by PDGF in human vascular smooth muscle cells. Int. J. Oncol. 2006, 28, 135–141. [Google Scholar] [CrossRef]
- Yu, H.S.; Wang, S.W.; Chang, A.C.; Tai, H.C.; Yeh, H.I.; Lin, Y.M.; Tang, C.H. Bradykinin promotes vascular endothelial growth factor expression and increases angiogenesis in human prostate cancer cells. Biochem. Pharmacol. 2014, 87, 243–253. [Google Scholar] [CrossRef]
- Hossain, M.A.; Bouton, C.M.; Pevsner, J.; Laterra, J. Induction of vascular endothelial growth factor in human astrocytes by lead. Involvement of a protein kinase C/activator protein-1 complex-dependent and hypoxia-inducible factor 1-independent signaling pathway. J. Biol. Chem. 2000, 275, 27874–27882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urata, Y.; Yamaguchi, M.; Higashiyama, Y.; Ihara, Y.; Goto, S.; Kuwano, M.; Horiuchi, S.; Sumikawa, K.; Kondo, T. Reactive oxygen species accelerate production of vascular endothelial growth factor by advanced glycation end products in RAW264.7 mouse macrophages. Free Radic. Biol. Med. 2002, 32, 688–701. [Google Scholar] [CrossRef]
- Fujita, M.; Hayashi, I.; Yamashina, S.; Fukamizu, A.; Itoman, M.; Majima, M. Angiotensin type 1a receptor signaling-dependent induction of vascular endothelial growth factor in stroma is relevant to tumor-associated angiogenesis and tumor growth. Carcinogenesis 2005, 26, 271–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itaya, H.; Imaizumi, T.; Yoshida, H.; Koyama, M.; Suzuki, S.; Satoh, K. Expression of vascular endothelial growth factor in human monocyte/macrophages stimulated with lipopolysaccharide. Thromb. Haemost. 2001, 85, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Textor, B.; Sator-Schmitt, M.; Richter, K.H.; Angel, P.; Schorpp-Kistner, M. c-Jun and JunB are essential for hypoglycemia-mediated VEGF induction. Ann. N. Y. Acad. Sci. 2006, 1091, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Abu El-Asrar, A.M.; Alam, K.; Nawaz, M.I.; Mohammad, G.; Van den Eynde, K.; Siddiquei, M.M.; Mousa, A.; De Hertogh, G.; Opdenakker, G. Upregulation of Thrombin/Matrix Metalloproteinase-1/Protease-Activated Receptor-1 Chain in Proliferative Diabetic Retinopathy. Curr. Eye Res. 2016, 41, 1590–1600. [Google Scholar] [CrossRef] [Green Version]
- Hariri, L.; Hardin, C.C. Covid-19, Angiogenesis, and ARDS Endotypes. N. Engl. J. Med. 2020, 383, 182–183. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Luscher, T. COVID-19 is, in the end, an endothelial disease. Eur. Heart J. 2020, 41, 3038–3044. [Google Scholar] [CrossRef] [PubMed]
- Pine, A.B.; Meizlish, M.L.; Goshua, G.; Chang, C.H.; Zhang, H.; Bishai, J.; Bahel, P.; Patel, A.; Gbyli, R.; Kwan, J.M.; et al. Circulating markers of angiogenesis and endotheliopathy in COVID-19. Pulm. Circ. 2020, 10, 2045894020966547. [Google Scholar] [CrossRef] [PubMed]
- Vassiliou, A.G.; Keskinidou, C.; Jahaj, E.; Gallos, P.; Dimopoulou, I.; Kotanidou, A.; Orfanos, S.E. ICU Admission Levels of Endothelial Biomarkers as Predictors of Mortality in Critically Ill COVID-19 Patients. Cells 2021, 10, 186. [Google Scholar] [CrossRef] [PubMed]
- Rovas, A.; Osiaevi, I.; Buscher, K.; Sackarnd, J.; Tepasse, P.R.; Fobker, M.; Kühn, J.; Braune, S.; Göbel, U.; Thölking, G.; et al. Microvascular dysfunction in COVID-19: The MYSTIC study. Angiogenesis 2021, 24, 145–157. [Google Scholar] [CrossRef]
- Sriram, K.; Insel, P.A. Proteinase-activated receptor 1: A target for repurposing in the treatment of COVID-19? Br. J. Pharmacol. 2020, 177, 4971–4974. [Google Scholar] [CrossRef]
- Pang, J.; Xu, F.; Aondio, G.; Li, Y.; Fumagalli, A.; Lu, M.; Valmadre, G.; Wei, J.; Bian, Y.; Canesi, M.; et al. Efficacy and tolerability of bevacizumab in patients with severe Covid-19. Nat. Commun. 2021, 12, 814. [Google Scholar] [CrossRef]
- Zhang, Z.; Lu, D.S.; Zhang, D.Q.; Wang, X.; Ming, Y.; Wu, Z.Y. Targeted Antagonism of Vascular Endothelial Growth Factor Reduces Mortality of Mice with Acute Respiratory Distress Syndrome. Curr. Med. Sci. 2020, 40, 671–676. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Catar, R.; Moll, G.; Hosp, I.; Simon, M.; Luecht, C.; Zhao, H.; Wu, D.; Chen, L.; Kamhieh-Milz, J.; Korybalska, K.; et al. Transcriptional Regulation of Thrombin-Induced Endothelial VEGF Induction and Proangiogenic Response. Cells 2021, 10, 910. https://doi.org/10.3390/cells10040910
Catar R, Moll G, Hosp I, Simon M, Luecht C, Zhao H, Wu D, Chen L, Kamhieh-Milz J, Korybalska K, et al. Transcriptional Regulation of Thrombin-Induced Endothelial VEGF Induction and Proangiogenic Response. Cells. 2021; 10(4):910. https://doi.org/10.3390/cells10040910
Chicago/Turabian StyleCatar, Rusan, Guido Moll, Isa Hosp, Michele Simon, Christian Luecht, Hongfan Zhao, Dashan Wu, Lei Chen, Julian Kamhieh-Milz, Katarzyna Korybalska, and et al. 2021. "Transcriptional Regulation of Thrombin-Induced Endothelial VEGF Induction and Proangiogenic Response" Cells 10, no. 4: 910. https://doi.org/10.3390/cells10040910
APA StyleCatar, R., Moll, G., Hosp, I., Simon, M., Luecht, C., Zhao, H., Wu, D., Chen, L., Kamhieh-Milz, J., Korybalska, K., Zickler, D., & Witowski, J. (2021). Transcriptional Regulation of Thrombin-Induced Endothelial VEGF Induction and Proangiogenic Response. Cells, 10(4), 910. https://doi.org/10.3390/cells10040910