
Dendritic Cells: Neglected Modulators of Peripheral Immune Responses and Neuroinflammation in Mood Disorders?

 , and
, and
Abstract
:1. Introduction
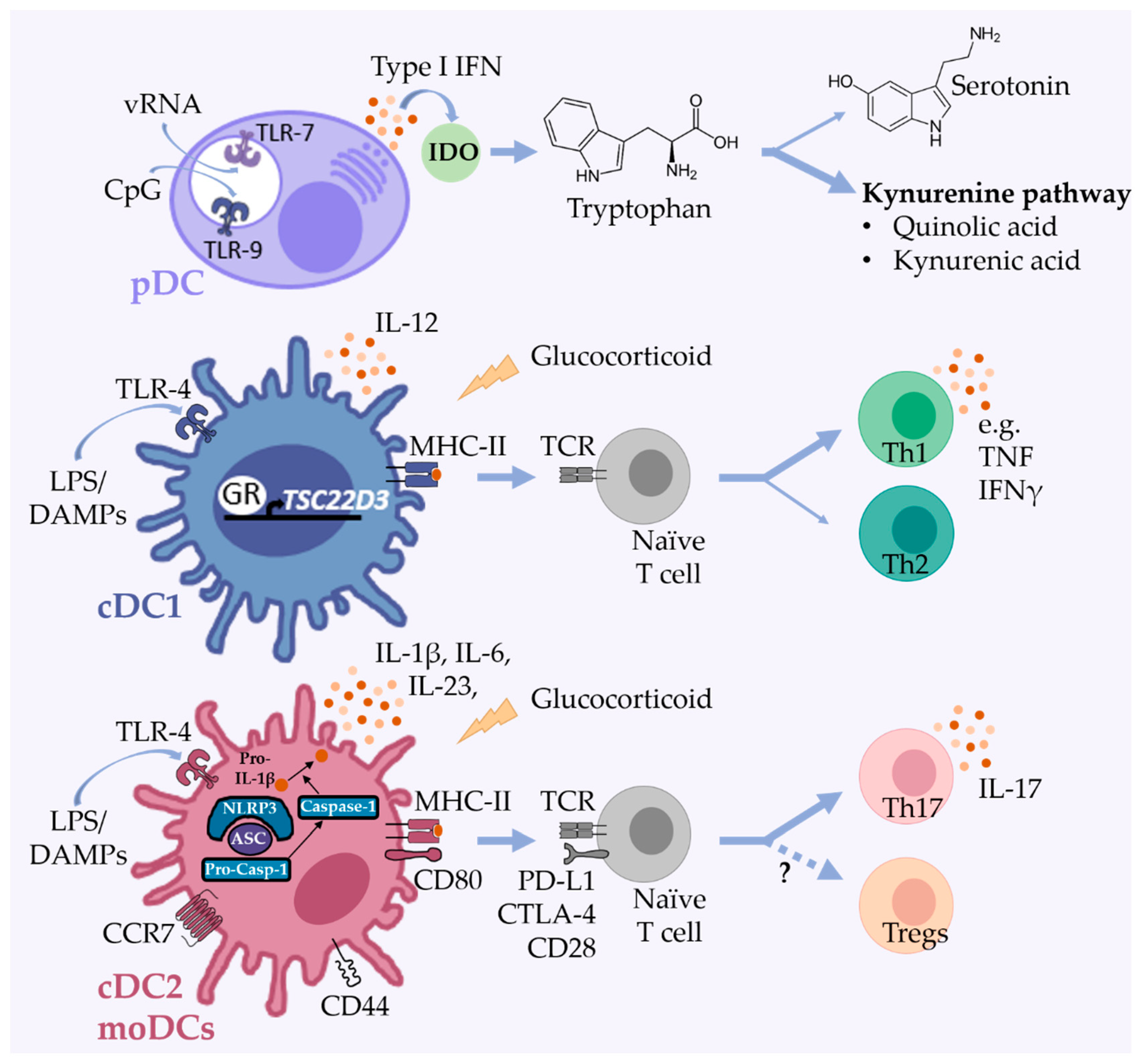
2. Selected Functions of DCs with Relevance for Mood Disorders
3. Human and Mouse DC Subsets
3.1. Plasmacytoid DCs
3.2. Conventional DCs

{kind=link}
{kind=link}
| DC Subset | Transcription Factors | Major Cytokines | Major Surface Makers | Major PRRs | Reference | ||
|---|---|---|---|---|---|---|---|
| Human | Mouse | Human | Mouse | ||||
| pDCs | IRF8, BCL11A, E2-2/TCF4 | type I IFN | CD123/IL-3RA, CD303/CLEC4C/BDCA-2, CD304/NRP1/BDCA-4 and HLADR low | CD11c low, B220, CD317, Siglec-H, CD172a, CD209, CCR2 low, CCR9, CXCR3 and MHC II low | TLR7 and TLR9 | TLR7 and TLR9 | [79,83,96] |
| cDC1s | BATF3, IRF8, ID2, Zbtb46 (BTBD4) | IL-12 | CD11c low, HLA-DR, CD141/BDCA1, XCR1, CLEC9A/DNGR1, DEC205, IDO | CD11c, MHC II, CD8α (resident), CD103 (migratory), CD24, XCR1, CLEC9A and DEC205 | TLR3 or CLEC12A | TLR4 or CLEC12A | [79,96,107,108] |
| cDC2s | ID2, Zeb2, NOTCH2, IRF4, KLF4, Zbtb46 (BTBD4) | IL-1β, IL-6, IL-10, IL-12, IL-23, and TNF | CD1c/BDCA-1, CD2, CD172a/SIRPA, CD11c, HLA-DR, CD11b, CD1a (migratory), FcεR1, ILT1, CD14 and CD5 (subset) | CD11c, MHC II, CD11b high, CD172a/SIRPA | TLRs 1-9 | TLRs 1-9 | [79,96] |
| moDCs | CSF1R, MAFB, KLF4, Zbtb46 (BTBD4) | IL-1β, IL-6, IL12, IL-23, and TNF | CD11c, HLA-DR, CD1c, CD11b, CD14, CD64, CD206, CD209, CD172a, CD1a, CCR2 | CD11c, MHC II, CD11b, Ly6C, CD64, CD206, CD209, CD14, CCR2 | - | - | [109,110] |
3.3. Monocyte-Derived DCs
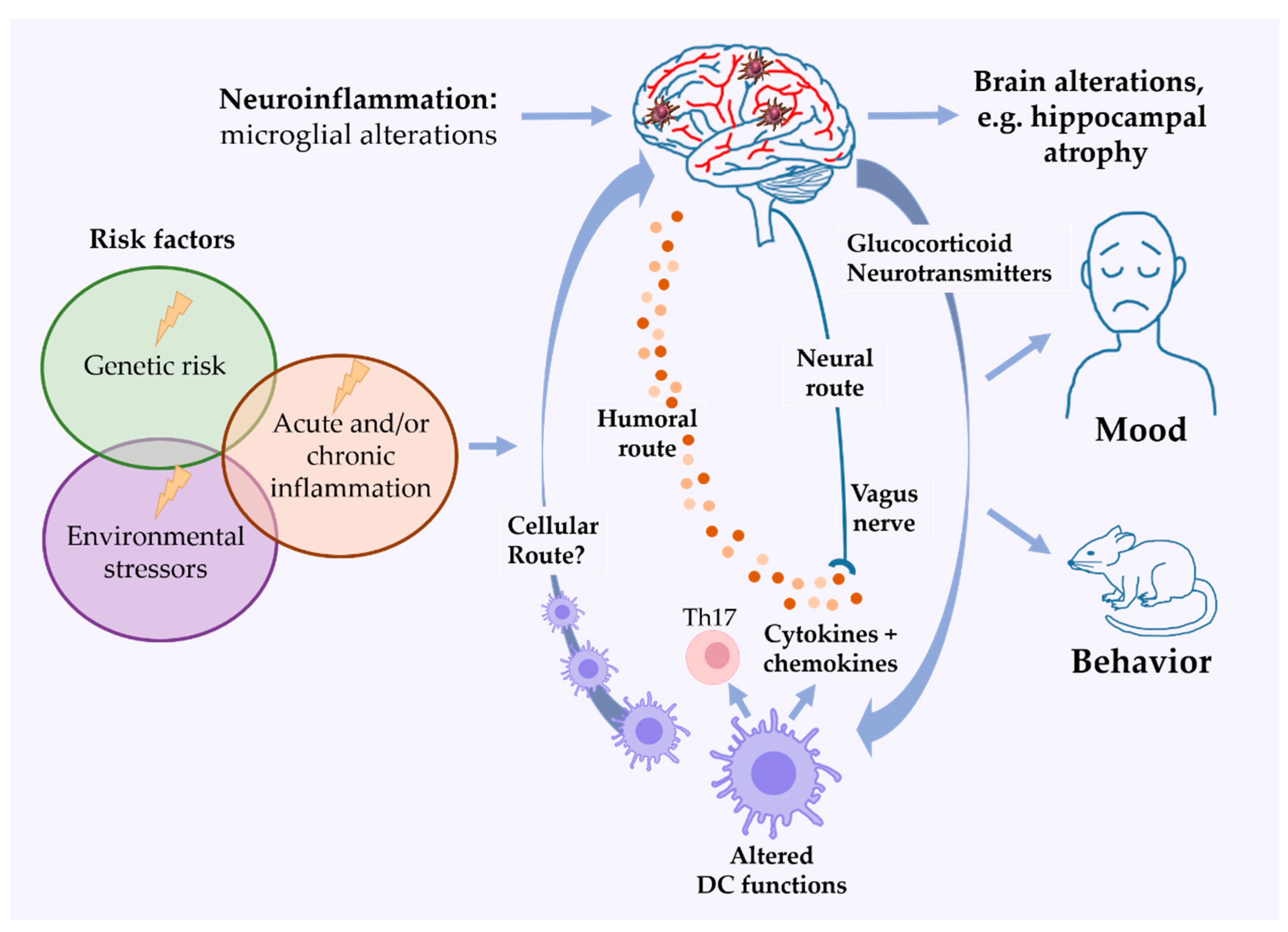
4. DCs in Mood Disorders and Depression-Like Behavior
4.1. Genetic Studies with a Relation to DCs in Mood Disorders
4.2. DC-Expressed Chemokines and Chemokine Receptors Involved in Mood Disorders and Depression-Like Behavior
4.3. DC-Derived Cytokines and Chemokines and Their Potential Influence on Microglia Function
4.4. DCs as Modulators of Adaptive Immune Responses in Mood Disorders
4.5. Effects of Antidepressant Treatment on Human and Murine DCs
5. DCs in Rodent Models of Mood Disorders
5.1. Models of Inflammation-Induced Depression Induced by Endotoxin Administration
5.2. DCs in Animal Models of Stress-Induced Behavioral Changes
6. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bromet, E.; Andrade, L.H.; Hwang, I.; Sampson, N.A.; Alonso, J.; De Girolamo, G.; De Graaf, R.; Demyttenaere, K.; Hu, C.; Iwata, N.; et al. Cross-National Epidemiology of Dsm-Iv Major Depressive Episode. BMC Med. 2011, 9, 90. [Google Scholar] [CrossRef]
- Kessler, R.C.; Bromet, E.J. The Epidemiology of Depression across Cultures. Annu. Rev. Public Health 2013, 34, 119–138. [Google Scholar] [CrossRef] [Green Version]
- Phillips, M.L.; Kupfer, D.J. Bipolar Disorder Diagnosis: Challenges and Future Directions. Lancet 2013, 381, 1663–1671. [Google Scholar] [CrossRef] [Green Version]
- Vieta, E.; Berk, M.; Schulze, T.G.; Carvalho, A.F.; Suppes, T.; Calabrese, J.R.; Gao, K.; Miskowiak, K.W.; Grande, I. Bipolar Disorders. Nat. Rev. Dis. Primers 2018, 4, 1–16. [Google Scholar] [CrossRef]
- Otte, C.; Gold, S.M.; Penninx, B.W.; Pariante, C.M.; Etkin, A.; Fava, M.; Mohr, D.C.; Schatzberg, A.F. Major Depressive Disorder. Nat. Rev. Dis. Primers 2016, 2, 16065. [Google Scholar] [CrossRef] [Green Version]
- Kupfer, D.J.; Frank, E.; Phillips, M.L. Major Depressive Disorder: New Clinical, Neurobiological, and Treatment Perspectives. Lancet 2012, 379, 1045–1055. [Google Scholar] [CrossRef] [Green Version]
- Shadrina, M.; Bondarenko, E.A.; Slominsky, P.A. Genetics Factors in Major Depression Disease. Front. Psychiatry 2018, 9, 334. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.H.; Raison, C.L. The Role of Inflammation in Depression: From Evolutionary Imperative to Modern Treatment Target. Nat. Rev. Immunol. 2016, 16, 22. [Google Scholar] [CrossRef] [Green Version]
- Maes, M.; Yirmyia, R.; Noraberg, J.; Brene, S.; Hibbeln, J.; Perini, G.; Kubera, M.; Bob, P.; Lerer, B.; Maj, M. The Inflammatory & Neurodegenerative (I&Nd) Hypothesis of Depression: Leads for Future Research and New Drug Developments in Depression. Metab. Brain Dis. 2009, 24, 27–53. [Google Scholar]
- Eyre, H.A.; Stuart, M.J.; Baune, B.T. A Phase-Specific Neuroimmune Model of Clinical Depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2014, 54, 265–274. [Google Scholar] [CrossRef]
- Gibney, S.M.; Drexhage, H.A. Evidence for a Dysregulated Immune System in the Etiology of Psychiatric Disorders. J. Neuroimmune Pharmacol. 2013, 8, 900–920. [Google Scholar] [CrossRef] [PubMed]
- Schiepers, O.J.G.; Wichers, M.C.; Maes, M. Cytokines and Major Depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2005, 29, 201–217. [Google Scholar] [CrossRef]
- Maes, M. A Review on the Acute Phase Response in Major Depression. Rev. Neurosci. 1993, 4, 407–416. [Google Scholar] [CrossRef]
- Capuron, L.; Miller, A.H. Cytokines and Psychopathology: Lessons from Interferon-Alpha. Biol. Psychiatry 2004, 56, 819–824. [Google Scholar] [CrossRef]
- Raison, C.L.; Capuron, L.; Miller, A.H. Cytokines Sing the Blues: Inflammation and the Pathogenesis of Depression. Trends Immunol. 2006, 27, 24–31. [Google Scholar] [CrossRef] [Green Version]
- Maes, M. Depression Is an Inflammatory Disease, but Cell-Mediated Immune Activation Is the Key Component of Depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2011, 35, 664–675. [Google Scholar] [CrossRef]
- Black, C.N.; Bot, M.; Scheffer, P.G.; Cuijpers, P.; Penninx, B.W. Is Depression Associated with Increased Oxidative Stress? A Systematic Review and Meta-Analysis. Psychoneuroendocrinology 2015, 51, 164–175. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.K.; Na, K.S.; Myint, A.M.; Leonard, B.E. The Role of Pro-Inflammatory Cytokines in Neuroinflammation, Neurogenesis and the Neuroendocrine System in Major Depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 64, 277–284. [Google Scholar] [CrossRef]
- Dowlati, Y.; Herrmann, N.; Swardfager, W.; Liu, H.; Sham, L.; Reim, E.K.; Lanctôt, K.L. A Meta-Analysis of Cytokines in Major Depression. Biol. Psychiatry 2010, 67, 446–457. [Google Scholar] [CrossRef]
- Howren, M.B.; Lamkin, D.M.; Suls, J. Associations of Depression with C-Reactive Protein, Il-1, and Il-6: A Meta-Analysis. Psychosom. Med. 2009, 71, 171–186. [Google Scholar] [CrossRef] [Green Version]
- Köhler, C.A.; Freitas, T.H.; Maes, M.; De Andrade, N.Q.; Liu, C.S.; Fernandes, B.S.; Stubbs, B.; Solmi, M.; Veronese, N.; Herrmann, N.; et al. Peripheral Cytokine and Chemokine Alterations in Depression: A Meta-Analysis of 82 Studies. Acta Psychiatr. Scand. 2017, 135, 373–387. [Google Scholar] [CrossRef]
- Rosenblat, J.D.; McIntyre, R.S. Bipolar Disorder and Immune Dysfunction: Epidemiological Findings, Proposed Pathophysiology and Clinical Implications. Brain Sci. 2017, 7, 144. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.H.; Haroon, E.; Raison, C.L.; Felger, J.C. Cytokine Targets in the Brain: Impact on Neurotransmitters and Neurocircuits. Depress. Anxiety 2013, 30, 297–306. [Google Scholar] [CrossRef] [Green Version]
- Swartz, J.R.; Prather, A.A.; Di Iorio, C.R.; Bogdan, R.; Hariri, A.R. A Functional Interleukin-18 Haplotype Predicts Depression and Anxiety through Increased Threat-Related Amygdala Reactivity in Women but Not Men. Neuropsychopharmacol. 2017, 42, 419–426. [Google Scholar] [CrossRef] [Green Version]
- Redlich, R.; Stacey, D.; Opel, N.; Grotegerd, D.; Dohm, K.; Kugel, H.; Heindel, W.; Arolt, V.; Baune, B.T.; Dannlowski, U. Evidence of an Ifn-Γ by Early Life Stress Interaction in the Regulation of Amygdala Reactivity to Emotional Stimuli. Psychoneuroendocrinology 2015, 62, 166–173. [Google Scholar] [CrossRef]
- Cattaneo, A.; Macchi, F.; Plazzotta, G.; Veronica, B.; Bocchio-Chiavetto, L.; Riva, M.A.; Pariante, C.M. Inflammation and Neuronal Plasticity: A Link between Childhood Trauma and Depression Pathogenesis. Front. Cell. Neurosci. 2015, 9, 40. [Google Scholar] [CrossRef] [Green Version]
- Dutcher, E.G.; Pama, E.A.C.; Lynall, M.-E.; Khan, S.; Clatworthy, M.R.; Robbins, T.W.; Bullmore, E.T.; Dalley, J.W. Early-Life Stress and Inflammation: A Systematic Review of a Key Experimental Approach in Rodents. Brain Neurosci. Adv. 2020, 4, 1–11. [Google Scholar] [CrossRef]
- Grosse, L.; Ambrée, O.; Jörgens, S.; Jawahar, M.C.; Singhal, G.; Stacey, D.; Arolt, V.; Baune, B.T. Cytokine Levels in Major Depression Are Related to Childhood Trauma but Not to Recent Stressors. Psychoneuroendocrinology 2016, 73, 24–31. [Google Scholar] [CrossRef]
- Kappelmann, N.; Lewis, G.; Dantzer, R.; Jones, P.B.; Khandaker, G.M. Antidepressant Activity of Anti-Cytokine Treatment: A Systematic Review and Meta-Analysis of Clinical Trials of Chronic Inflammatory Conditions. Mol. Psychiatry 2018, 23, 335–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köhler, C.A.; Freitas, T.H.; Stubbs, B.; Maes, M.; Solmi, M.; Veronese, N.; De Andrade, N.Q.; Morris, G.; Fernandes, B.S.; Brunoni, A.R.; et al. Peripheral Alterations in Cytokine and Chemokine Levels after Antidepressant Drug Treatment for Major Depressive Disorder: Systematic Review and Meta-Analysis. Mol. Neurobiol. 2018, 55, 4195–4206. [Google Scholar] [CrossRef] [Green Version]
- Müller, N.; Schwarz, M.J.; Dehning, S.; Douhe, A.; Cerovecki, A.; Goldstein-Müller, B.; Spellmann, I.; Hetzel, G.; Maino, K.; Kleindienst, N.; et al. The Cyclooxygenase-2 Inhibitor Celecoxib Has Therapeutic Effects in Major Depression: Results of a Double-Blind, Randomized, Placebo Controlled, Add-on Pilot Study to Reboxetine. Mol. Psychiatry 2006, 11, 680–684. [Google Scholar] [CrossRef] [PubMed]
- Raison, C.L.; Rutherford, R.E.; Woolwine, B.J.; Shuo, C.; Schettler, P.; Drake, D.F.; Haroon, E.; Miller, A.H. A Randomized Controlled Trial of the Tumor Necrosis Factor Antagonist Infliximab for Treatment-Resistant Depression: The Role of Baseline Inflammatory Biomarkers. JAMA Psychiatry 2013, 70, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Dantzer, R.; Kelley, K.W. Twenty Years of Research on Cytokine-Induced Sickness Behavior. Brain Behav. Immun. 2007, 21, 153–160. [Google Scholar] [CrossRef] [Green Version]
- Horrobin, D.F.; Lieb, J. A Biochemical Basis for the Actions of Lithium on Behaviour and on Immunity: Relapsing and Remitting Disorders of Inflammation and Immunity Such as Multiple Sclerosis or Recurrent Herpes as Manic-Depression of the Immune System. Med. Hypotheses 1981, 7, 891–905. [Google Scholar] [CrossRef]
- Munkholm, K.; Braüner, J.V.; Kessing, L.V.; Vinberg, M. Cytokines in Bipolar Disorder vs. Healthy Control Subjects: A Systematic Review and Meta-Analysis. J. Psychiatr. Res. 2013, 47, 1119–1133. [Google Scholar] [CrossRef]
- Goldsmith, D.R.; Rapaport, M.H.; Miller, B.J. A Meta-Analysis of Blood Cytokine Network Alterations in Psychiatric Patients: Comparisons between Schizophrenia, Bipolar Disorder and Depression. Mol. Psychiatry 2016, 21, 1696–1709. [Google Scholar] [CrossRef]
- Sowa-Kućma, M.; Styczeń, K.; Siwek, M.; Misztak, P.; Nowak, R.J.; Dudek, D.; Rybakowski, J.K.; Nowak, G.; Maes, M. Are There Differences in Lipid Peroxidation and Immune Biomarkers between Major Depression and Bipolar Disorder: Effects of Melancholia, Atypical Depression, Severity of Illness, Episode Number, Suicidal Ideation and Prior Suicide Attempts. Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 81, 372–383. [Google Scholar] [CrossRef]
- Beumer, W.; Gibney, S.M.; Drexhage, R.C.; Pont-Lezica, L.; Doorduin, J.; Klein, H.C.; Steiner, J.; Connor, T.J.; Harkin, A.; Versnel, M.A.; et al. The Immune Theory of Psychiatric Diseases: A Key Role for Activated Microglia and Circulating Monocytes. J. Leukoc. Biol. 2012, 92, 959–975. [Google Scholar] [CrossRef]
- Takahashi, Y.; Yu, Z.; Sakai, M.; Tomita, H. Linking Activation of Microglia and Peripheral Monocytic Cells to the Pathophysiology of Psychiatric Disorders. Front. Cell. Neurosci. 2016, 10, 144. [Google Scholar] [CrossRef]
- Ramirez, K.; Fornaguera-Trías, J.; Sheridan, J.F. Stress-Induced Microglia Activation and Monocyte Trafficking to the Brain Underlie the Development of Anxiety and Depression. Curr. Top. Behav. Neurosci. 2017, 31, 155–172. [Google Scholar]
- Wohleb, E.S.; Delpech, J.C. Dynamic Cross-Talk between Microglia and Peripheral Monocytes Underlies Stress-Induced Neuroinflammation and Behavioral Consequences. Prog. Neuropsychopharmacol. Biol. Psychiatry 2017, 79, 40–48. [Google Scholar] [CrossRef]
- Dey, A.; Hankey Giblin, P.A. Insights into Macrophage Heterogeneity and Cytokine-Induced Neuroinflammation in Major Depressive Disorder. Pharmaceuticals 2018, 11, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roman, A.; Kreiner, G.; Nalepa, I. Macrophages and Depression-a Misalliance or Well-Arranged Marriage? Pharmacol. Rep. 2013, 65, 1663–1672. [Google Scholar] [CrossRef]
- Prinz, M.; Priller, J. Microglia and Brain Macrophages in the Molecular Age: From Origin to Neuropsychiatric Disease. Nat. Rev. Neurosci. 2014, 15, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, I.G.; Rocha, N.P.; Assis, F.; Vieira, É.L.M.; Soares, J.C.; Bauer, M.E.; Teixeira, A.L. Monocyte and Lymphocyte Activation in Bipolar Disorder: A New Piece in the Puzzle of Immune Dysfunction in Mood Disorders. Int. J. Neuropsychopharmacol. 2015, 18, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Engler, H.; Bailey, M.T.; Engler, A.; Sheridan, J.F. Effects of Repeated Social Stress on Leukocyte Distribution in Bone Marrow, Peripheral Blood and Spleen. J. Neuroimmunol. 2004, 148, 106–115. [Google Scholar] [CrossRef]
- Steinman, R.M. Decisions about Dendritic Cells: Past, Present, and Future. Annu. Rev. Immunol. 2012, 30, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Scheu, S.; Ali, S.; Ruland, C.; Arolt, V.; Alferink, J. The Cc Chemokines Ccl17 and Ccl22 and Their Receptor Ccr4 in Cns Autoimmunity. Int. J. Mol. Sci. 2017, 18, 2306. [Google Scholar] [CrossRef] [Green Version]
- Steinman, R.M.; Cohn, Z.A. Identification of a Novel Cell Type in Peripheral Lymphoid Organs of Mice: I.; Morphology, Quantitation, Tissue Distribution. J. Exp. Med. 1973, 137, 1142–1162. [Google Scholar] [CrossRef]
- Steinman, R.M.; Cohn, Z.A. Identification of a Novel Cell Type in Peripheral Lymphoid Organs of Mice: II. Functional Properties in Vitro. J. Exp. Med. 1974, 139, 380–397. [Google Scholar] [CrossRef] [Green Version]
- Guilliams, M.; Dutertre, C.A.; Scott, C.L.; McGovern, N.; Sichien, D.; Chakarov, S.; Van Gassen, S.; Chen, J.; Poidinger, M.; De Prijck, S.; et al. Unsupervised High-Dimensional Analysis Aligns Dendritic Cells across Tissues and Species. Immunity 2016, 45, 669–684. [Google Scholar] [CrossRef] [Green Version]
- Merad, M.; Sathe, P.; Helft, J.; Miller, J.; Mortha, A. The Dendritic Cell Lineage: Ontogeny and Function of Dendritic Cells and Their Subsets in the Steady State and the Inflamed Setting. Annu. Rev. Immunol. 2013, 31, 563–604. [Google Scholar] [CrossRef] [Green Version]
- Mildner, A.; Jung, S. Development and Function of Dendritic Cell Subsets. Immunity 2014, 40, 642–656. [Google Scholar] [CrossRef] [Green Version]
- Murphy, T.L.; Grajales-Reyes, G.E.; Wu, X.; Tussiwand, R.; Briseño, C.G.; Iwata, A.; Kretzer, N.M.; Durai, V.; Murphy, K.M. Transcriptional Control of Dendritic Cell Development. Annu. Rev. Immunol. 2016, 34, 93–119. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Akira, S. The Role of Pattern-Recognition Receptors in Innate Immunity: Update on Toll-Like Receptors. Nat. Immunol. 2010, 11, 373. [Google Scholar] [CrossRef]
- Iwasaki, A.; Medzhitov, R. Control of Adaptive Immunity by the Innate Immune System. Nat. Immunol. 2015, 16, 343–353. [Google Scholar] [CrossRef]
- Dantzer, R.; O’Connor, J.C.; Freund, G.G.; Johnson, R.W.; Kelley, K.W. From Inflammation to Sickness and Depression: When the Immune System Subjugates the Brain. Nat. Rev. Neurosci. 2008, 9, 46–56. [Google Scholar] [CrossRef] [Green Version]
- DellaGioia, N.; Hannestad, J. A Critical Review of Human Endotoxin Administration as an Experimental Paradigm of Depression. Neurosci. Biobehav. Rev. 2010, 34, 130–143. [Google Scholar] [CrossRef] [Green Version]
- Schedlowski, M.; Engler, H.; Grigoleit, J.-S. Endotoxin-Induced Experimental Systemic Inflammation in Humans: A Model to Disentangle Immune-to-Brain Communication. Brain Behav. Immun. 2014, 35, 1–8. [Google Scholar] [CrossRef]
- Hemmi, H.; Akira, S. TLR signalling and the function of dendritic cells. Chem. Immunol. Allergy 2005, 86, 120–135. [Google Scholar]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. Damp-Sensing Receptors in Sterile Inflammation and Inflammatory Diseases. Nat. Rev. Immunol. 2020, 20, 95–112. [Google Scholar] [CrossRef] [PubMed]
- Fucikova, J.; Palova-Jelinkova, L.; Bartunkova, J.; Spisek, R. Induction of Tolerance and Immunity by Dendritic Cells: Mechanisms and Clinical Applications. Front. Immunol. 2019, 10, 2393. [Google Scholar] [CrossRef]
- Chen, G.Y.; Nuñez, G. Sterile Inflammation: Sensing and Reacting to Damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franklin, T.C.; Xu, C.; Duman, R.S. Depression and Sterile Inflammation: Essential Role of Danger Associated Molecular Patterns. Brain Behav. Immun. 2018, 72, 2–13. [Google Scholar] [CrossRef]
- Iwata, M.; Ota, K.T.; Li, X.-Y.; Sakaue, F.; Li, N.; Dutheil, S.; Banasr, M.; Duric, V.; Yamanashi, T.; Kaneko, K.; et al. Psychological Stress Activates the Inflammasome Via Release of Adenosine Triphosphate and Stimulation of the Purinergic Type 2 × 7 Receptor. Biol. Psychiatry 2016, 80, 12–22. [Google Scholar] [CrossRef]
- Kaufmann, F.N.; Costa, A.P.; Ghisleni, G.; Diaz, A.P.; Rodrigues, A.L.S.; Peluffo, H.; Kaster, M.P. Nlrp3 Inflammasome-Driven Pathways in Depression: Clinical and Preclinical Findings. Brain Behav. Immun. 2017, 64, 367–383. [Google Scholar] [CrossRef]
- Netea, M.G.; Quintin, J.; Van Der Meer, J.W.M. Trained Immunity: A Memory for Innate Host Defense. Cell Host Microbe 2011, 9, 355–361. [Google Scholar] [CrossRef] [Green Version]
- Netea, M.G.; Domínguez-Andrés, J.; Barreiro, L.B.; Chavakis, T.; Divangahi, M.; Fuchs, E.; Joosten, L.A.B.; Van Der Meer, J.W.M.; Mhlanga, M.M.; Mulder, W.J.M. Defining Trained Immunity and Its Role in Health and Disease. Nat. Rev. Immunol. 2020, 20, 375–388. [Google Scholar] [CrossRef] [Green Version]
- Horowitz, M.A.; Zunszain, P.A. Neuroimmune and Neuroendocrine Abnormalities in Depression: Two Sides of the Same Coin. Ann. N. Y. Acad. Sci. 2015, 1351, 68–79. [Google Scholar] [CrossRef] [PubMed]
- Colbert, J.D.; Cruz, F.M.; Rock, K.L. Cross-Presentation of Exogenous Antigens on Mhc I Molecules. Curr. Opin. Immunol. 2020, 64, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kurts, C.; Cannarile, M.; Klebba, I.; Brocker, T. Cutting Edge: Dendritic Cells Are Sufficient to Cross-Present Self-Antigens to Cd8 T Cells in Vivo. J. Immunol. 2001, 166, 1439–1442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neefjes, J.; Jongsma, M.L.M.; Paul, P.; Bakke, O. Towards a Systems Understanding of Mhc Class I and Mhc Class II Antigen Presentation. Nat. Rev. Immunol. 2011, 11, 823–836. [Google Scholar] [CrossRef]
- Mellor, A.L.; Munn, D.H. Ido Expression by Dendritic Cells: Tolerance and Tryptophan Catabolism. Nat. Rev. Immunol. 2004, 4, 762–774. [Google Scholar] [CrossRef]
- Worthen, R.J.; Zighelboim, S.S.G.; Jaramillo, C.S.T.; Beurel, E. Anti-Inflammatory Il-10 Administration Rescues Depression-Associated Learning and Memory Deficits in Mice. J. Neuroinflamm. 2020, 17, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Roque, S.; Correia-Neves, M.; Mesquita, A.R.; Palha, J.A.; Sousa, N. Interleukin-10: A Key Cytokine in Depression? Cardiovasc. Psychiatry Neurol. 2009, 2009, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grosse, L.; Hoogenboezem, T.; Ambrée, O.; Bellingrath, S.; Jörgens, S.; De Wit, H.J.; Wijkhuijs, A.M.; Arolt, V.; Drexhage, H.A. Deficiencies of the T and Natural Killer Cell System in Major Depressive Disorder: T Regulatory Cell Defects Are Associated with Inflammatory Monocyte Activation. Brain Behav. Immun. 2016, 54, 38–44. [Google Scholar] [CrossRef]
- O’Connor, J.C.; Lawson, M.A.; Andre, C.; Moreau, M.; Lestage, J.; Castanon, N.; Kelley, K.W.; Dantzer, R. Lipopolysaccharide-Induced Depressive-Like Behavior Is Mediated by Indoleamine 2, 3-Dioxygenase Activation in Mice. Mol. Psychiatry 2009, 14, 511–522. [Google Scholar] [CrossRef] [Green Version]
- Segura, E. Review of Mouse and Human Dendritic Cell Subsets. Methods Mol. Biol. 2016, 1423, 3–15. [Google Scholar]
- Collin, M.; Bigley, V. Human Dendritic Cell Subsets: An Update. Immunology 2018, 154, 3–20. [Google Scholar] [CrossRef]
- Reizis, B. Plasmacytoid Dendritic Cells: Development, Regulation, and Function. Immunity 2019, 50, 37–50. [Google Scholar] [CrossRef] [Green Version]
- Ali, S.; Mann-Nüttel, R.; Schulze, A.; Richter, L.; Alferink, J.; Scheu, S. Sources of Type I Interferons in Infectious Immunity: Plasmacytoid Dendritic Cells Not Always in the Driver’s Seat. Front. Immunol. 2019, 10, 778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segura, E.; Coillard, A. In Vivo Differentiation of Human Monocytes. Front. Immunol. 2019, 10, 1907. [Google Scholar]
- Villar, J.; Segura, E. Recent Advances towards Deciphering Human Dendritic Cell Development. Mol. Immunol. 2020, 122, 109–115. [Google Scholar] [CrossRef]
- Hemmati, S.; Sadeghi, M.A.; Jafari, R.M.; Yousefi-Manesh, H.; Dehpour, A.R. The Antidepressant Effects of Gm-Csf Are Mediated by the Reduction of Tlr4/Nf-ĸb-Induced Ido Expression. J. Neuroinflamm. 2019, 16, 117. [Google Scholar] [CrossRef]
- Swiecki, M.; Colonna, M. The Multifaceted Biology of Plasmacytoid Dendritic Cells. Nat. Rev. Immunol. 2015, 15, 471–485. [Google Scholar] [CrossRef]
- Grajkowska, L.T.; Ceribelli, M.; Lau, C.M.; Warren, M.E.; Tiniakou, I.; Higa, S.N.; Bunin, A.; Haecker, H.; Mirny, L.A.; Staudt, L.M. Isoform-Specific Expression and Feedback Regulation of E Protein Tcf4 Control Dendritic Cell Lineage Specification. Immunity 2017, 46, 65–77. [Google Scholar] [CrossRef] [Green Version]
- Musumeci, A.; Lutz, K.; Winheim, E.; Krug, A.B. What Makes a Pdc: Recent Advances in Understanding Plasmacytoid Dc Development and Heterogeneity. Front. Immunol. 2019, 10, 1222. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Fu, T.; Yin, R.; Zhang, Q.; Shen, B. Prevalence of Depression and Anxiety in Systemic Lupus Erythematosus: A Systematic Review and Meta-Analysis. BMC Psychiatry 2017, 17, 70. [Google Scholar] [CrossRef] [Green Version]
- Cella, M.; Jarrossay, D.; Facchetti, F.; Alebardi, O.; Nakajima, H.; Lanzavecchia, A.; Colonna, M. Plasmacytoid Monocytes Migrate to Inflamed Lymph Nodes and Produce Large Amounts of Type I Interferon. Nat. Med. 1999, 5, 919–923. [Google Scholar] [CrossRef] [PubMed]
- Siegal, F.P.; Kadowaki, N.; Shodell, M.; Fitzgerald-Bocarsly, P.A.; Shah, K.; Ho, S.; Antonenko, S.; Liu, Y.-J. The Nature of the Principal Type 1 Interferon-Producing Cells in Human Blood. Science 1999, 284, 1835–1837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, J.; Dress, R.J.; Schulze, A.; Dresing, P.; Ali, S.; Deenen, R.; Alferink, J.; Scheu, S. Cutting Edge: Ifn-Β Expression in the Spleen Is Restricted to a Subpopulation of Plasmacytoid Dendritic Cells Exhibiting a Specific Immune Modulatory Transcriptome Signature. J. Immunol. 2016, 196, 4447–4451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbas, A.; Manh, T.-P.V.; Valente, M.; Collinet, N.; Attaf, N.; Dong, C.; Naciri, K.; Chelbi, R.; Brelurut, G.; Cervera-Marzal, I. The Activation Trajectory of Plasmacytoid Dendritic Cells in Vivo during a Viral Infection. Nat. Immunol. 2020, 21, 983–997. [Google Scholar] [CrossRef]
- Zhang, H.; Gregorio, J.D.; Iwahori, T.; Zhang, X.; Choi, O.; Tolentino, L.L.; Prestwood, T.; Carmi, Y.; Engleman, E.G. A Distinct Subset of Plasmacytoid Dendritic Cells Induces Activation and Differentiation of B and T Lymphocytes. Proc. Natl. Acad. Sci. USA 2017, 114, 1988–1993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amon, L.; Lehmann, C.H.; Heger, L.; Heidkamp, G.F.; Dudziak, D. The Ontogenetic Path of Human Dendritic Cells. Mol. Immunol. 2020, 120, 122–129. [Google Scholar] [CrossRef]
- Anderson, D.A.; Dutertre, C.-A.; Ginhoux, F.; Murphy, K.M. Genetic Models of Human and Mouse Dendritic Cell Development and Function. Nat. Rev. Immunol. 2020, 21, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic Cells in Cancer Immunology and Immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef]
- Eisenbarth, S. Dendritic Cell Subsets in T Cell Programming: Location Dictates Function. Nat. Rev. Immunol. 2019, 19, 89–103. [Google Scholar] [CrossRef]
- Hildner, K.; Edelson, B.T.; Purtha, W.E.; Diamond, M.; Matsushita, H.; Kohyama, M.; Calderon, B.; Schraml, B.U.; Unanue, E.R.; Diamond, M.S. Batf3 Deficiency Reveals a Critical Role for Cd8α+ Dendritic Cells in Cytotoxic T Cell Immunity. Science 2008, 322, 1097–1100. [Google Scholar] [CrossRef] [Green Version]
- Joffre, O.P.; Segura, E.; Savina, A.; Amigorena, S. Cross-Presentation by Dendritic Cells. Nat. Rev. Immunol. 2012, 12, 557–569. [Google Scholar] [CrossRef] [PubMed]
- Wichers, M.C.; Koek, G.H.; Robaeys, G.; Verkerk, R.; Scharpe, S.; Maes, M. Ido and Interferon-A-Induced Depressive Symptoms: A Shift in Hypothesis from Tryptophan Depletion to Neurotoxicity. Mol. Psychiatry 2005, 10, 538–544. [Google Scholar] [CrossRef] [Green Version]
- Leventhal, D.S.; Gilmore, D.C.; Berger, J.M.; Nishi, S.; Lee, V.; Malchow, S.; Kline, D.E.; Kline, J.; Vander Griend, D.J.; Huang, H.; et al. Dendritic Cells Coordinate the Development and Homeostasis of Organ-Specific Regulatory T Cells. Immunity 2016, 44, 847–859. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Jeong, Y.; Ashraf, M.U.; Bae, Y.-S. Dendritic Cell-Mediated Th2 Immunity and Immune Disorders. Int. J. Mol. Sci. 2019, 20, 2159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balan, S.; Saxena, M.; Bhardwaj, N. International Review of Cell and Molecular Biology, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 1–68. [Google Scholar]
- Korn, T.; Bettelli, E.; Oukka, M.; Kuchroo, V.K. Il-17 and Th17 Cells. Annu. Rev. Immunol. 2009, 27, 485–517. [Google Scholar] [CrossRef] [PubMed]
- Burkett, P.R.; Meyer zu Horste, G.; Kuchroo, V.K. Pouring Fuel on the Fire: Th17 Cells, the Environment, and Autoimmunity. J. Clin. Investig. 2015, 125, 2211–2219. [Google Scholar] [CrossRef]
- Beurel, E.; Lowell, J.A. Th17 Cells in Depression. Brain Behav. Immun. 2018, 69, 28–34. [Google Scholar] [CrossRef]
- Poulin, L.F.; Reyal, Y.; Uronen-Hansson, H.; Schraml, B.U.; Sancho, D.; Murphy, K.M.; Håkansson, U.K.; Ferreira Moita, L.; Agace, W.W.; Bonnet, D. Dngr-1 Is a Specific and Universal Marker of Mouse and Human Batf3-Dependent Dendritic Cells in Lymphoid and Nonlymphoid Tissues. Blood 2012, 119, 6052–6062. [Google Scholar] [CrossRef]
- Dorner, B.G.; Dorner, M.B.; Zhou, X.; Opitz, C.; Mora, A.; Güttler, S.; Hutloff, A.; Mages, H.W.; Ranke, K.; Schaefer, M.; et al. Selective Expression of the Chemokine Receptor Xcr1 on Cross-Presenting Dendritic Cells Determines Cooperation with Cd8+ T Cells. Immunity 2009, 31, 823–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang-Huau, T.-L.; Segura, E. Seminars in Cell & Developmental Biology, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2019; Volume 86, pp. 44–49. [Google Scholar]
- Pruenster, M.; Vogl, T.; Roth, J.; Sperandio, M. S100a8/A9: From Basic Science to Clinical Application. Pharmacol. Ther. 2016, 167, 120–131. [Google Scholar] [CrossRef]
- Briseño, C.G.; Haldar, M.; Kretzer, N.M.; Wu, X.; Theisen, D.J.; Kc, W.; Durai, V.; Grajales-Reyes, G.E.; Iwata, A.; Bagadia, P.; et al. Distinct Transcriptional Programs Control Cross-Priming in Classical and Monocyte-Derived Dendritic Cells. Cell Rep. 2016, 15, 2462–2474. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, P.F.; Neale, M.C.; Kendler, K.S. Genetic Epidemiology of Major Depression: Review and Meta-Analysis. Am. J. Psychiatry 2000, 157, 1552–1562. [Google Scholar] [CrossRef]
- Howard, D.M.; Adams, M.J.; Clarke, T.-K.; Hafferty, J.D.; Gibson, J.; Shirali, M.; Coleman, J.R.I.; Hagenaars, S.P.; Ward, J.; Wigmore, E.M. Genome-Wide Meta-Analysis of Depression Identifies 102 Independent Variants and Highlights the Importance of the Prefrontal Brain Regions. Nat. Neurosci. 2019, 22, 343–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lokki, M.L.; Paakkanen, R. The Complexity and Diversity of Major Histocompatibility Complex Challenge Disease Association Studies. Hla 2019, 93, 3–15. [Google Scholar]
- De Bakker, P.I.; McVean, G.; Sabeti, P.C.; Miretti, M.M.; Green, T.; Marchini, J.; Ke, X.; Monsuur, A.J.; Whittaker, P.; Delgado, M.; et al. A High-Resolution Hla and Snp Haplotype Map for Disease Association Studies in the Extended Human Mhc. Nat Genet 2006, 38, 1166–1172. [Google Scholar] [CrossRef]
- Glanville, K.P.; Coleman, J.R.I.; Hanscombe, K.B.; Euesden, J.; Choi, S.W.; Purves, K.L.; Breen, G.; Air, T.M.; Andlauer, T.F.M.; Baune, B.T.; et al. Classical Human Leukocyte Antigen Alleles and C4 Haplotypes Are Not Significantly Associated with Depression. Biol. Psychiatry 2020, 87, 419–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wray, N.R.; Ripke, S.; Mattheisen, M.; Trzaskowski, M.; Byrne, E.M.; Abdellaoui, A.; Adams, M.J.; Agerbo, E.; Air, T.M.; Andlauer, T.M.F. Genome-Wide Association Analyses Identify 44 Risk Variants and Refine the Genetic Architecture of Major Depression. Nat. Genet. 2018, 50, 668–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullins, N.; Forstner, A.J.; O’Connell, K.S.; Coombes, B.; Coleman, J.R.I.; Qiao, Z.; Als, T.D.; Bigdeli, T.B.; Børte, S.; Bryois, J. Genome-Wide Association Study of over 40,000 Bipolar Disorder Cases Provides Novel Biological Insights. medRxiv 2020, 1, 1–30. [Google Scholar]
- Finucane, H.K.; Bulik-Sullivan, B.; Gusev, A.; Trynka, G.; Reshef, Y.; Loh, P.R.; Anttila, V.; Xu, H.; Zang, C.; Farh, K.; et al. Partitioning Heritability by Functional Annotation Using Genome-Wide Association Summary Statistics. Nat. Genet. 2015, 47, 1228–1235. [Google Scholar] [CrossRef] [Green Version]
- Finucane, H.K.; Reshef, Y.A.; Anttila, V.; Slowikowski, K.; Gusev, A.; Byrnes, A.; Gazal, S.; Loh, P.R.; Lareau, C.; Shoresh, N.; et al. Heritability Enrichment of Specifically Expressed Genes Identifies Disease-Relevant Tissues and Cell Types. Nat. Genet. 2018, 50, 621–629. [Google Scholar] [CrossRef]
- Okbay, A.; Baselmans, B.M.; De Neve, J.E.; Turley, P.; Nivard, M.G.; Fontana, M.A.; Meddens, S.F.; Linnér, R.K.; Rietveld, C.A.; Derringer, J.; et al. Genetic Variants Associated with Subjective Well-Being, Depressive Symptoms, and Neuroticism Identified through Genome-Wide Analyses. Nat. Genet. 2016, 48, 624–633. [Google Scholar] [CrossRef] [Green Version]
- Ripke, S.M.; Neale, B.; Corvin, A.; Walters, J.T.R.; Holmans, P.A.; Lee, P.; Bulik-Sullivan, B.; Collier, D.A.; Huang, H.; Pers, T.H.; et al. Biological Insights from 108 Schizophrenia-Associated Genetic Loci. Nature 2014, 511, 421–427. [Google Scholar]
- Cisse, B.; Caton, M.L.; Lehner, M.; Maeda, T.; Scheu, S.; Locksley, R.; Holmberg, D.; Zweier, C.; Den Hollander, N.S.; Kant, S.G. Transcription Factor E2-2 Is an Essential and Specific Regulator of Plasmacytoid Dendritic Cell Development. Cell 2008, 135, 37–48. [Google Scholar] [CrossRef] [Green Version]
- Bhat, S.; Dao, D.T.; Terrillion, C.E.; Arad, M.; Smith, R.J.; Soldatov, N.M.; Gould, T.D. Cacna1c (Cav1. 2) in the Pathophysiology of Psychiatric Disease. Prog. Neurobiol. 2012, 99, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Vukcevic, M.; Spagnoli, G.C.; Iezzi, G.; Zorzato, F.; Treves, S. Ryanodine Receptor Activation by Cav1. 2 Is Involved in Dendritic Cell Major Histocompatibility Complex Class Ii Surface Expression. J. Biol. Chem. 2008, 283, 34913–34922. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Becker, J.; Bechheim, M.; Kaiser, V.; Noursadeghi, M.; Fricker, N.; Beier, E.; Klaschik, S.; Boor, P.; Hess, T.; et al. Characterizing the Genetic Basis of Innate Immune Response in Tlr4-Activated Human Monocytes. Nat. Commun. 2014, 5, 5236. [Google Scholar] [CrossRef]
- Schultze, J.L.; Aschenbrenner, A.C. Systems Immunology Allows a New View on Human Dendritic Cells. Semin. Cell Dev. Biol. 2019, 86, 15–23. [Google Scholar] [CrossRef]
- Tang, P.; Wang, J.M. Chemokines: The Past, the Present and the Future. Cell. Mol. Immunol. 2018, 15, 295–298. [Google Scholar] [CrossRef]
- Mackay, C.R. Chemokines: Immunology’s High Impact Factors. Nat. Immunol. 2001, 2, 95–101. [Google Scholar] [CrossRef] [PubMed]
- De Haas, A.H.; Van Weering, H.R.J.; De Jong, E.K.; Boddeke, H.; Biber, K.P.H. Neuronal Chemokines: Versatile Messengers in Central Nervous System Cell Interaction. Mol. Neurobiol. 2007, 36, 137–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, P.B.; Miller, R.J. Chemokine Receptors: Signposts to Brain Development and Disease. Nat. Rev. Neurosci. 2003, 4, 444–455. [Google Scholar] [CrossRef] [PubMed]
- Milenkovic, V.M.; Stanton, E.H.; Nothdurfter, C.; Rupprecht, R.; Wetzel, C.H. The Role of Chemokines in the Pathophysiology of Major Depressive Disorder. Int. J. Mol. Sci. 2019, 20, 2283. [Google Scholar] [CrossRef] [Green Version]
- Förster, R.; Davalos-Misslitz, A.C.; Rot, A. Ccr7 and Its Ligands: Balancing Immunity and Tolerance. Nat. Rev. Immunol. 2008, 8, 362–371. [Google Scholar] [CrossRef]
- Sánchez-Sánchez, N.; Riol-Blanco, L.; Rodríguez-Fernández, J.L. The Multiple Personalities of the Chemokine Receptor Ccr7 in Dendritic Cells. J. Immunol. 2006, 176, 5153–5159. [Google Scholar] [CrossRef] [PubMed]
- Noor, S.; Wilson, E.H. Role of C-C Chemokine Receptor Type 7 and Its Ligands during Neuroinflammation. J. Neuroinflamm. 2012, 9, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaehne, E.J.; Baune, B.T. Effects of Chemokine Receptor Signalling on Cognition-Like, Emotion-Like and Sociability Behaviours of Ccr6 and Ccr7 Knockout Mice. Behav. Brain Res. 2014, 261, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Ambree, O.; Klassen, I.; Forster, I.; Arolt, V.; Scheu, S.; Alferink, J. Reduced Locomotor Activity and Exploratory Behavior in Cc Chemokine Receptor 4 Deficient Mice. Behav. Brain Res. 2016, 314, 87–95. [Google Scholar] [CrossRef]
- Hellwig, S.; Brioschi, S.; Dieni, S.; Frings, L.; Masuch, A.; Blank, T.; Biber, K. Altered Microglia Morphology and Higher Resilience to Stress-Induced Depression-Like Behavior in Cx3cr1-Deficient Mice. Brain Behav. Immun. 2016, 55, 126–137. [Google Scholar] [CrossRef]
- Dichmann, S.; Herouy, Y.; Purlis, D.; Rheinen, H.; Gebicke-Härter, P.; Norgauer, J. Fractalkine Induces Chemotaxis and Actin Polymerization in Human Dendritic Cells. Inflamm. Res. 2001, 50, 529–533. [Google Scholar] [CrossRef]
- Drexhage, R.C.; Hoogenboezem, T.H.; Versnel, M.A.; Berghout, A.; Nolen, W.A.; Drexhage, H.A. The Activation of Monocyte and T Cell Networks in Patients with Bipolar Disorder. Brain Behav. Immun. 2011, 25, 1206–1213. [Google Scholar] [CrossRef]
- Leighton, S.P.; Nerurkar, L.; Krishnadas, R.; Johnman, C.; Graham, G.J.; Cavanagh, J. Chemokines in Depression in Health and in Inflammatory Illness: A Systematic Review and Meta-Analysis. Mol. Psychiatry 2018, 23, 48–58. [Google Scholar] [CrossRef] [Green Version]
- Stuart, M.J.; Singhal, G.; Baune, B.T. Systematic Review of the Neurobiological Relevance of Chemokines to Psychiatric Disorders. Front. Cell. Neurosci. 2015, 9, 357. [Google Scholar] [CrossRef]
- Sozzani, S.; Allavena, P.; Mantovani, A. Dendritic Cells: Biology and Clinical Applications, 2nd ed.; Academic Press: Sa Diego, CA, USA, 2001; pp. 203–211. [Google Scholar]
- Russo, R.C.; Garcia, C.C.; Teixeira, M.M.; Amaral, F.A. The Cxcl8/Il-8 Chemokine Family and Its Receptors in Inflammatory Diseases. Expert Rev. Clin. Immunol. 2014, 10, 593–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.Y.S.; Körner, H. The Ccr6-Ccl20 Axis in Humoral Immunity and Tb Cell Immunobiology. Immunobiology 2019, 224, 449–454. [Google Scholar] [CrossRef]
- Ranasinghe, R.; Eri, R. Pleiotropic Immune Functions of Chemokine Receptor 6 in Health and Disease. Medicines 2018, 5, 69. [Google Scholar] [CrossRef] [Green Version]
- Kabashima, K.; Shiraishi, N.; Sugita, K.; Mori, T.; Onoue, A.; Kobayashi, M.; Sakabe, J.; Yoshiki, R.; Tamamura, H.; Fujii, N.; et al. Cxcl12-Cxcr4 Engagement Is Required for Migration of Cutaneous Dendritic Cells. Am. J. Pathol. 2007, 171, 1249–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiberio, L.; Del Prete, A.; Schioppa, T.; Sozio, F.; Bosisio, D.; Sozzani, S. Chemokine and Chemotactic Signals in Dendritic Cell Migration. Cell. Mol. Immunol. 2018, 15, 346–352. [Google Scholar] [CrossRef]
- Guyon, A. Cxcl12 Chemokine and Its Receptors as Major Players in the Interactions between Immune and Nervous Systems. Front. Cell. Neurosci. 2014, 8, 65. [Google Scholar] [CrossRef] [Green Version]
- Guyon, A. Cxcl12 Chemokine and Gaba Neurotransmitter Systems Crosstalk and Their Putative Roles. Front. Cell. Neurosci. 2014, 5, 115. [Google Scholar] [CrossRef] [Green Version]
- Stuart, M.J.; Baune, B.T. Chemokines and Chemokine Receptors in Mood Disorders, Schizophrenia, and Cognitive Impairment: A Systematic Review of Biomarker Studies. Neurosci. Biobehav. Rev. 2014, 42, 93–115. [Google Scholar] [CrossRef]
- Heinisch, S.; Kirby, L.G. Sdf-1α/Cxcl12 Enhances Gaba and Glutamate Synaptic Activity at Serotonin Neurons in the Rat Dorsal Raphe Nucleus. Neuropharmacology 2010, 58, 501–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Peng, H.; Cui, M.; Whitney, N.P.; Huang, Y.; Zheng, J.C. Cxcl12 Increases Human Neural Progenitor Cell Proliferation through Akt-1/Foxo3a Signaling Pathway. J. Neurochem. 2009, 109, 1157–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.; Lee, Y.; Song, J.; Lee, J.; Chang, S.-Y. Tissue-Specific Role of Cx3cr1 Expressing Immune Cells and Their Relationships with Human Disease. Immune Netw. 2018, 18, 1–19. [Google Scholar] [CrossRef]
- Singhal, G.; Baune, B.T. Inflammation and Immunity in Depression, 1st ed.; Academic Press: San Diego, CA, USA, 2018; pp. 135–159. [Google Scholar]
- Ness, T.L.; Ewing, J.L.; Hogaboam, C.M.; Kunkel, S.L. Ccr4 Is a Key Modulator of Innate Immune Responses. J. Immunol. 2006, 177, 7531–7539. [Google Scholar] [CrossRef] [Green Version]
- Yoshie, O.; Matsushima, K. Ccr4 and Its Ligands: From Bench to Bedside. Int. Immunol. 2015, 27, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Alferink, J.; Lieberam, I.; Reindl, W.; Behrens, A.; Weiss, S.; Huser, N.; Gerauer, K.; Ross, R.; Reske-Kunz, A.B.; Ahmad-Nejad, P.; et al. Compartmentalized Production of Ccl17 in Vivo: Strong Inducibility in Peripheral Dendritic Cells Contrasts Selective Absence from the Spleen. J. Exp. Med. 2003, 197, 585–599. [Google Scholar] [CrossRef] [Green Version]
- Poppensieker, K.; Otte, D.M.; Schurmann, B.; Limmer, A.; Dresing, P.; Drews, E.; Schumak, B.; Klotz, L.; Raasch, J.; Mildner, A.; et al. Cc Chemokine Receptor 4 Is Required for Experimental Autoimmune Encephalomyelitis by Regulating Gm-Csf and Il-23 Production in Dendritic Cells. Proc. Natl. Acad. Sci. USA 2012, 109, 3897–3902. [Google Scholar] [CrossRef] [Green Version]
- Ruland, C.; Renken, H.; Kuzmanov, I.; Fattahi Mehr, A.; Schwarte, K.; Cerina, M.; Herrmann, A.; Otte, D.M.; Zimmer, A.; Schwab, N.; et al. Chemokine Ccl17 Is Expressed by Dendritic Cells in the Cns During Experimental Autoimmune Encephalomyelitis and Promotes Pathogenesis of Disease. Brain Behav. Immun. 2017, 66, 382–393. [Google Scholar] [CrossRef]
- Fülle, L.; Offermann, N.; Hansen, J.N.; Breithausen, B.; Erazo, A.B.; Schanz, O.; Radau, L.; Gondorf, F.; Knöpper, K.; Alferink, J. Ccl17 Exerts a Neuroimmune Modulatory Function and Is Expressed in Hippocampal Neurons. Glia 2018, 66, 2246–2261. [Google Scholar] [CrossRef] [PubMed]
- Milenkovic, V.M.; Sarubin, N.; Hilbert, S.; Baghai, T.C.; Stöffler, F.; Lima-Ojeda, J.M.; Manook, A.; Almeqbaali, K.; Wetzel, C.H.; Rupprecht, R.; et al. Macrophage-Derived Chemokine: A Putative Marker of Pharmacological Therapy Response in Major Depression? Neuroimmunomodulation 2017, 24, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Herz, J.; Filiano, A.J.; Smith, A.; Yogev, N.; Kipnis, J. Myeloid Cells in the Central Nervous System. Immunity 2017, 46, 943–956. [Google Scholar] [CrossRef] [Green Version]
- Ransohoff, R.M.; Brown, M.A. Innate Immunity in the Central Nervous System. J. Clin. Investig. 2012, 122, 1164–1171. [Google Scholar] [CrossRef]
- Singhal, G.; Baune, B.T. Microglia: An Interface between the Loss of Neuroplasticity and Depression. Front. Cell. Neurosci. 2017, 11, 270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tremblay, M.; Lowery, R.L.; Majewska, A.K. Microglial Interactions with Synapses Are Modulated by Visual Experience. PLoS Biol. 2010, 8, e1000527. [Google Scholar] [CrossRef] [Green Version]
- Eyre, H.; Baune, B.T. Neuroplastic Changes in Depression: A Role for the Immune System. Psychoneuroendocrinology 2012, 37, 1397–1416. [Google Scholar] [CrossRef]
- Setiawan, E.; Attwells, S.; Wilson, A.A.; Mizrahi, R.; Rusjan, P.M.; Miler, L.; Xu, C.; Sharma, S.; Kish, S.; Houle, S.; et al. Association of Translocator Protein Total Distribution Volume with Duration of Untreated Major Depressive Disorder: A Cross-Sectional Study. Lancet Psychiatry 2018, 5, 339–347. [Google Scholar] [CrossRef]
- Wohleb, E.S.; McKim, D.B.; Sheridan, J.F.; Godbout, J.P. Monocyte Trafficking to the Brain with Stress and Inflammation: A Novel Axis of Immune-to-Brain Communication That Influences Mood and Behavior. Front. Neurosci. 2014, 8, 447. [Google Scholar] [CrossRef]
- Bowley, M.P.; Drevets, W.C.; Ongür, D.; Price, J.L. Low Glial Numbers in the Amygdala in Major Depressive Disorder. Biol. Psychiatry 2002, 52, 404–412. [Google Scholar] [CrossRef]
- Yirmiya, R.; Rimmerman, N.; Reshef, R. Depression as a Microglial Disease. Trends Neurosci. 2015, 38, 637–658. [Google Scholar] [CrossRef]
- Böttcher, C.; Fernández-Zapata, C.; Snijders, G.J.L.; Schlickeiser, S.; Sneeboer, M.A.M.; Kunkel, D.; De Witte, L.D.; Priller, J. Single-Cell Mass Cytometry of Microglia in Major Depressive Disorder Reveals a Non-Inflammatory Phenotype with Increased Homeostatic Marker Expression. Transl. Psychiatry 2020, 10, 310. [Google Scholar] [CrossRef] [PubMed]
- Morante-Palacios, O.; Fondelli, F.; Ballestar, E.; Martínez-Cáceres, E.M. Tolerogenic Dendritic Cells in Autoimmunity and Inflammatory Diseases. Trends Immunol. 2021, 42, 59–75. [Google Scholar] [CrossRef]
- Jha, M.K.; Lee, W.-H.; Suk, K. Functional Polarization of Neuroglia: Implications in Neuroinflammation and Neurological Disorders. Biochem. Pharmacol. 2016, 103, 1–16. [Google Scholar] [CrossRef]
- Hilligan, K.L.; Ronchese, F. Antigen Presentation by Dendritic Cells and Their Instruction of Cd4+ T Helper Cell Responses. Cell. Mol. Immunol. 2020, 17, 587–599. [Google Scholar] [CrossRef] [PubMed]
- Agalioti, T.; Villablanca, E.J.; Huber, S.; Gagliani, N. Th17Cell Plasticity: The Role of Dendritic Cells and Molecular Mechanisms. J. Autoimmun. 2018, 87, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Sakaguchi, N.; Asano, M.; Itoh, M.; Toda, M. Immunologic Self-Tolerance Maintained by Activated T Cells Expressing Il-2 Receptor Alpha-Chains (Cd25). Breakdown of a Single Mechanism of Self-Tolerance Causes Various Autoimmune Diseases. J. Immunol. 1995, 155, 1151–1164. [Google Scholar] [PubMed]
- Reber, S.O.; Siebler, P.H.; Donner, N.C.; Morton, J.T.; Smith, D.G.; Kopelman, J.M.; Lowe, K.R.; Wheeler, K.J.; Fox, J.H.; Hassell, J.E. Immunization with a Heat-Killed Preparation of the Environmental Bacterium Mycobacterium Vaccae Promotes Stress Resilience in Mice. Proc. Natl. Acad. Sci. USA 2016, 113, E3130–E3139. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.H. Depression and Immunity: A Role for T Cells? Brain Behav. Immun. 2010, 24, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, D.; Reber, S.O.; Botteron, C.; Barth, T.; Peterlik, D.; Uschold, N.; Männel, D.N.; Lechner, A. Chronic Psychosocial Stress Promotes Systemic Immune Activation and the Development of Inflammatory Th Cell Responses. Brain Behav. Immun. 2010, 24, 1097–1104. [Google Scholar] [CrossRef]
- Beurel, E.; Harrington, L.E.; Jope, R.S. Inflammatory T Helper 17 Cells Promote Depression-Like Behavior in Mice. Biol. Psychiatry 2013, 73, 622–630. [Google Scholar] [CrossRef] [Green Version]
- Hong, M.; Zheng, J.; Ding, Z.Y.; Chen, J.H.; Yu, L.; Niu, Y.; Hua, Y.Q.; Wang, L.L. Imbalance between Th17 and Treg Cells May Play an Important Role in the Development of Chronic Unpredictable Mild Stress-Induced Depression in Mice. Neuroimmunomodulation 2013, 20, 39–50. [Google Scholar] [CrossRef]
- Slyepchenko, A.; Maes, M.; Köhler, C.A.; Anderson, G.; Quevedo, J.; Alves, G.S.; Berk, M.; Fernandes, B.S.; Carvalho, A.F. T Helper 17 Cells May Drive Neuroprogression in Major Depressive Disorder: Proposal of an Integrative Model. Neurosci. Biobehav. Rev. 2016, 64, 83–100. [Google Scholar] [CrossRef]
- Patas, K.; Willing, A.; Demiralay, C.; Engler, J.B.; Lupu, A.; Ramien, C.; Schäfer, T.; Gach, C.; Stumm, L.; Chan, K. T Cell Phenotype and T Cell Receptor Repertoire in Patients with Major Depressive Disorder. Front. Immunol. 2018, 9, 291. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Xiao, B.; Qiu, W.; Yang, L.; Hu, B.; Tian, X.; Yang, H. Altered Expression of Cd4+Cd25+ Regulatory T Cells and Its 5-Ht1a Receptor in Patients with Major Depression Disorder. J. Affect. Disord. 2010, 124, 68–75. [Google Scholar] [CrossRef]
- Chen, Y.; Jiang, T.; Chen, P.; Ouyang, J.; Xu, G.; Zeng, Z.; Sun, Y. Emerging Tendency Towards Autoimmune Process in Major Depressive Patients: A Novel Insight from Th17 Cells. Psychiatry Res. 2011, 188, 224–230. [Google Scholar] [CrossRef]
- Becking, K.; Haarman, B.C.M.; Grosse, L.; Nolen, W.A.; Claes, S.; Arolt, V.; Schoevers, R.A.; Drexhage, H.A. The Circulating Levels of Cd4+ T Helper Cells Are Higher in Bipolar Disorder as Compared to Major Depressive Disorder. J. Neuroimmunol. 2018, 319, 28–36. [Google Scholar] [CrossRef]
- Schwarz, M.J.; Chiang, S.; Müller, N.; Ackenheil, M. T-Helper-1 and T-Helper-2 Responses in Psychiatric Disorders. Brain Behav. Immun. 2001, 15, 340–370. [Google Scholar] [CrossRef] [Green Version]
- Toben, C.; Baune, B.T. An Act of Balance between Adaptive and Maladaptive Immunity in Depression: A Role for T Lymphocytes. J. Neuroimmune Pharmacol. 2015, 10, 595–609. [Google Scholar] [CrossRef]
- Allan, S. Tailoring T-Helper-Cell Responses. Nat. Rev. Immunol. 2009, 9, 76. [Google Scholar] [CrossRef]
- Moser, M.; Murphy, K.M. Dendritic Cell Regulation of Th 1-Th 2 Development. Nat. Immunol. 2000, 1, 199–205. [Google Scholar] [CrossRef]
- Noble, A.; Thomas, M.J.; Kemeny, D.M. Early Th1/Th2 Cell Polarization in the Absence of Il-4 and Il-12: T Cell Receptor Signaling Regulates the Response to Cytokines in Cd4 and Cd8 T Cells. Eur. J. Immunol. 2001, 31, 2227–2235. [Google Scholar] [CrossRef]
- Lippens, C.; Duraes, F.V.; Dubrot, J.; Brighouse, D.; Lacroix, M.; Irla, M.; Aubry-Lachainaye, J.-P.; Reith, W.; Mandl, J.N.; Hugues, S. Ido-Orchestrated Crosstalk between Pdcs and Tregs Inhibits Autoimmunity. J. Autoimmun. 2016, 75, 39–49. [Google Scholar] [CrossRef]
- Nakano, R.; Yoshida, O.; Kimura, S.; Nakao, T.; Yokota, S.; Ono, Y.; Minervini, M.I.; Geller, D.A.; Thomson, A.W. Donor Plasmacytoid Dendritic Cells Modulate Effector and Regulatory T Cell Responses in Mouse Spontaneous Liver Transplant Tolerance. Am. J. Transplant. 2020. [Google Scholar] [CrossRef]
- Tkach, M.; Kowal, J.; Zucchetti, A.E.; Enserink, L.; Jouve, M.; Lankar, D.; Saitakis, M.; Martin-Jaular, L.; Théry, C. Qualitative Differences in T-Cell Activation by Dendritic Cell-Derived Extracellular Vesicle Subtypes. EMBO J. 2017, 36, 3012–3028. [Google Scholar] [CrossRef] [PubMed]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B.; Dingli, F.; Loew, D.; Tkach, M.; Théry, C. Proteomic Comparison Defines Novel Markers to Characterize Heterogeneous Populations of Extracellular Vesicle Subtypes. Proc. Natl. Acad. Sci. USA 2016, 113, E968–E977. [Google Scholar] [CrossRef] [Green Version]
- Eyre, H.; Siddarth, P.; Cyr, N.; Yang, H.; Cole, S.; Forbes, M.; Lavretsky, H. Comparing the Immune-Genomic Effects of Vilazodone and Paroxetine in Late-Life Depression: A Pilot Study. Pharmacopsychiatry 2017, 50, 256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hooli, B.; Tanzi, R.E. Genomics, Circuits, and Pathways in Clinical Neuropsychiatry, 1st ed.; Academic Press: San Diego, CA, USA, 2016; pp. 547–571. [Google Scholar]
- Sawcer, S.; Hellenthal, G.; Pirinen, M.; Spencer, C.C.; Patsopoulos, N.A.; Moutsianas, L.; Dilthey, A.; Su, Z.; Freeman, C.; Hunt, S.E.; et al. Genetic Risk and a Primary Role for Cell-Mediated Immune Mechanisms in Multiple Sclerosis. Nature 2011, 476, 214–219. [Google Scholar]
- Witoelar, A.; Jansen, I.E.; Wang, Y.; Desikan, R.S.; Gibbs, J.R.; Blauwendraat, C.; Thompson, W.K.; Hernandez, D.G.; Djurovic, S.; Schork, A.J.; et al. Genome-Wide Pleiotropy between Parkinson Disease and Autoimmune Diseases. JAMA Neurol. 2017, 74, 780–792. [Google Scholar] [CrossRef]
- Eyre, H.A.; Eskin, A.; Nelson, S.F.; St. Cyr, N.M.; Siddarth, P.; Baune, B.T.; Lavretsky, H. Genomic Predictors of Remission to Antidepressant Treatment in Geriatric Depression Using Genome-Wide Expression Analyses: A Pilot Study. Int. J. Geriatr. Psychiatry 2016, 31, 510–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katrinli, S.; Lori, A.; Kilaru, V.; Carter, S.; Powers, A.; Gillespie, C.F.; Wingo, A.P.; Michopoulos, V.; Jovanovic, T.; Ressler, K.J. Association of Hla Locus Alleles with Posttraumatic Stress Disorder. Brain Behav. Immun. 2019, 81, 655–658. [Google Scholar] [CrossRef] [PubMed]
- James, L.M.; Christova, P.; Engdahl, B.E.; Lewis, S.M.; Carpenter, A.F.; Georgopoulos, A.P. Human Leukocyte Antigen (Hla) and Gulf War Illness (Gwi): Hla-Drb1*13:02 Spares Subcortical Atrophy in Gulf War Veterans. EBioMedicine 2017, 26, 126–131. [Google Scholar] [CrossRef] [Green Version]
- Vroman, H.; Bergen, I.M.; Van Hulst, J.A.C.; Van Nimwegen, M.; Van Uden, D.; Schuijs, M.J.; Pillai, S.Y.; Van Loo, G.; Hammad, H.; Lambrecht, B.N. Tnf-A–Induced Protein 3 Levels in Lung Dendritic Cells Instruct Th2 or Th17 Cell Differentiation in Eosinophilic or Neutrophilic Asthma. J. Allergy Clin. Immunol. 2018, 141, 1620–1633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, T.-N.; Lee, C.-S.; Wu, B.-J.; Sun, H.-J.; Chang, C.-H.; Chen, C.-Y.; Chen, C.-K.; Wu, L.S.-H.; Cheng, A.T.-A. Immunophenotypes Associated with Bipolar Disorder and Lithium Treatment. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef]
- Knijff, E.M.; Ruwhof, C.; de Wit, H.J.; Kupka, R.W.; Vonk, R.; Akkerhuis, G.W.; Nolen, W.A.; Drexhage, H.A. Monocyte-Derived Dendritic Cells in Bipolar Disorder. Biol. Psychiatry 2006, 59, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Van Kaer, L.; Wu, L.; Joyce, S. Mechanisms and Consequences of Antigen Presentation by Cd1. Trends Immunol. 2016, 37, 738–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, S.-J.; Kim, J.W.; Kim, B.G.; Lee, K.L.; Im, J.P.; Kim, J.S. Fluoxetine Inhibits Hyperresponsive Lamina Propria Mononuclear Cells and Bone Marrow-Derived Dendritic Cells, and Ameliorates Chronic Colitis in Il-10-Deficient Mice. Dig. Dis. Sci. 2015, 60, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Branco-de-Almeida, L.S.; Kajiya, M.; Cardoso, C.R.; Silva, M.J.B.; Ohta, K.; Rosalen, P.L.; Franco, G.C.N.; Han, X.; Taubman, M.A.; Kawai, T. Selective Serotonin Reuptake Inhibitors Attenuate the Antigen Presentation from Dendritic Cells to Effector T Lymphocytes. FEMS Immunol. Med. Microbiol. 2011, 62, 283–294. [Google Scholar] [CrossRef]
- Cryan, J.F.; Mombereau, C. In Search of a Depressed Mouse: Utility of Models for Studying Depression-Related Behavior in Genetically Modified Mice. Mol. Psychiatry 2004, 9, 326–357. [Google Scholar] [CrossRef]
- Krishnan, V.; Nestler, E.J. Molecular and Functional Models in Neuropsychiatry, 7th ed.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 121–147. [Google Scholar]
- Blanchard, D.C.; Griebel, G.; Blanchard, R.J. Mouse Defensive Behaviors: Pharmacological and Behavioral Assays for Anxiety and Panic. Neurosci. Biobehav. Rev. 2001, 25, 205–218. [Google Scholar] [CrossRef]
- Ahmetspahic, D.; Brinker, D.; Alferink, J. Inflammation and Immunity in Depression, 1st ed.; Academic Press: San Diego, CA, USA, 2018; pp. 1–16. [Google Scholar]
- Weber, M.D.; Godbout, J.P.; Sheridan, J.F. Repeated Social Defeat, Neuroinflammation, and Behavior: Monocytes Carry the Signal. Neuropsychopharmacology 2017, 42, 46–61. [Google Scholar] [CrossRef] [Green Version]
- Ménard, C.; Pfau, M.L.; Hodes, G.E.; Russo, S.J. Immune and Neuroendocrine Mechanisms of Stress Vulnerability and Resilience. Neuropsychopharmacology 2017, 42, 62–80. [Google Scholar] [CrossRef] [Green Version]
- Castagné, V.; Moser, P.; Roux, S.; Porsolt, R.D. Rodent Models of Depression: Forced Swim and Tail Suspension Behavioral Despair Tests in Rats and Mice. Curr. Protoc. Neurosci. 2011, 55, 8–10. [Google Scholar] [CrossRef] [PubMed]
- Rygula, R.; Abumaria, N.; Flügge, G.; Fuchs, E.; Rüther, E.; Havemann-Reinecke, U. Anhedonia and Motivational Deficits in Rats: Impact of Chronic Social Stress. Behav. Brain Res. 2005, 162, 127–134. [Google Scholar] [CrossRef]
- Porsolt, R.D.; Brossard, G.; Hautbois, C.; Roux, S. Rodent Models of Depression: Forced Swimming and Tail Suspension Behavioral Despair Tests in Rats and Mice. Curr. Protoc. Neurosci. 2001, 14, 8–10. [Google Scholar] [CrossRef] [PubMed]
- Golden, S.A.; Covington Iii, H.E.; Berton, O.; Russo, S.J. A Standardized Protocol for Repeated Social Defeat Stress in Mice. Nat. Protoc. 2011, 6, 1183. [Google Scholar] [CrossRef]
- Nestler, E.J.; Hyman, S.E. Animal Models of Neuropsychiatric Disorders. Nat. Neurosci. 2010, 13, 1161–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slattery, D.A.; Cryan, J.F. Modelling Depression in Animals: At the Interface of Reward and Stress Pathways. Psychopharmacology 2017, 234, 1451–1465. [Google Scholar] [CrossRef]
- Muir, J.; Lopez, J.; Bagot, R.C. Wiring the Depressed Brain: Optogenetic and Chemogenetic Circuit Interrogation in Animal Models of Depression. Neuropsychopharmacology 2019, 44, 1013–1026. [Google Scholar] [CrossRef]
- Remus, J.L.; Dantzer, R. Inflammation Models of Depression in Rodents: Relevance to Psychotropic Drug Discovery. Int. J. Neuropsychopharmacol. 2016, 19, 9. [Google Scholar] [CrossRef] [PubMed]
- Capuron, L.; Miller, A.H. Immune System to Brain Signaling: Neuropsychopharmacological Implications. Pharmacol. Ther. 2011, 130, 226–238. [Google Scholar] [CrossRef] [Green Version]
- Goehler, L.E.; Gaykema, R.P.; Nguyen, K.T.; Lee, J.E.; Tilders, F.J.; Maier, S.F.; Watkins, L.R. Interleukin-1beta in Immune Cells of the Abdominal Vagus Nerve: A Link between the Immune and Nervous Systems? J. Neurosci. 1999, 19, 2799–2806. [Google Scholar] [CrossRef] [Green Version]
- Reichenberg, A.; Yirmiya, R.; Schuld, A.; Kraus, T.; Haack, M.; Morag, A.; Pollmächer, T. Cytokine-Associated Emotional and Cognitive Disturbances in Humans. Arch. Gen. Psychiatry 2001, 58, 445–452. [Google Scholar] [CrossRef]
- Grigoleit, J.-S.; Oberbeck, J.R.; Lichte, P.; Kobbe, P.; Wolf, O.T.; Montag, T.; Rey, A.D.; Gizewski, E.R.; Engler, H.; Schedlowski, M. Lipopolysaccharide-Induced Experimental Immune Activation Does Not Impair Memory Functions in Humans. Neurobiol. Learn. Mem. 2010, 94, 561–567. [Google Scholar] [CrossRef]
- Wright, C.E.; Strike, P.C.; Brydon, L.; Steptoe, A. Acute Inflammation and Negative Mood: Mediation by Cytokine Activation. Brain Behav. Immun. 2005, 19, 345–350. [Google Scholar] [CrossRef]
- Yirmiya, R. Endotoxin Produces a Depressive-Like Episode in Rats. Brain Res. 1996, 711, 163–174. [Google Scholar] [CrossRef]
- Leonard, B.; Maes, M. Mechanistic Explanations How Cell-Mediated Immune Activation, Inflammation and Oxidative and Nitrosative Stress Pathways and Their Sequels and Concomitants Play a Role in the Pathophysiology of Unipolar Depression. Neurosci. Biobehav. Rev. 2012, 36, 764–785. [Google Scholar] [CrossRef] [PubMed]
- Lasselin, J.; Schedlowski, M.; Karshikoff, B.; Engler, H.; Lekander, M.; Konsman, J.P. Neuroscience & Biobehavioral Reviews, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 15–24. [Google Scholar]
- Gibney, S.M.; McGuinness, B.; Prendergast, C.; Harkin, A.; Connor, T.J. Poly I:C-Induced Activation of the Immune Response Is Accompanied by Depression and Anxiety-Like Behaviours, Kynurenine Pathway Activation and Reduced Bdnf Expression. Brain Behav. Immun. 2013, 28, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Mayerhofer, R.; Fröhlich, E.E.; Reichmann, F.; Farzi, A.; Kogelnik, N.; Fröhlich, E.; Sattler, W.; Holzer, P. Diverse Action of Lipoteichoic Acid and Lipopolysaccharide on Neuroinflammation, Blood-Brain Barrier Disruption, and Anxiety in Mice. Brain Behav. Immun. 2017, 60, 174–187. [Google Scholar] [CrossRef] [Green Version]
- Takeda, K.; Akira, S. Tlr Signaling Pathways. Semin. Immunol. 2004, 16, 3–9. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Signaling to Nf-Kappab by Toll-Like Receptors. Trends Mol. Med. 2007, 13, 460–469. [Google Scholar] [CrossRef]
- Miller, A.H.; Maletic, V.; Raison, C.L. Inflammation and Its Discontents: The Role of Cytokines in the Pathophysiology of Major Depression. Biol. Psychiatry 2009, 65, 732–741. [Google Scholar] [CrossRef] [Green Version]
- Moon, Y.W.; Hajjar, J.; Hwu, P.; Naing, A. Targeting the Indoleamine 2,3-Dioxygenase Pathway in Cancer. J. Immunother. Cancer 2015, 3, 51. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Wu, T.; Shao, S.; Shi, B.; Zhao, Y. Phenotype, Development, and Biological Function of Myeloid-Derived Suppressor Cells. Oncoimmunology 2016, 5, e1004983. [Google Scholar] [CrossRef] [Green Version]
- Prendergast, G.C.; Malachowski, W.J.; Mondal, A.; Scherle, P.; Muller, A.J. Indoleamine 2,3-Dioxygenase and Its Therapeutic Inhibition in Cancer. Int. Rev. Cell Mol. Biol. 2018, 336, 175–203. [Google Scholar] [PubMed]
- Mellor, A.L.; Lemos, H.; Huang, L. Indoleamine 2,3-Dioxygenase and Tolerance: Where Are We Now? Front. Immunol. 2017, 8, 1360. [Google Scholar] [CrossRef] [PubMed]
- Hunt, C.; Macedo, E.C.T.; Suchting, R.; De Dios, C.; Cuellar Leal, V.A.; Soares, J.C.; Dantzer, R.; Teixeira, A.L.; Selvaraj, S. Effect of Immune Activation on the Kynurenine Pathway and Depression Symptoms-a Systematic Review and Meta-Analysis. Neurosci. Biobehav. Rev. 2020, 118, 514–523. [Google Scholar] [CrossRef]
- Raison, C.L.; Dantzer, R.; Kelley, K.W.; Lawson, M.A.; Woolwine, B.J.; Vogt, G.; Spivey, J.R.; Saito, K.; Miller, A.H. Csf Concentrations of Brain Tryptophan and Kynurenines During Immune Stimulation with Ifn-A: Relationship to Cns Immune Responses and Depression. Mol. Psychiatry 2010, 15, 393–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonaccorso, S.; Marino, V.; Puzella, A.; Pasquini, M.; Biondi, M.; Artini, M.; Almerighi, C.; Verkerk, R.; Meltzer, H.; Maes, M. Increased Depressive Ratings in Patients with Hepatitis C Receiving Interferon-A—Based Immunotherapy Are Related to Interferon-A–Induced Changes in the Serotonergic System. J. Clin. Psychopharmacol. 2002, 22, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.L.; Llenos, I.C.; Dulay, J.R.; Weis, S. Upregulation of the Initiating Step of the Kynurenine Pathway in Postmortem Anterior Cingulate Cortex from Individuals with Schizophrenia and Bipolar Disorder. Brain Res. 2006, 1073, 25–37. [Google Scholar] [CrossRef]
- Bartoli, F.; Misiak, B.; Callovini, T.; Cavaleri, D.; Cioni, R.M.; Crocamo, C.; Savitz, J.B.; Carrà, G. The Kynurenine Pathway in Bipolar Disorder: A Meta-Analysis on the Peripheral Blood Levels of Tryptophan and Related Metabolites. Mol. Psychiatry 2020, 1, 1–11. [Google Scholar] [CrossRef]
- Mellor, A.L.; Munn, D.H. Creating Immune Privilege: Active Local Suppression That Benefits Friends, but Protects Foes. Nat. Rev. Immunol. 2008, 8, 74–80. [Google Scholar] [CrossRef]
- Pryce, C.R.; Fontana, A. Depression in Autoimmune Diseases. Curr. Top. Behav. Neurosci. 2017, 31, 139–154. [Google Scholar]
- Hellmuth, J.; Colby, D.; Valcour, V.; Suttichom, D.; Spudich, S.; Ananworanich, J.; Prueksakaew, P.; Sailasuta, N.; Allen, I.; Jagodzinski, L.L. Depression and Anxiety Are Common in Acute Hiv Infection and Associate with Plasma Immune Activation. AIDS Behav. 2017, 21, 3238–3246. [Google Scholar] [CrossRef]
- Yang, H.; Xia, L.; Chen, J.; Zhang, S.; Martin, V.; Li, Q.; Lin, S.; Chen, J.; Calmette, J.; Lu, M. Stress–Glucocorticoid–Tsc22d3 Axis Compromises Therapy-Induced Antitumor Immunity. Nat. Med. 2019, 25, 1428–1441. [Google Scholar] [CrossRef] [PubMed]
- Powell, N.D.; Mays, J.W.; Bailey, M.T.; Hanke, M.L.; Sheridan, J.F. Immunogenic Dendritic Cells Primed by Social Defeat Enhance Adaptive Immunity to Influenza a Virus. Brain Behav. Immun. 2011, 25, 46–52. [Google Scholar] [CrossRef] [Green Version]
- Ambree, O.; Ruland, C.; Scheu, S.; Arolt, V.; Alferink, J. Alterations of the Innate Immune System in Susceptibility and Resilience after Social Defeat Stress. Front. Behav. Neurosci. 2018, 12, 141. [Google Scholar] [CrossRef]
- Powell, N.D.; Bailey, M.T.; Mays, J.W.; Stiner-Jones, L.M.; Hanke, M.L.; Padgett, D.A.; Sheridan, J.F. Repeated Social Defeat Activates Dendritic Cells and Enhances Toll-Like Receptor Dependent Cytokine Secretion. Brain Behav. Immun. 2009, 23, 225–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stark, J.L.; Avitsur, R.; Padgett, D.A.; Campbell, K.A.; Beck, F.M.; Sheridan, J.F. Social Stress Induces Glucocorticoid Resistance in Macrophages. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001, 280, R1799–R1805. [Google Scholar] [CrossRef]
- Ambree, O.; Ruland, C.; Zwanzger, P.; Klotz, L.; Baune, B.T.; Arolt, V.; Scheu, S.; Alferink, J. Social Defeat Modulates T Helper Cell Percentages in Stress Susceptible and Resilient Mice. Int. J. Mol. Sci. 2019, 20, 3512. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, Y.; Hayakawa, K.; Fujishiro, M.; Ikeda, K.; Tsushima, H.; Hirai, T.; Kawasaki, M.; Tominaga, M.; Suga, Y.; Takamori, K. Social Defeat Stress Exacerbates Atopic Dermatitis through Downregulation of DNA Methyltransferase 1 and Upregulation of C–C Motif Chemokine Receptor 7 in Skin Dendritic Cells. Biochem. Biophys. Res. Commun. 2020, 529, 1073–1079. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Kitaoka, S.; Kawano, Y.; Ishii, S.; Suzuki, T.; Wakahashi, K.; Kato, T.; Katayama, Y.; Furuyashiki, T. Repeated Social Defeat Stress Induces Neutrophil Mobilization in Mice: Maintenance after Cessation of Stress and Strain-Dependent Difference in Response. Br. J. Pharmacol. 2020, 178, 1–18. [Google Scholar]
- Krishnan, V.; Han, M.H.; Graham, D.L.; Berton, O.; Renthal, W.; Russo, S.J.; Laplant, Q.; Graham, A.; Lutter, M.; Lagace, D.C.; et al. Molecular Adaptations Underlying Susceptibility and Resistance to Social Defeat in Brain Reward Regions. Cell 2007, 131, 391–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watford, W.T.; Moriguchi, M.; Morinobu, A.; O’Shea, J.J. The Biology of Il-12: Coordinating Innate and Adaptive Immune Responses. Cytokine Growth Factor Rev. 2003, 14, 361–368. [Google Scholar] [CrossRef]
- Tait Wojno, E.D.; Hunter, C.A.; Stumhofer, J.S. The Immunobiology of the Interleukin-12 Family: Room for Discovery. Immunity 2019, 50, 851–870. [Google Scholar] [CrossRef]
- Dong, C.; Flavell, R.A. Cell Fate Decision: T-Helper 1 and 2 Subsets in Immune Responses. Arthritis Res. 2000, 2, 179–188. [Google Scholar] [CrossRef] [Green Version]
- Truckenmiller, M.E.; Bonneau, R.H.; Norbury, C.C. Stress Presents a Problem for Dendritic Cells: Corticosterone and the Fate of Mhc Class I Antigen Processing and Presentation. Brain Behav. Immun. 2006, 20, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Elftman, M.D.; Norbury, C.C.; Bonneau, R.H.; Truckenmiller, M.E. Corticosterone Impairs Dendritic Cell Maturation and Function. Immunology 2007, 122, 279–290. [Google Scholar] [CrossRef]
- Cao, Y.; Bender, I.K.; Konstantinidis, A.K.; Shin, S.C.; Jewell, C.M.; Cidlowski, J.A.; Schleimer, R.P.; Lu, N.Z. Glucocorticoid Receptor Translational Isoforms Underlie Maturational Stage-Specific Glucocorticoid Sensitivities of Dendritic Cells in Mice and Humans. Blood 2013, 121, 1553–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vétillard, M.; Schlecht-Louf, G. Glucocorticoid-Induced Leucine Zipper: Fine-Tuning of Dendritic Cells Function. Front. Immunol. 2018, 9, 1232. [Google Scholar] [CrossRef] [PubMed]
- Cohen, N.; Mouly, E.; Hamdi, H.; Maillot, M.C.; Pallardy, M.; Godot, V.; Capel, F.; Balian, A.; Naveau, S.; Galanaud, P.; et al. Gilz Expression in Human Dendritic Cells Redirects Their Maturation and Prevents Antigen-Specific T Lymphocyte Response. Blood 2006, 107, 2037–2044. [Google Scholar] [CrossRef] [PubMed]
- Mohan, K.N.; Chaillet, J.R. Cell and Molecular Biology of DNA Methyltransferase 1. Int. Rev. Cell Mol. Biol. 2013, 306, 1–42. [Google Scholar] [PubMed]


| Chemokine Receptor | Ligand | Function in DCs | Impact on Behavior | Reference |
| CCR4 | CCL17, CCL22 | Multiple functions including migration and secretion of GM-CSF and IL-23 | CCR4 knockout mice show reduced locomotor activity, less anxiety-related behavior, and diminished social exploration | [137] |
| CCR6 | CCL20 | Chemotaxis of DCs to inflammatory sites and the brain | CCR6 knockout mice show higher locomotor activity, lower anxiety, and reduced preference for saccharin (in weekly testing) | [136] |
| CCR7 | CCL19, CCL21 | Migration, differentiation, endocytosis, release of cytokines | CCR7 knockout mice show impaired learning (Barnes maze), higher anxiety, and reduced preference for saccharin (in weekly testing) | [136] |
| CX3CR1 | CX3CL1 | Induces e.g., actin polymerization and migration of DCs, independent of their maturation status | CX3CR1 knockout mice show increased resilience to stress-induced depression-like behavior | [138,139] |
| Ligand | Chemokine Receptor | Function in DCs | Clinical Studies | Reference |
| CCL2 | CCR2 | Migration, maturation, and production of IL-12 | Increased CCL2 serum levels in patients with affective disorders | [140] |
| CXCL8 | CXCR1, CXCR2 | Chemotaxis of immature DCs to inflammatory sites | Increased CXCL8 blood levels in depressed individuals | [141] |
| CXCL12 | CXCR4 | Migration of DCs from the skin into the regional lymph nodes | Reduced CXCL12 plasma levels in patients with non-affective psychosis | [142] |
| Animal Model | Duration | Tissues Analyzed | Alterations Found in DCs | Reference |
|---|---|---|---|---|
| SDR | 6 days | Spleen | Increased MHC I, CD80 and CD44 expression and glucocorticoid resistance ex vivo and IL-6 and TNF productionafter in vitro stimulation with LPS | [252] |
| SDR | 6 days | Spleen, lung | Enhanced maturation and capacity to induce antiviral T cell responses, adoptive transfer of splenic DCs from SDR exposed mice confers immunity towards influenza A virus, glucocorticoid resistance | [250] |
| SDS | 10 days | Spleen | Increased MHC II and CD80 expression by DCs of susceptible mice, higher IL-12+ DC proportions in resilient mice | [251,254] |
| SDS | 10 days | Spleen, LN, tumor | Upregulated TSC22D3 expression and reduced capability to produce type I IFN in tumor-infiltrating DCs after SDS and reduced capability to induce IFN-γ secretion in tumor-infiltrating T cells | [249] |
| SDS | 10 days | Skin | Downregulated DNMT1 and upregulated CCR7 expression in skin DCs, exacerbated experimentally-induced atopic dermatitis | [255] |
| SDR and SDS | 6 (SDR) and 10 (SDS) days | Spleen, blood, bone marrow | Reduced cDC1 and cDC2 cell percentages in bone marrow after SDR and SDS; reduced DC percentages in peripheral blood of subordinate animals after SDR | [256] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leite Dantas, R.; Freff, J.; Ambrée, O.; Beins, E.C.; Forstner, A.J.; Dannlowski, U.; Baune, B.T.; Scheu, S.; Alferink, J. Dendritic Cells: Neglected Modulators of Peripheral Immune Responses and Neuroinflammation in Mood Disorders? Cells 2021, 10, 941. https://doi.org/10.3390/cells10040941
Leite Dantas R, Freff J, Ambrée O, Beins EC, Forstner AJ, Dannlowski U, Baune BT, Scheu S, Alferink J. Dendritic Cells: Neglected Modulators of Peripheral Immune Responses and Neuroinflammation in Mood Disorders? Cells. 2021; 10(4):941. https://doi.org/10.3390/cells10040941
Chicago/Turabian StyleLeite Dantas, Rafael, Jana Freff, Oliver Ambrée, Eva C. Beins, Andreas J. Forstner, Udo Dannlowski, Bernhard T. Baune, Stefanie Scheu, and Judith Alferink. 2021. "Dendritic Cells: Neglected Modulators of Peripheral Immune Responses and Neuroinflammation in Mood Disorders?" Cells 10, no. 4: 941. https://doi.org/10.3390/cells10040941
APA StyleLeite Dantas, R., Freff, J., Ambrée, O., Beins, E. C., Forstner, A. J., Dannlowski, U., Baune, B. T., Scheu, S., & Alferink, J. (2021). Dendritic Cells: Neglected Modulators of Peripheral Immune Responses and Neuroinflammation in Mood Disorders? Cells, 10(4), 941. https://doi.org/10.3390/cells10040941